
Hierarchically Ordered Supramolecular Protein-Polymer Composites with Thermoresponsive Properties

Abstract
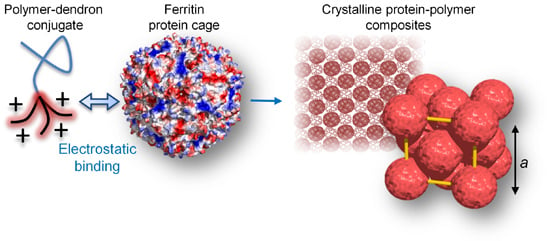
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
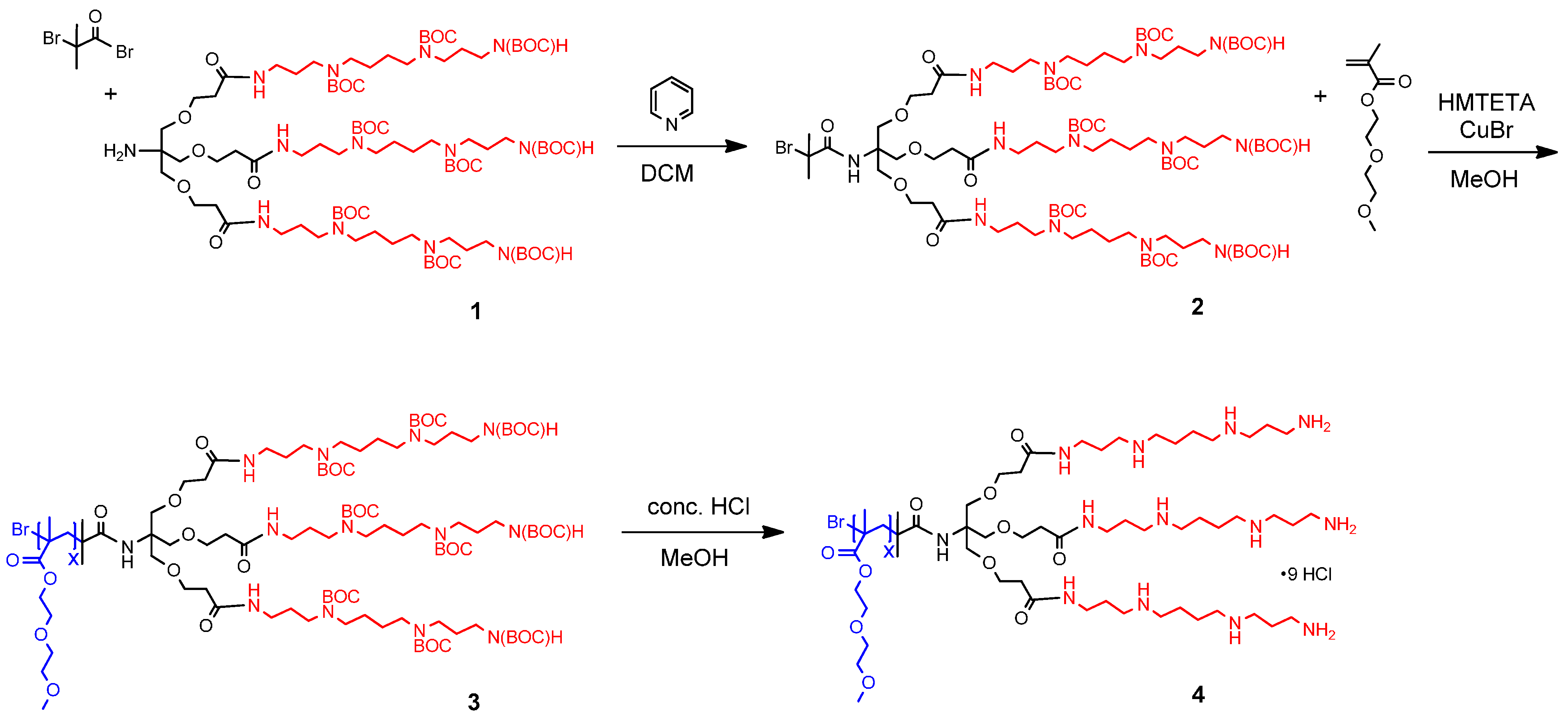
2.1. Synthesis

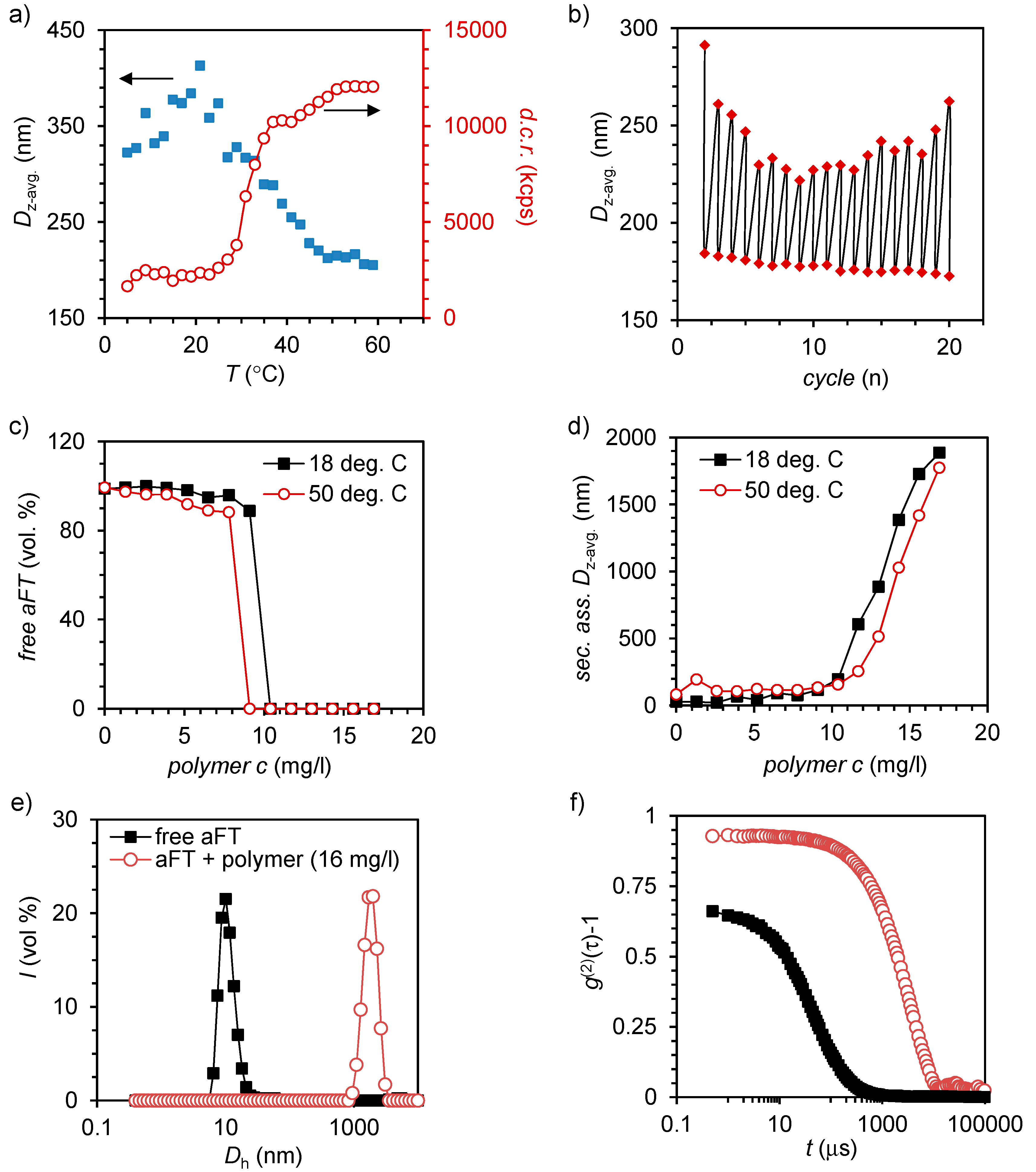
2.2. Self-Assembly of Large Protein-Polymer Complexes

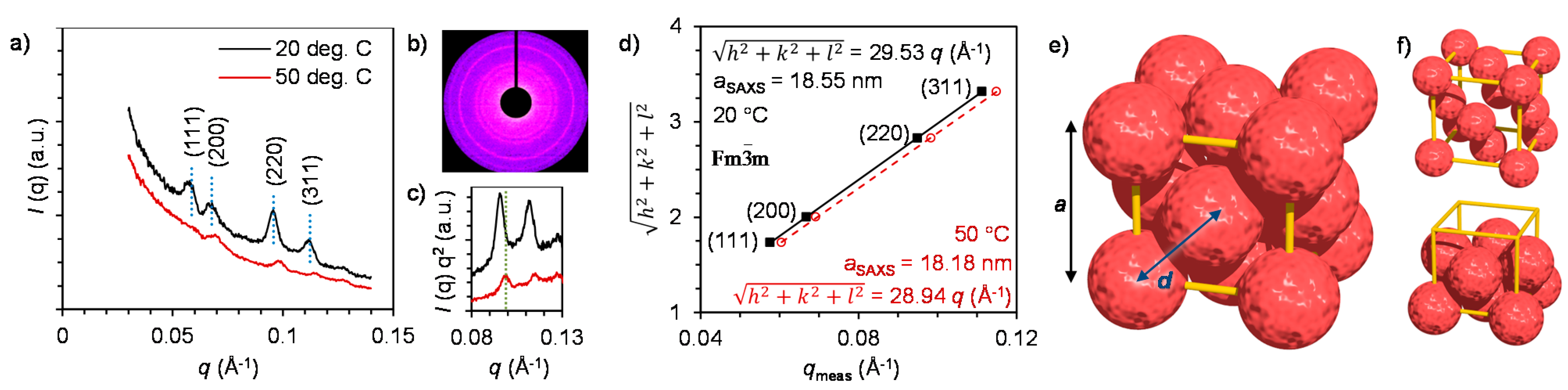
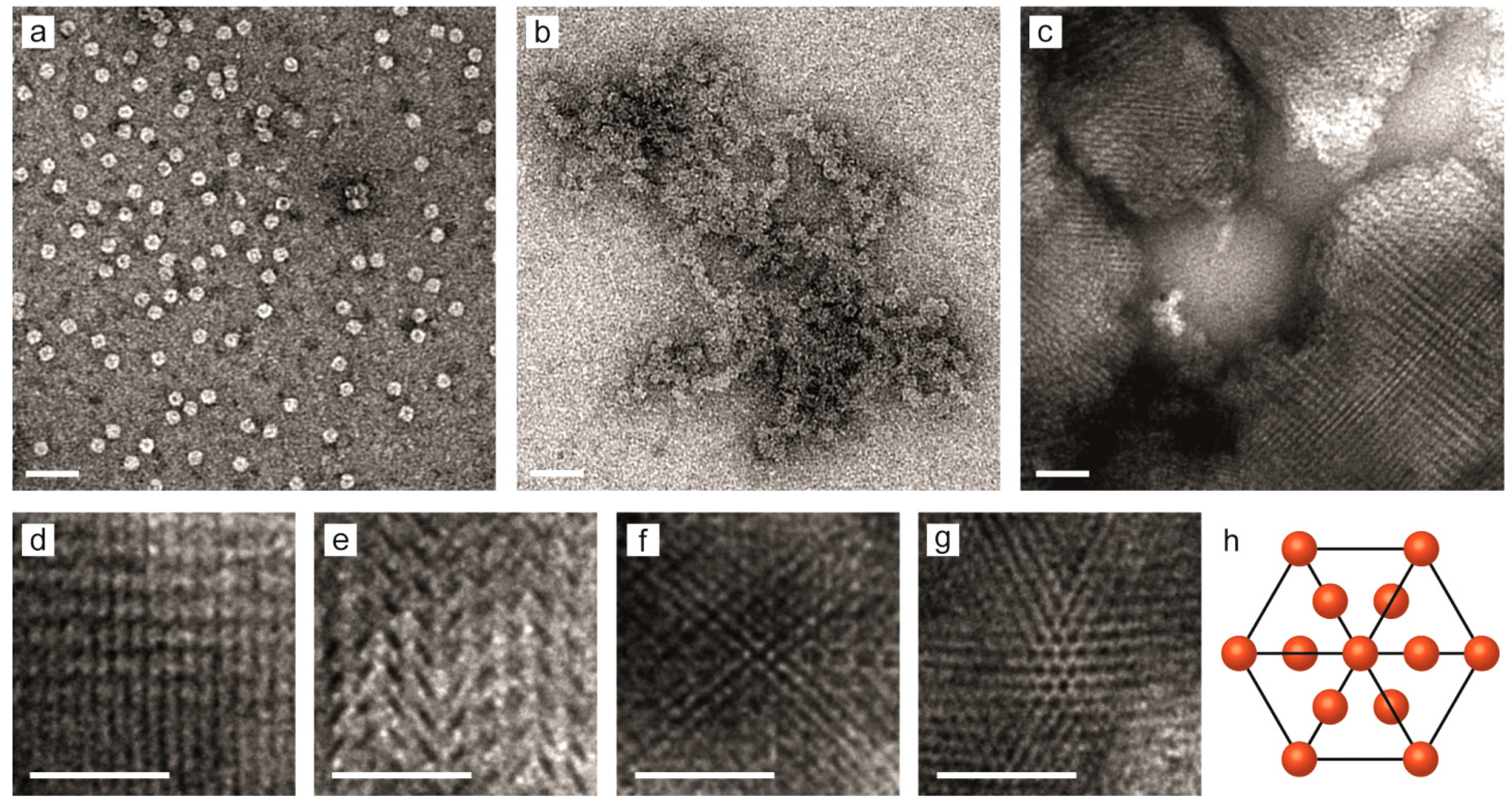
2.3. Nanostructure of the Crystalline Protein-Polymer Complexes
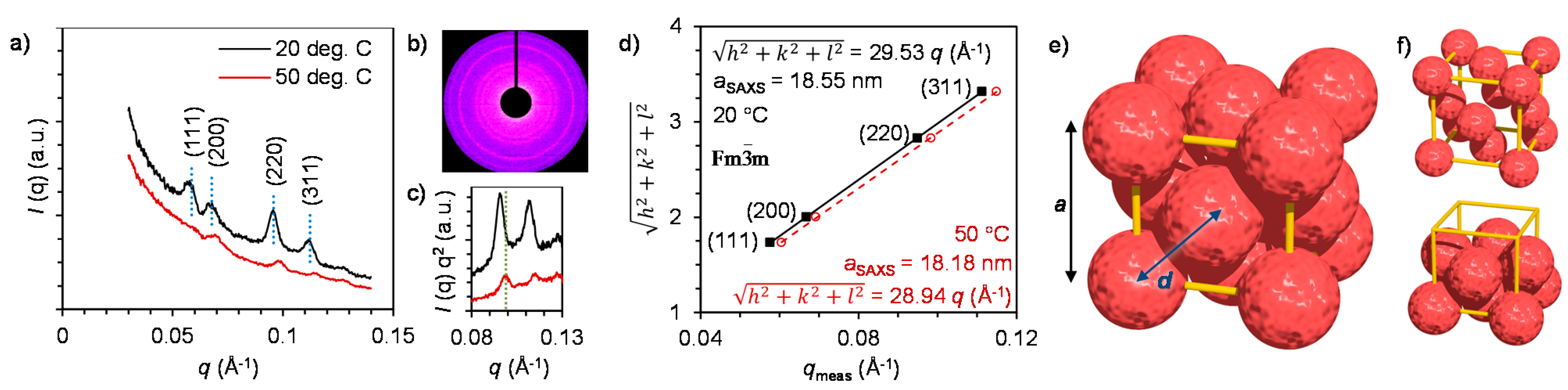
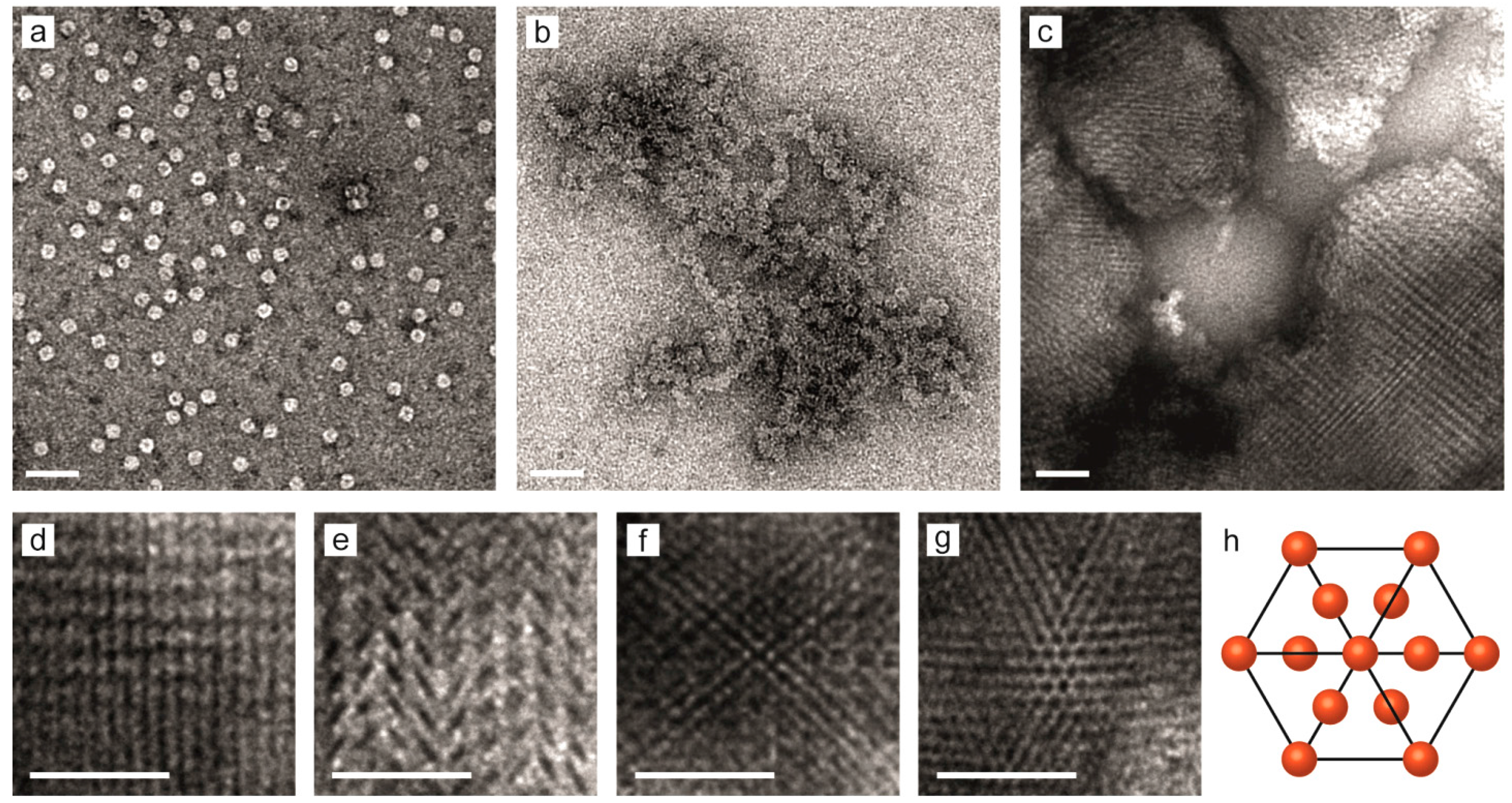
3. Experimental Section
3.1. Dynamic Light Scattering
3.2. Small-Angle X-ray Scattering
3.3. Transmission Electron Microscopy Imaging
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Van Rijn, P. Polymer directed protein assemblies. Polymers 2013, 5, 576–599. [Google Scholar]
- Kochendoerfer, G.G.; Chen, S.-Y.; Mao, F.; Cressman, S.; Traviglia, S.; Shao, H.Y.; Hunter, C.L.; Low, D.W.; Cagle, E.N.; Carnivali, M.; et al. Design and chemical synthesis of a homogenous polymer-modified erytropoiesis protein. Science 2003, 299, 884–887. [Google Scholar] [CrossRef] [PubMed]
- Shachaf, Y.; Gonen-Wadmany, M.; Seliktar, D. The biocompatibility of Pluronic F127 fibrinogen-based hydrogels. Biomaterials 2010, 31, 2836–2847. [Google Scholar] [CrossRef] [PubMed]
- Perrault, S.D.; Shih, W.M. Virus-inspired membrane encapsulation of DNA nanostructures to achieve in vivo stability. ACS Nano 2014, 8, 5132–5140. [Google Scholar] [CrossRef] [PubMed]
