
Determination of Oxidized Phosphatidylcholines by Hydrophilic Interaction Liquid Chromatography Coupled to Fourier Transform Mass Spectrometry


Abstract
:1. Introduction
2. Results and Discussion
2.1. HPLC and Mass Spectrometry Development
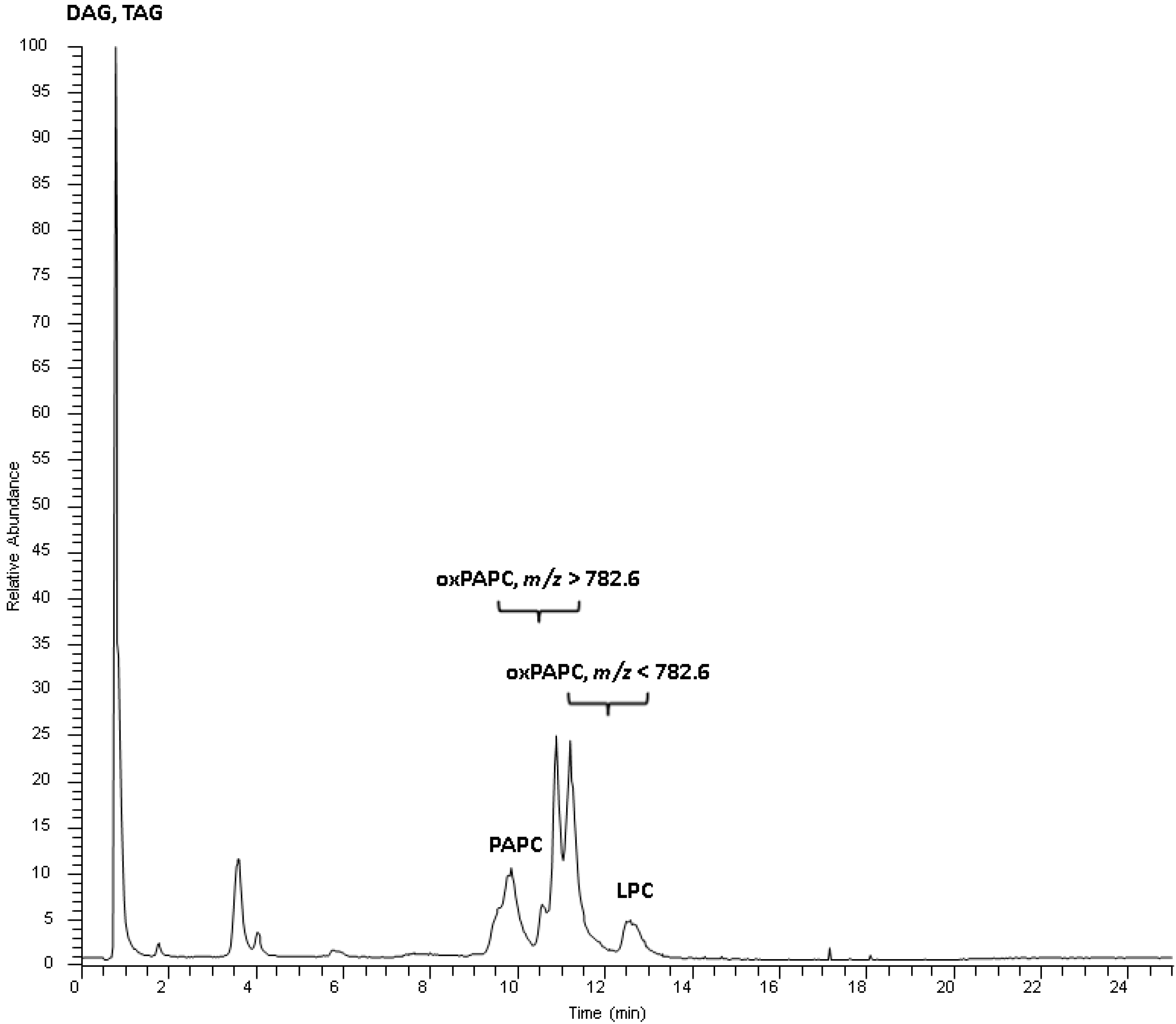
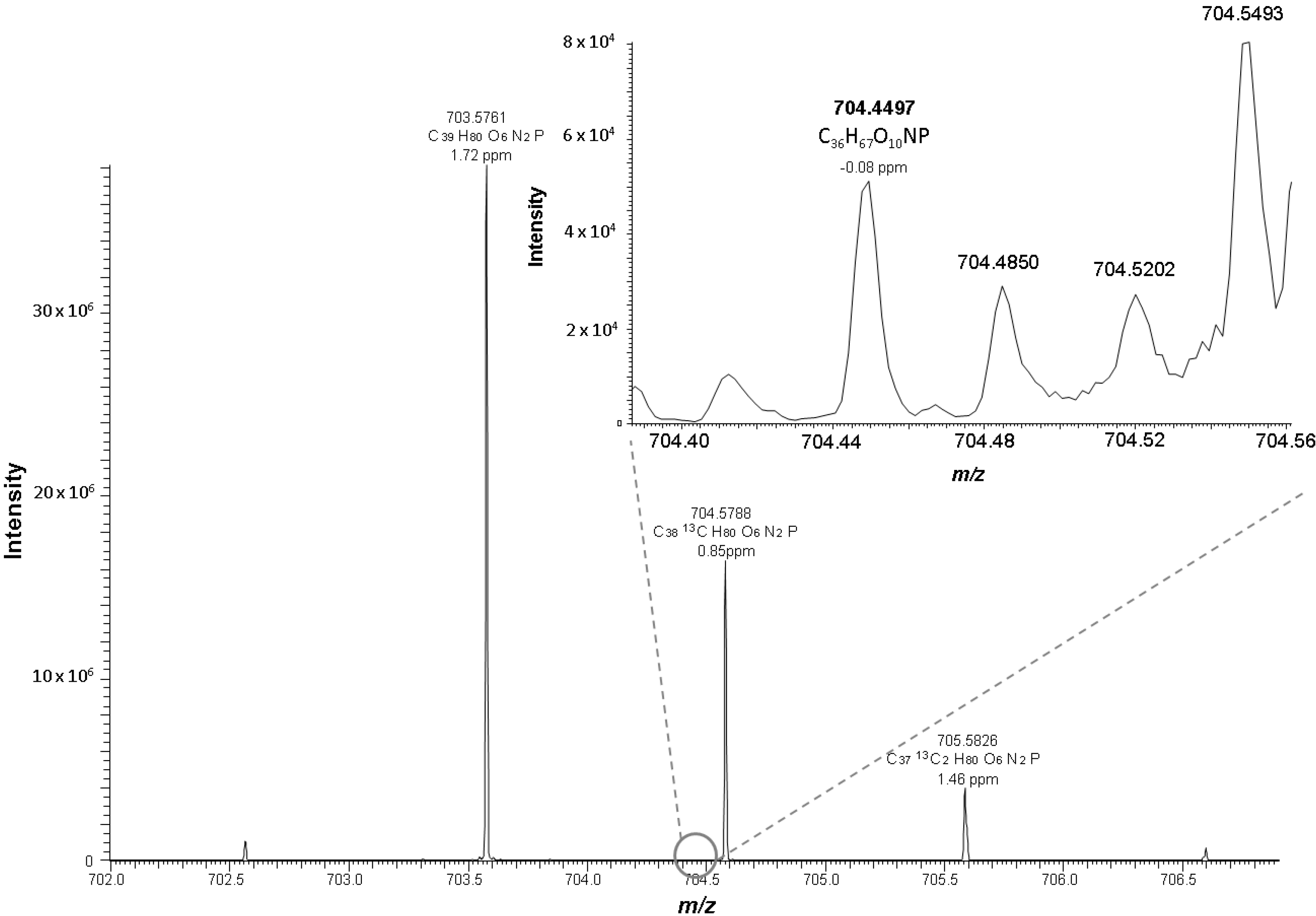
2.2. Identification Strategy for Oxidized Phospholipids

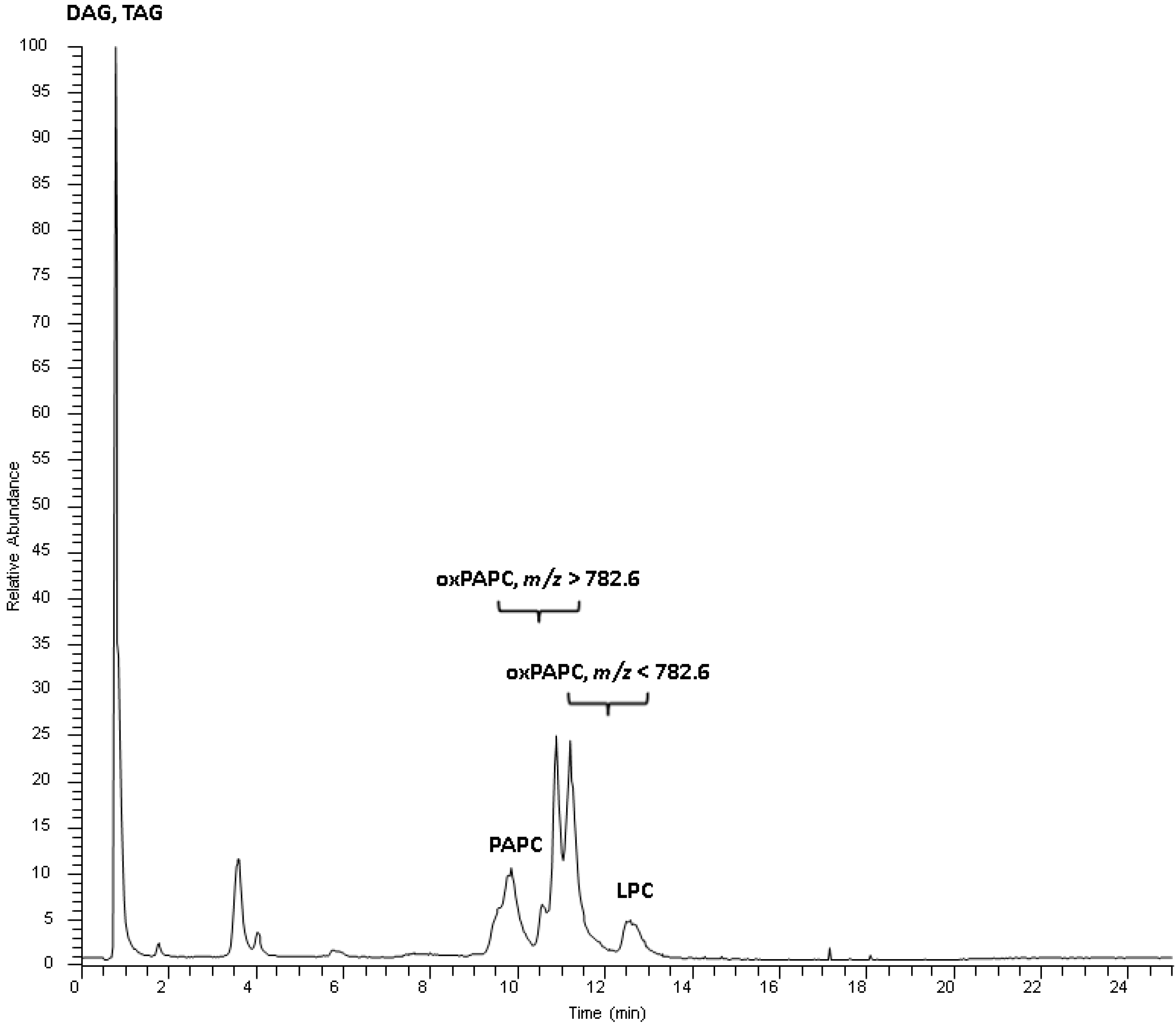{kind=link}
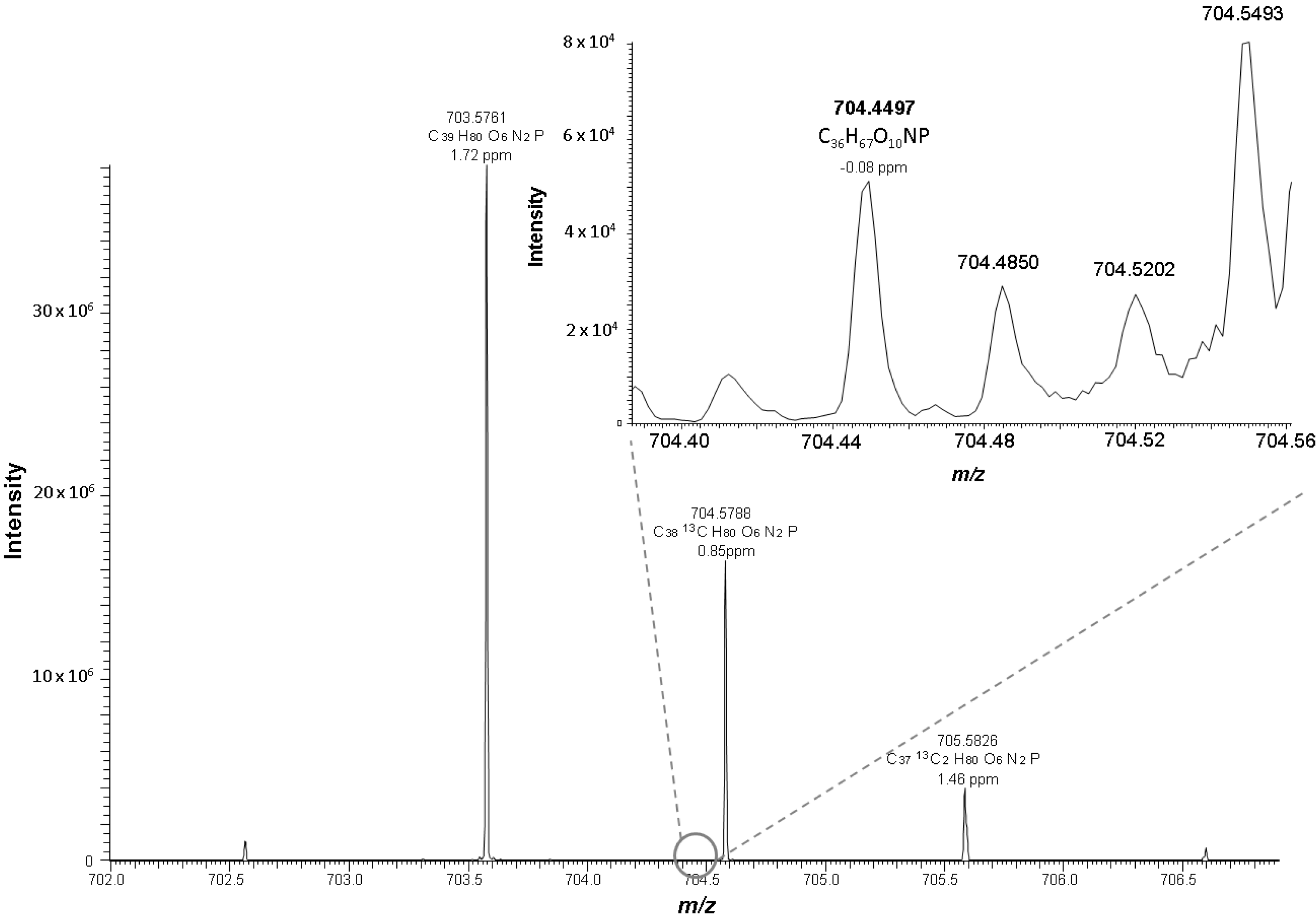{kind=link}
| Requirement | Fragment | Scoring Points |
|---|---|---|
| Exact mass < 2 ppm | 1.0 | |
| Headgroup fragment | [PChol]+ or [M-59]+ | 1.0 |
| Fatty acyl fragment | [M-Rox]+ | 1.0 |
| Fatty acyl fragment | [M-Rox=C=O]+ | 1.0 |
| Water loss at oxidized moiety | [M-H2O]+ | 0.5 |
| Water loss at oxidized moiety | [M-2H2O]+ | 0.5 |
2.3. Analysis of Oxidized Phosphatidylcholine Species in Oxidized Low Density Lipoprotein
| Species | Elemental Composition | m/z Observed [M+H]+ | Δ ppm | RT [min] | Relative Contribution [%] | Score |
|---|---|---|---|---|---|---|
| PC 16:0_20:4 [4O] | C44H80O12NP | 846.5502 | 1.30 | 10.23 | 0.91 | 5.0 |
| PC 16:0_20:3 [4O] | C44H82O12NP | 848.5655 | 0.93 | 10.38 | 0.71 | 5.0 |
| PC 18:0_20:5 [2O] | C46H82O10NP | 840.5750 | 0.15 | 10.09 | 0.54 | 5.0 |
| PC 18:0_20:4 [3O] | C46H84O11NP | 858.5850 | 0.57 | 9.64 | 3.40 | 5.0 |
| PC 18:0_20:4 [4O] | C46H84O12NP | 874.5793 | −1.11 | 10.06 | 0.83 | 5.0 |
| PC 16:0_5:1 [O] | C29H56O9NP | 594.3770 | 0.82 | 11.82 | 1.48 | 4.5 |
| PC 16:0_8:3 [3O] | C32H58O11NP | 664.3829 | 1.33 | 12.20 | 0.01 | 4.5 |
| PC 16:0_8:2 [3O] | C32H60O11NP | 666.3986 | 1.33 | 11.48 | 0.03 | 4.5 |
| PC 16:0_22:4 [O] | C46H84O9NP | 826.5960 | −1.79 | 12.03 | 0.56 | 4.5 |
| PC 16:0_4:1 [O] | C30H58O9NP | 608.3920 | 0.30 | 11.82 | 0.60 | 4.5 |
| PC 16:0_6:1 [O] | C33H62O10NP | 664.4178 | 0.73 | 11.08 | 0.09 | 4.5 |
| PC 16:0_9:2 [2O] | C46H84O9NP | 826.5960 | 0.52 | 9.65 | 0.16 | 4.5 |
| PC 16:0_9:0 [O] | C33H64O9NP | 650.4402 | −1.69 | 10.86 | 7.38 | 4.0 |
| PC 16:0_8:2 [2O] | C32H60O10NP | 650.4036 | 1.43 | 11.22 | 0.13 | 4.0 |