- Mikkilä, J.; Eskelinen, A.-P.; Niemelä, E.H.; Linko, V.; Frilander, M.J.; Törmä, P.; Kostiainen, M.A. Virus-encapsulated DNA origami nanostructures for cellular delivery. Nano Lett. 2014, 14, 2196–2200. [Google Scholar] [CrossRef] [PubMed]
- Stuart, M.A.C.; Huck, W.T.S.; Genzer, J.; Muller, M.; Ober, C.; Stamm, M.; Sukhorukov, G.B.; Szleifer, I.; Tsukruk, V.V.; Urban, M.; et al. Emerging applications of stimuli-responsive polymer materials. Nat. Mater. 2010, 9, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, A.S. Bioconjugates of intelligent polymers and recognition proteins for use in diagnostics and affinity separations. Clin. Chem. 2000, 46, 1478–1486. [Google Scholar] [PubMed]
- Pelegri-O’Day, E.M.; Lin, E.-W.; Maynard, H.D. Therapeutic protein-polymer conjugates: Advancing beyond PEGylation. J. Am. Chem. Soc. 2014, 136, 14323–14332. [Google Scholar] [CrossRef] [PubMed]
- Cobo, I.; Li, M.; Sumerlin, B.S.; Perrier, S. Smart hybrid materials by conjugation of responsive polymers to biomacromolecules. Nat. Mater. 2014, 14, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Bebis, K.; Jones, M.W.; Haddleton, D.M.; Gibson, M.I. Thermoresponsive behaviour of poly(oligo(ethyleneglycol methacrylate))s and their protein conjugates: Importance of concentration and solvent system. Polym. Chem. 2011, 2, 975–982. [Google Scholar] [CrossRef]
- Yaşayan, G.; Saeed, A.O.; Fernández-Trillo, F.; Allen, S.; Davies, M.C.; Jangher, A.; Paul, A.; Thurecht, K.J.; King, S.M.; Schweins, R.; et al. Responsive hybrid block co-polymer conjugates of proteins-controlled architecture to modulate substrate specificity and solution behaviour. Polym. Chem. 2011, 2, 1567–1578. [Google Scholar] [CrossRef]