| PC 16:0_20:6 [2O] | C44H76O10NP | 810.5290 | 1.24 | 10.23 | 1.71 | 4.0 |
| PC 16:0_20:5 [3O] | C44H78O11NP | 828.5393 | 0.91 | 10.06 | 3.28 | 4.0 |
| PC 16:0_20:4 [3O] | C44H80O11NP | 830.5552 | 1.27 | 9.81 | 6.08 | 4.0 |
| PC 16:0_20:3 [3O] | C44H82O11NP | 832.5687 | −1.25 | 10.01 | 3.02 | 4.0 |
| PC 16:0_22:6 [2O] | C46H80O10NP | 838.5590 | 0.29 | 10.06 | 1.41 | 4.0 |
| PC 16:0_12:2 [2O] | C36H68O10NP | 706.4650 | 0.52 | 10.97 | 0.06 | 3.5 |
| PC 16:0_18:2 [2O] | C42H80O10NP | 790.5577 | 1.85 | 10.04 | 15.86 | 3.5 |
| PC 18:0_9:0 [O] | C35H68O9NP | 678.4714 | −1.53 | 10.68 | 5.08 | 3.5 |
| PC 18:0_18:2 [O] | C44H84O9NP | 802.5946 | 1.22 | 9.81 | 6.12 | 3.5 |
| PC 16:0_20:4 [O] | C44H80O9NP | 798.5651 | 1.03 | 9.81 | 0.94 | 3.5 |
| PC 18:0_5:1 [O] | C31H60O9NP | 622.4080 | −0.29 | 11.62 | 1.90 | 3.5 |
| PC 18:0_5:1 [2O] | C31H60O10NP | 638.4027 | 0.00 | 10.74 | 0.20 | 3.5 |
| PC 16:0_10:3 [2O] | C34H62O10NP | 676.4193 | −1.44 | 11.10 | 0.08 | 3.5 |
| PC 18:0_10:3 [2O] | C36H66O10NP | 704.4497 | −0.08 | 10.49 | 0.18 | 3.5 |
| PC 36:5 [2O] | C44H78O10NP | 812.5440 | −0.53 | 10.23 | 1.57 | 3.0 |
| PC 38:6 [2O] | C46H80O10NP | 838.5590 | −0.29 | 10.10 | 1.61 | 3.0 |
| PC 40:6 [O] | C48H84O9NP | 850.5960 | −0.50 | 10.70 | 0.10 | 2.5 |
| PC 34:2 [O] | C42H80O9NP | 774.5643 | −0.08 | 9.98 | 20.03 | 2.5 |
| PC 27:0 [2O] | C35H68O10NP | 694.4650 | 0.53 | 10.87 | 1.30 | 2.5 |
| PC 36:4 [2O] | C44H80O10NP | 814.5590 | 0.30 | 9.75 | 5.02 | 2.5 |
| PC 26:3 [2O] | C34H62O10NP | 676.4183 | 0.00 | 10.86 | 0.08 | 2.5 |
| PC 26:3 [3O] | C34H62O11NP | 692.4130 | 0.44 | 12.75 | 0.03 | 2.5 |
| PC 26:2 [3O] | C34H64O11NP | 694.4284 | 0.70 | 10.94 | 0.06 | 2.5 |
| PC 20:1 [2O] | C28H54O10NP | 596.3548 | 1.64 | 11.73 | 0.03 | 2.5 |
| PC 22:1 [2O] | C30H58O10NP | 624.3871 | 0.00 | 11.56 | 0.09 | 2.5 |
| PC 27:3 [O] | C35H64O9NP | 674.4381 | 1.45 | 11.75 | 0.07 | 2.5 |
| PC 36:6 [O] | C44H76O9NP | 794.5325 | 0.62 | 11.24 | 0.11 | 2.5 |
| PC 25:0 [2O] | C33H64O10NP | 666.4351 | −1.56 | 10.55 | 1.31 | 2.0 |
| PC 28:3 [3O] | C36H66O11NP | 720.4446 | 0.00 | 10.38 | 0.06 | 2.0 |
| PC 21:1 [2O] | C29H56O10NP | 610.3720 | 0.91 | 12.43 | 0.11 | 2.0 |
| PC 34:6 [O] | C42H72O9NP | 766.5020 | −0.40 | 12.74 | 0.03 | 2.0 |
| PC 24:2 [O] | C32H60O9NP | 634.4088 | −1.64 | 11.45 | 0.06 | 2.0 |
| Species | Possible Oxidized Structure | Possible Precursor | [PChol]+ [M-59]+ | [M-Rox]+ [M-Rox=C=O]+ | [M-H2O]+ [M-2H2O]+ | Score |
|---|---|---|---|---|---|---|
| PC 16:0_20:4 [4O] | isoPGG2 | PC 16:0/20:4 | 787 | 496, 478 | 828, 810 | 5.0 |
| PC 16:0_20:3 [4O] | isoTxA2 | PC 16:0/20:4 | 789 | 496, 478 | 830, 812 | 5.0 |
| PC 18:0_20:5 [2O] | isoPGJ2/A2 | PC 18:0/20:4 | 781 | 524, 506 | 822, 804 | 5.0 |
| PC 18:0_20:4 [3O] | isoPGE2/D2 isoLGE2/D2 isoTXB2 | PC 18:0/20:4 | 799 | 524, 506 | 840, 822 | 5.0 |
| PC 18:0_20:4 [4O] | isoPGG2 | PC 18:0/20:4 | 815 | 524, 506 | 856, 838 | 5.0 |
| PC 16:0_5:1 [O] | OV | PC 16:0/20:4 | 184 | 496, 478 | 576 | 4.5 |
| PC 16:0_8:3 [3O] | KOdiA | PC 16:0/20:4 | 184 | 496, 478 | 646 | 4.5 |
| PC 16:0_8:2 [3O] | HOdiA | PC 16:0/20:4 | 184 | 496, 478 | 648 | 4.5 |
| PC 16:0_22:4 [O] | PC 16:0/22:4 | 767 | 496, 478 | 808 | 4.5 | |
| PC 16:0_4:1 [O] | PC 16:0/22:6 | 184 | 496, 478 | 562 | 4.5 | |
| PC 16:0_6:1 [O] | PC 18:0/22:6 | 184 | 496, 478 | 590 | 4.5 | |
| PC 16:0_9:2 [2O] | PC 18:0/22:6 | 184 | 496, 478 | 646 | 4.5 | |
| PC 16:0_9:0 [O] | ON | PC 16:0/18:2 | 184 | 496, 478 | 4.0 | |
| PC 16:0_8:2 [2O] | HOOA | PC 16:0/20:4 | 184 | 496, 478 | 4.0 | |
| PC 16:0_20:6 [2O] | EC | PC 16:0/20:4 | 184 | 496 | 792, 774 | 4.0 |
| PC 16:0_20:5 [3O] | EI | PC 16:0/20:4 | 769 | 496 | 810, 792 | 4.0 |
| PC 16:0_20:4 [3O] | isoPGE2/D2 isoLGE2/D2 isoTXB2 | PC 16:0/20:4 | 771 | 496 | 812, 794 | 4.0 |
| PC 16:0_20:3 [3O] | isoPGF2α | PC 16:0/20:4 | 773 | 496 | 814, 796 | 4.0 |
| PC 16:0_22:6 [2O] | PC 16:0/22:6 | 779 | 496 | 820, 802 | 4.0 | |
| PC 16:0_12:2 [2O] | HODA | PC 16:0/18:2 | 184 | 478 | 688 | 3.5 |
| PC 16:0_18:2 [2O] | HpODE | PC 16:0/18:2 | 184 | 496 | 772 | 3.5 |
| PC 18:0_9:0 [O] | ON | PC 18:0/18:2 | 184 | 524 | 660 | 3.5 |
| PC 18:0_18:2 [O] | HODE | PC 18:0/18:2 | 184 | 524 | 784 | 3.5 |
| PC 16:0_20:4 [O] | HETE | PC 16:0/20:4 | 184 | 478 | 680 | 3.5 |