- Deiters, A.; Cropp, T.A.; Summerer, D.; Mukherji, M.; Schultz, P.G. Site-specific PEGylation of proteins containing unnatural amino acids. Bioorg. Med. Chem. Lett. 2004, 14, 5743–5745. [Google Scholar] [CrossRef] [PubMed]
- Stayton, P.S.; Shimoboji, T.; Long, C.; Chilkoti, A.; Ghen, G.; Harris, J.M.; Hoffman, A.S. Control of protein-ligand recognition using a stimuli-responsive polymer. Nature 1995, 378, 472–474. [Google Scholar] [CrossRef] [PubMed]
- Shimoboji, T.; Larenas, E.; Fowler, T.; Hoffman, A.S.; Stayton, P.S. Temperature-induced switching of enzyme activity with smart polymer-enzyme conjugates. Bioconj. Chem. 2003, 14, 517–525. [Google Scholar] [CrossRef]
- Shimoboji, T.; Larenas, E.; Fowler, T.; Kulkarni, S.; Hoffman, A.S.; Stayton, P.S. Photoresponsive polymer-enzyme switches. Proc. Natl. Acad. Sci. USA 2002, 99, 16592–16596. [Google Scholar] [CrossRef] [PubMed]
- Shimoboji, T.; Ding, Z.L.; Stayton, P.S.; Hoffman, A.S. Photoswitching of ligand association with a photoresponsive polymer–protein conjugate. Bioconj. Chem. 2002, 13, 915–919. [Google Scholar] [CrossRef]
- Kostiainen, M.A.; Hiekkataipale, P.; de la Torre, J.A.; Nolte, R.J.M.; Cornelissen, J.J.L.M. Electrostatic self-assembly of virus polymer complexes. J. Mater. Chem. 2011, 21, 2112–2117. [Google Scholar] [CrossRef]
- Kostiainen, M.A.; Pietsch, C.; Hoogenboom, R.; Nolte, R.J.M.; Cornelissen, J.J.L.M. Temperature-switchable assembly of supramolecular virus-polymer complexes. Adv. Funct. Mater. 2011, 21, 2012–2019. [Google Scholar] [CrossRef]