| PC 18:0_5:1 [O] | OV | PC 18:0/20:4 | 184 | 524 | 604 | 3.5 |
| PC 18:0_5:1 [2O] | G | PC 18:0/20:4 | 184 | 506 | 620 | 3.5 |
| PC 16:0_10:3 [2O] | PC 16:0/22:6 | 184 | 496 | 658 | 3.5 | |
| PC 18:0_10:3 [2O] | PC 18:0/22:6 | 184 | 506 | 686 | 3.5 | |
| PC 36:5 [2O] | PC 16:0/20:4 | 753 | 794, 772 | 3.0 | ||
| PC 38:6 [2O] | PC 18:0/20:4 | 779 | 820, 802 | 3.0 | ||
| PC 40:6 [O] | PC 18:0/22:6 | 791 | 832 | 2.5 | ||
| PC 34:2 [O] | PC 16:0/18:2 | 184 | 756 | 2.5 | ||
| PC 27:0 [2O] | PC 18:0/18:2 | 184 | 676 | 2.5 | ||
| PC 36:4 [2O] | PC 16:0/20:4 | 755 | 796 | 2.5 | ||
| PC 26:3 [2O] | PC 18:0/20:4 | 184 | 658 | 2.5 | ||
| PC 26:3 [3O] | PC 18:0/20:4 | 184 | 674 | 2.5 | ||
| PC 26:2 [3O] | PC 18:0/20:4 | 184 | 676 | 2.5 | ||
| PC 20:1 [2O] | PC 16:0/22:6 | 184 | 578 | 2.5 | ||
| PC 22:1 [2O] | PC 18:0/22:6 | 184 | 606 | 2.5 | ||
| PC 27:3 [O] | PC 18:0/22:6 | 184 | 656 | 2.5 | ||
| PC 36:6 [O] | PC 18:0/22:6 | 184 | 776 | 2.5 | ||
| PC 25:0 [2O] | PC 16:0/18:2 | 184 | 2.0 | |||
| PC 28:3 [3O] | PC 16:0/18:2 | 184 | 2.0 | |||
| PC 21:1 [2O] | PC 16:0/20:4 | 184 | 2.0 | |||
| PC 34:6 [O] | PC 16:0/22:6 | 184 | 2.0 | |||
| PC 24:2 [O] | PC 18:0/22:6 | 184 | 2.0 |
3. Material and Methods
3.1. Materials
3.2. Oxidation of PAPC
3.3. Isolation and Oxidation of LDL
3.4. High Performance Liquid Chromatography-Mass Spectrometry
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lushchak, V.I. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem. Biol. Interact. 2014, 224, 164–175. [Google Scholar] [CrossRef]
- Fruhwirth, G.O.; Loidl, A.; Hermetter, A. Oxidized phospholipids: From molecular properties to disease. Biochim. Biophys. Acta Mol. Basis Dis. 2007, 1772, 718–736. [Google Scholar] [CrossRef]
- Esterbauer, H.; Gebicki, J.; Puhl, H.; Jurgens, G. The role of lipid-peroxidation and antioxidants in oxidative modification of ldl. Free Radic. Biol. Med. 1992, 13, 341–390. [Google Scholar] [CrossRef]
- Bochkov, V.N.; Oskolkova, O.V.; Birukov, K.G.; Levonen, A.L.; Binder, C.J.; Stockl, J. Generation and biological activities of oxidized phospholipids. Antioxid. Redox Signal. 2010, 12, 1009–1059. [Google Scholar] [CrossRef]