- Kostiainen, M.A.; Hiekkataipale, P.; Laiho, A.; Lemieux, V.; Seitsonen, J.; Ruokolainen, J.; Ceci, P. Electrostatic assembly of binary nanoparticle superlattices using protein cages. Nat. Nanotechnol. 2013, 8, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Koskela, J.E.; Liljeström, V.; Lim, J.; Simanek, E.E.; Ras, R.H.A.; Priimagi, A.; Kostiainen, M.A. Light-fuelled transport of large dendrimers and proteins. J. Am. Chem. Soc. 2014, 136, 6850–6853. [Google Scholar] [CrossRef] [PubMed]
- Liljeström, V.; Mikkilä, J.; Kostiainen, M.A. Self-assembly and modular functionalization of three-dimensional crystals from oppositely charged proteins. Nat. Commun. 2014, 5, 4445. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; MacKay, J.A.; Frechet, J.M. J.; Szoka, F.C. Designing dendrimers for biological applications. Nat. Biotechnol. 2005, 23, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Boas, U.; Christensen, J.B.; Heegard, P.M.H. Dendrimers in Medicine and Biotechnology; RSC: Cambridge, UK, 2006. [Google Scholar]
- Mammen, M.; Choi, S.-K.; Whitesides, G.M. Polyvalent interactions in biological systems: Implications for design and use of multivalent ligands and inhibitors. Angew. Chem. Int. Ed. 1998, 37, 2754–2794. [Google Scholar] [CrossRef]
- Kostiainen, M.A.; Hardy, J.G.; Smith, D.K. High-affinity multivalent DNA binding by using low-molecular-weight dendrons. Angew. Chem. Int. Ed. 2005, 44, 2556–2559. [Google Scholar] [CrossRef]
- Kostiainen, M.A.; Smith, D.K.; Ikkala, O. Optically triggered release of DNA from multivalent dendrons by degrading and charge-switching multivalency. Angew. Chem. Int. Ed. 2007, 46, 7600–7604. [Google Scholar] [CrossRef]