- Leitinger, N. The Role of Phospholipid Oxidation Products in Inflammatory and Autoimmune Diseases; Springer: New York, NY, USA, 2008; Volume 49, pp. 325–350. [Google Scholar]
- Brame, C.J.; Salomon, R.G.; Morrow, J.D.; Roberts, L.J. Identification of extremely reactive gamma-ketoaldehydes (isolevuglandins) as products of the isoprostane pathway and characterization of their lysyl protein adducts. J. Biol. Chem. 1999, 274, 13139–13146. [Google Scholar] [CrossRef]
- Kofeler, H.C.; Fauland, A.; Rechberger, G.N.; Trotzmuller, M. Mass spectrometry based lipidomics: An overview of technological platforms. Metabolites 2012, 2, 19–38. [Google Scholar] [CrossRef]
- Uchikata, T.; Matsubara, A.; Nishiumi, S.; Yoshida, M.; Fukusaki, E.; Bamba, T. Development of oxidized phosphatidylcholine isomer profiling method using supercritical fluid chromatography/tandem mass spectrometry. J. Chromatogr. A 2012, 1250, 205–211. [Google Scholar] [CrossRef]
- Lee, J.Y.; Lim, S.; Park, S.; Moon, M.H. Characterization of oxidized phospholipids in oxidatively modified low density lipoproteins by nanoflow liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2013, 1288, 54–62. [Google Scholar] [CrossRef]
- Gruber, F.; Bicker, W.; Oskolkova, O.V.; Tschachler, E.; Bochkov, V.N. A simplified procedure for semi-targeted lipidomic analysis of oxidized phosphatidylcholines induced by uva irradiation. J. Lipid Res. 2012, 53, 1232–1242. [Google Scholar] [CrossRef]
- Watson, A.D.; Leitinger, N.; Navab, M.; Faull, K.F.; Horkko, S.; Witztum, J.L.; Palinski, W.; Schwenke, D.; Salomon, R.G.; Sha, W.; et al. Structural identification by mass spectrometry of oxidized phospholipids in minimally oxidized low density lipoprotein that induce monocyte/endothelial interactions and evidence for their presence in vivo. J. Biol. Chem. 1997, 272, 13597–13607. [Google Scholar]
- Watson, A.D.; Subbanagounder, G.; Welsbie, D.S.; Faull, K.F.; Navab, M.; Jung, M.E.; Fogelman, A.M.; Berliner, J.A. Structural identification of a novel pro-inflammatory epoxyisoprostane phospholipid in mildly oxidized low density lipoprotein. J. Biol. Chem. 1999, 274, 24787–24798. [Google Scholar] [CrossRef] [PubMed]
- Samhan-Arias, A.K.; Ji, J.; Demidova, O.M.; Sparvero, L.J.; Feng, W.; Tyurin, V.; Tyurina, Y.Y.; Epperly, M.W.; Shvedova, A.A.; Greenberger, J.S.; et al. Oxidized phospholipids as biomarkers of tissue and cell damage with a focus on cardiolipin. Biochim. Biophys. Acta Biomembr. 2012, 1818, 2413–2423. [Google Scholar]
- Liebisch, G.; Scherer, M. Quantification of bioactive sphingo- and glycerophospholipid species by electrospray ionization tandem mass spectrometry in blood. J. Chromatogr. B 2012, 883, 141–146. [Google Scholar] [CrossRef]
- Lisa, M.; Cifkova, E.; Holcapek, M. Lipidomic profiling of biological tissues using off-line two-dimensional high-performance liquid chromatography mass spectrometry. J. Chromatogr. A 2011, 1218, 5146–5156. [Google Scholar] [CrossRef] [PubMed]
- Schwalbe-Herrmann, M.; Willmann, J.; Leibfritz, D. Separation of phospholipid classes by hydrophilic interaction chromatography detected by electrospray ionization mass spectrometry. J. Chromatogr. A 2010, 1217, 5179–5183. [Google Scholar] [CrossRef] [PubMed]
- Reis, A.; Domingues, P.; Domingues, M.R.M. Structural motifs in primary oxidation products of palmitoyl-arachidonoyl-phosphatidylcholines by LC-MS/MS. J. Mass. Spectrom. 2013, 48, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Stubiger, G.; Belgacem, O.; Rehulka, P.; Bicker, W.; Binder, B.R.; Bochkov, V. Analysis of oxidized phospholipids by maldi mass spectrometry using 6-aza-2-thiothymine together with matrix additives and disposable target surfaces. Anal. Chem. 2010, 82, 5502–5510. [Google Scholar] [CrossRef] [PubMed]
- Jonasdottir, H.S.; Nicolardi, S.; Jonker, W.; Derks, R.; Palmblad, M.; Ioan-Facsinay, A.; Toes, R.; van der Burgt, Y.E.M.; Deelder, A.M.; Mayboroda, O.A.; et al. Detection and structural elucidation of esterified oxylipids in human synovial fluid by electrospray ionization-fourier transform ion-cyclotron mass spectrometry and liquid chromatography-ion trap-ms3: Detection of esterified hydroxylated docosapentaenoic acid containing phospholipids. Anal. Chem. 2013, 85, 6003–6010. [Google Scholar]