- Kostiainen, M.A.; Rosilo, H. Low-molecular-weight dendrons for DNA binding and release by reduction-triggered degradation of multivalent interactions. Chem. Eur. J. 2009, 15, 5656–5660. [Google Scholar] [CrossRef] [PubMed]
- Kostiainen, M.A.; Kasyutich, O.; Cornelissen, J.J.L.M.; Nolte, R.J.M. Self-assembly and optically triggered disassembly of hierarchical dendron-virus complexes. Nat. Chem. 2010, 2, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Kostiainen, M.A.; Ceci, P.; Fornara, M.; Hiekkataipale, P.; Kasyutich, O.; Nolte, R.J.M.; Cornelissen, J.J.L.M.; Desautels, R.D.; van Lierop, J. Hierarchical self-assembly and optical disassembly for controlled switching of magnetoferritin nanoparticle magnetism. ACS Nano 2011, 5, 6394–6402. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Kostiainen, M.; Maly, J.; da Costa, V.C.P.; Annunziata, O.; Pavan, G.M.; Simanek, E.E. Synthesis of large dendrimers with the dimensions of small viruses. J. Am. Chem. Soc. 2013, 135, 4660–4663. [Google Scholar] [CrossRef] [PubMed]
- Mikkilä, J.; Rosilo, H.; Nummelin, S.; Seitsonen, J.; Ruokolainen, J.; Kostiainen, M.A. Janus-dendrimer-mediated formation of crystalline virus assemblies. ACS Macro Lett. 2013, 2, 720–724. [Google Scholar] [CrossRef]
- Tähkä, S.; Laiho, A.; Kostiainen, M.A. Diblock-copolymer-mediated self-assembly of protein-stabilized iron oxide nanoparticle clusters for magnetic resonance imaging. Chem. Eur. J. 2014, 20, 2718–2722. [Google Scholar] [CrossRef] [PubMed]
- Brasch, M.; de la Escosura, A.; Ma, Y.; Uetrecht, C.; Heck, A.J.R.; Torres, T.; Cornelissen, J.J.L.M. Encapsulation of phthalocyanine supramolecular stacks into virus-like particles. J. Am. Chem. Soc. 2011, 133, 6878–6881. [Google Scholar] [CrossRef] [PubMed]
- De la Escosura, A.; Verwegen, M.; Sikkema, F.D.; Comellas-Aragonès, M.; Kirilyuk, A.; Rasing, T.; Nolte, R.J.M.; Cornelissen, J.J.L.M. Viral capsids as templates for the production of monodisperse Prussian blue nanoparticles. Chem. Commun. 2008, 44, 1542–1544. [Google Scholar] [CrossRef]
- Setaro, F.; Brasch, M.; Hahn, U.; Koay, M.S.T.; Cornelissen, J.J.L.M.; de la Escosura, A.; Torres, T. Generation-dependent templated self-assembly of biohybrid protein nanoparticles around photosensitizer dendrimers. Nano Lett. 2015, 15, 1245–1251. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Z.; Tang, W.; Guo, C.; Chen, H.; Lin, X.; Liu, G.; Fei, B.; Chen, X.; Xu, B.; Xie, J. Ferritin nanocages to encapsulate and deliver photosensitizers for efficient photodynamic therapy against cancer. ACS Nano 2013, 7, 6988–6996. [Google Scholar] [CrossRef] [PubMed]
- Vannucci, L.; Falvo, E.; Fornara, M.; di Micco, P.; Benada, O.; Krizan, J.; Svoboda, J.; Hulikova-Capkova, K.; Morea, V.; Boffi, A.; et al. Selective targeting of melanoma by PEG-masked protein-based multifunctional nanoparticles. Int. J. Nanomed. 2012, 7, 1489–1509. [Google Scholar]
- Jutz, G.; van Rijn, P.; Santos Miranda, B.; Böker, A. Ferritin: A versatile building block for bionanotechnology. Chem. Rev. 2015, 115, 1653–1701. [Google Scholar] [CrossRef] [PubMed]
- Uchida, M.; Klem, M.T.; Allen, M.; Suci, P.; Flenniken, M.; Gillitzer, E.; Varpness, Z.; Liepold, L.O.; Young, M.; Douglas, T. Biological containers: Protein cages as multifunctional nanoplatforms. Adv. Mater. 2007, 19, 1025–1042. [Google Scholar] [CrossRef]