- Milic, I.; Hoffmann, R.; Fedorova, M. Simultaneous detection of low and high molecular weight carbonylated compounds derived from lipid peroxidation by electrospray ionization-tandem mass spectrometry. Anal. Chem. 2013, 85, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.; Koster, G.; Douet, L.J.; Scigelova, M.; Woffendin, G.; Ward, J.M.; Smith, A.; Humphries, J.; Burnand, K.G.; Macphee, C.H.; et al. Electrospray ionization mass spectrometry identifies substrates and products of lipoprotein-associated phospholipase A2 in oxidized human low density lipoprotein. J. Biol. Chem. 2008, 283, 6428–6437. [Google Scholar]
- Ishida, M.; Yamazaki, T.; Houjou, T.; Imagawa, M.; Harada, A.; Inoue, K.; Taguchi, R. High-resolution analysis by nano-electrospray ionization fourier transform ion cyclotron resonance mass spectrometry for the identification of molecular species of phospholipids and their oxidized metabolites. Rapid Commun. Mass Spectrom. 2004, 18, 2486–2494. [Google Scholar] [CrossRef] [PubMed]
- Hartler, J.; Trotzmuller, M.; Chitraju, C.; Spener, F.; Kofeler, H.C.; Thallinger, G.G. Lipid data analyzer: Unattended identification and quantitation of lipids in LC-MS data. Bioinformatics 2011, 27, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.L. Charge-Remote fragmentations—Method, mechanism and applications. Int. J. Mass Spectrom. Ion Process. 1992, 118, 137–165. [Google Scholar] [CrossRef]
- Pittenauer, E.; Allmaier, G. The renaissance of high-energy cid for structural elucidation of complex lipids: MALDI-TOF/RTOF-MS of alkali cationized triacylglycerols. J. Am. Soc. Mass Spectrom. 2009, 20, 1037–1047. [Google Scholar] [CrossRef] [PubMed]