- Bulte, J.W.; Douglas, T.; Mann, S.; Frankel, R.B.; Moskowitz, B.M.; Brooks, R.A.; Baumgarner, C.D.; Vymazal, J.; Strub, M.P.; Frank, J.A. Magnetoferritin: Characterization of a novel superparamagnetic MR contrast agent. J. Magn. Reson. Imaging 1994, 4, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Meldrum, F.C.; Heywood, B.R.; Mann, S. Magnetoferritin: In vitro synthesis of a novel magnetic protein. Science 1992, 257, 522–523. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.C.; Fassler, J.D.; Meade, T. Mimicking liver iron overload using liposomal ferritin preparations. Magn. Reson. Med. 2004, 51, 607–611. [Google Scholar] [CrossRef] [PubMed]
- Aime, S.; Frullano, L.; Geninatti Crich, S. Compartmentalization of a gadolinium complex in the apoferritin cavity: A route to obtain high relaxivity contrast agents for magnetic resonance imaging. Angew. Chem. Int. Ed. 2002, 41, 1017–1019. [Google Scholar] [CrossRef]
- Wong, K.K.W.; Douglas, T.; Gider, S.; Awschalom, D.D.; Mann, S. Biomimetic synthesis and characterization of magnetic proteins (magnetoferritin). Chem. Mater. 1998, 10, 279–285. [Google Scholar] [CrossRef]
- Ueno, T.; Suzuki, M.; Goto, T.; Matsumoto, T.; Nagayama, K.; Watanabe, Y. Size-selective olefin hydrogenation by a Pd nanocluster provided in an apo-ferritin cage. Angew. Chem. Int. Ed. 2004, 43, 2527–2530. [Google Scholar] [CrossRef]
- Srivastava, S.; Samanta, B.; Jordan, B.J.; Hong, R.; Xiao, Q.; Tuominen, M.T.; Rotello, V.M. Integrated magnetic bionanocomposites through nanoparticle-mediated assembly of ferritin. J. Am. Chem. Soc. 2007, 129, 11776–11780. [Google Scholar] [CrossRef] [PubMed]
- Uchida, M.; Kang, S.; Reichhardt, C.; Harlen, K.; Douglas, T. The ferritin superfamily: Supramolecular templates for materials synthesis. BBA Gen. Subj. 2010, 1800, 834–845. [Google Scholar] [CrossRef]
- Kasyutich, O.; Sarua, A.; Schwarzacher, W. Bioengineered magnetic crystals. J. Phys. D Appl. Phys. 2008, 41, 134022. [Google Scholar] [CrossRef]
- Maye, M.M.; Kumara, M.T.; Nykypanchuk, D.; Sherman, W.B.; Gang, O. Switching binary states of nanoparticle superlattices and dimer clusters by DNA strands. Nat. Nanotechnol. 2010, 5, 116–120. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Välimäki, S.; Mikkilä, J.; Liljeström, V.; Rosilo, H.; Ora, A.; Kostiainen, M.A. Hierarchically Ordered Supramolecular Protein-Polymer Composites with Thermoresponsive Properties. Int. J. Mol. Sci. 2015, 16, 10201-10213. https://doi.org/10.3390/ijms160510201
Välimäki S, Mikkilä J, Liljeström V, Rosilo H, Ora A, Kostiainen MA. Hierarchically Ordered Supramolecular Protein-Polymer Composites with Thermoresponsive Properties. International Journal of Molecular Sciences. 2015; 16(5):10201-10213. https://doi.org/10.3390/ijms160510201
Chicago/Turabian StyleVälimäki, Salla, Joona Mikkilä, Ville Liljeström, Henna Rosilo, Ari Ora, and Mauri A. Kostiainen. 2015. "Hierarchically Ordered Supramolecular Protein-Polymer Composites with Thermoresponsive Properties" International Journal of Molecular Sciences 16, no. 5: 10201-10213. https://doi.org/10.3390/ijms160510201