- Harrison, K.A.; Davies, S.S.; Marathe, G.K.; McIntyre, T.; Prescott, S.; Reddy, K.M.; Falck, J.R.; Murphy, R.C. Analysis of oxidized glycerophosphocholine lipids using electrospray ionization mass spectrometry and microderivatization techniques. J. Mass Spectrom. 2000, 35, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Liebisch, G.; Vizcaino, J.A.; Kofeler, H.; Trotzmuller, M.; Griffiths, W.J.; Schmitz, G.; Spener, F.; Wakelam, M.J.O. Shorthand notation for lipid structures derived from mass spectrometry. J. Lipid Res. 2013, 54, 1523–1530. [Google Scholar] [CrossRef] [PubMed]
- Fauland, A.; Kofeler, H.; Trotzmuller, M.; Knopf, A.; Hartler, J.; Eberl, A.; Chitraju, C.; Lankmayr, E.; Spener, F. A comprehensive method for lipid profiling by liquid chromatography-ion cyclotron resonance mass spectrometry. J. Lipid Res. 2011, 52, 2314–2322. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Lees, M.; Stanley, G.H.S. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [PubMed]
- Ho, Y.L.; Chiu, J.H.; Wu, C.Y.; Liu, M.Y. Separation and determination of in vitro oxidized phospholipids by capillary zone electrophoresis. Anal. Biochem. 2007, 367, 210–218. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sala, P.; Pötz, S.; Brunner, M.; Trötzmüller, M.; Fauland, A.; Triebl, A.; Hartler, J.; Lankmayr, E.; Köfeler, H.C. Determination of Oxidized Phosphatidylcholines by Hydrophilic Interaction Liquid Chromatography Coupled to Fourier Transform Mass Spectrometry. Int. J. Mol. Sci. 2015, 16, 8351-8363. https://doi.org/10.3390/ijms16048351
Sala P, Pötz S, Brunner M, Trötzmüller M, Fauland A, Triebl A, Hartler J, Lankmayr E, Köfeler HC. Determination of Oxidized Phosphatidylcholines by Hydrophilic Interaction Liquid Chromatography Coupled to Fourier Transform Mass Spectrometry. International Journal of Molecular Sciences. 2015; 16(4):8351-8363. https://doi.org/10.3390/ijms16048351
Chicago/Turabian StyleSala, Pia, Sandra Pötz, Martina Brunner, Martin Trötzmüller, Alexander Fauland, Alexander Triebl, Jürgen Hartler, Ernst Lankmayr, and Harald C. Köfeler. 2015. "Determination of Oxidized Phosphatidylcholines by Hydrophilic Interaction Liquid Chromatography Coupled to Fourier Transform Mass Spectrometry" International Journal of Molecular Sciences 16, no. 4: 8351-8363. https://doi.org/10.3390/ijms16048351