
Recent Developments of Engineered Translational Machineries for the Incorporation of Non-Canonical Amino Acids into Polypeptides

Abstract
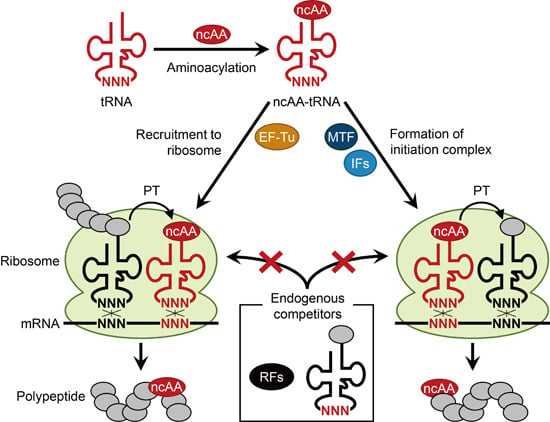
:
{kind=link}
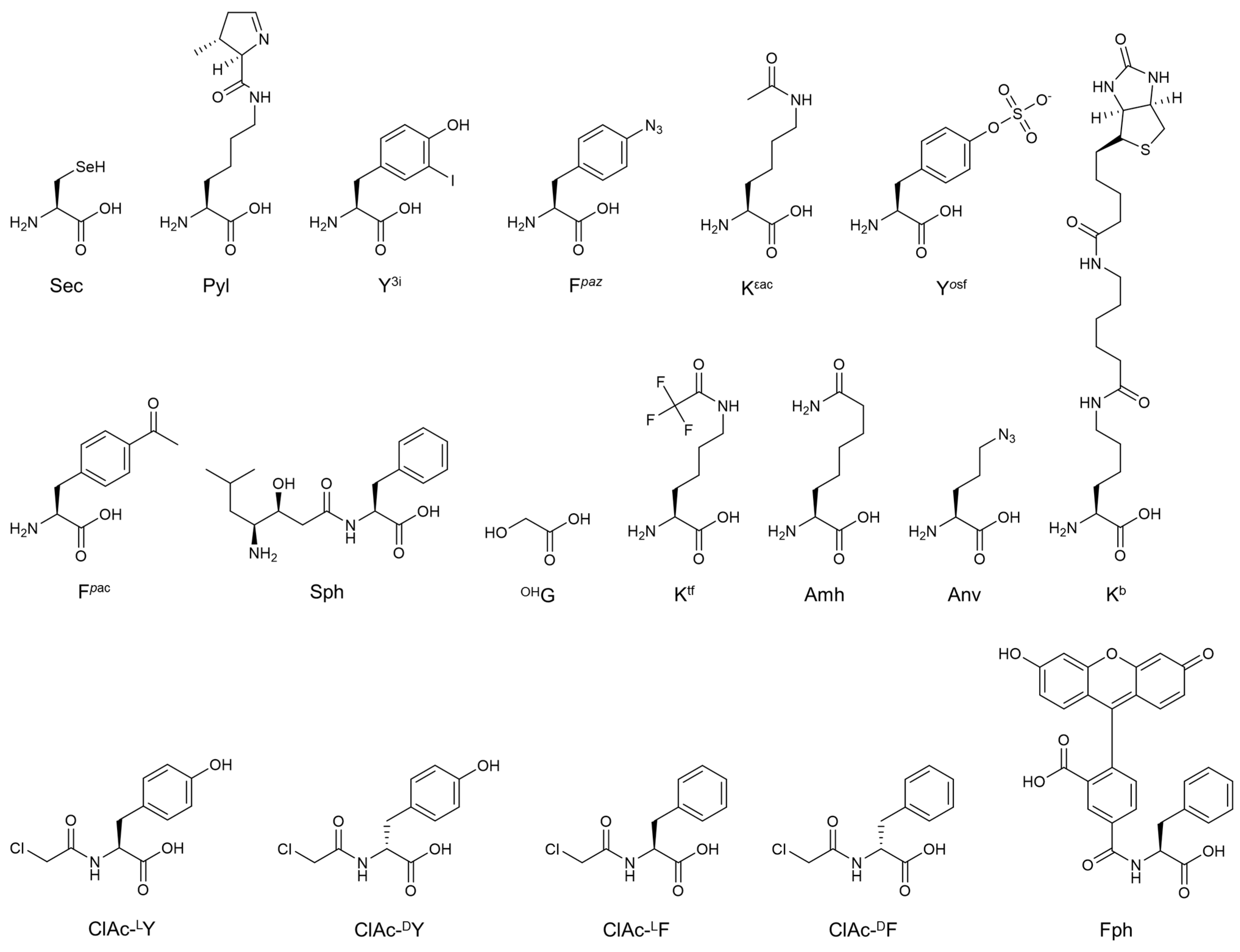{kind=link}
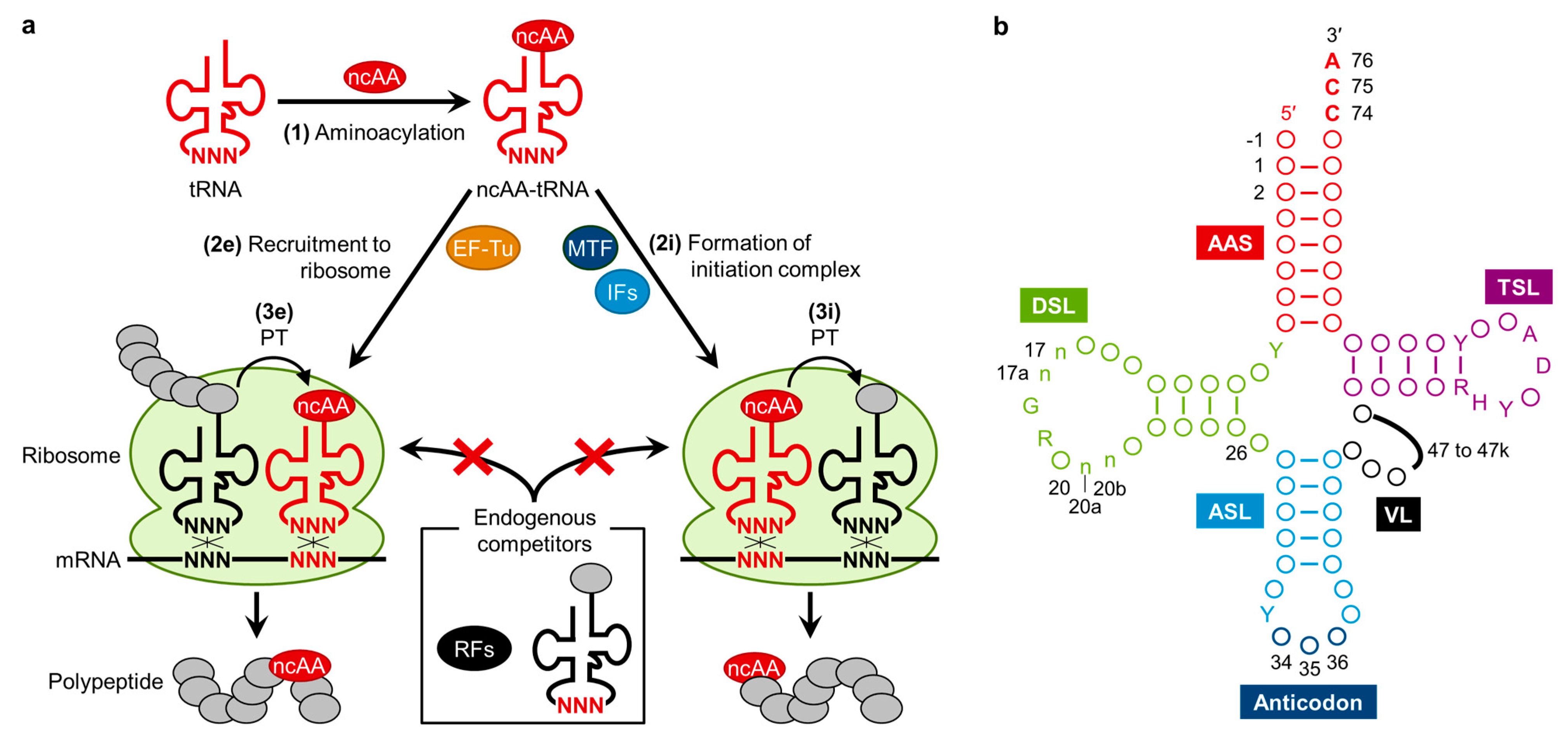{kind=link}
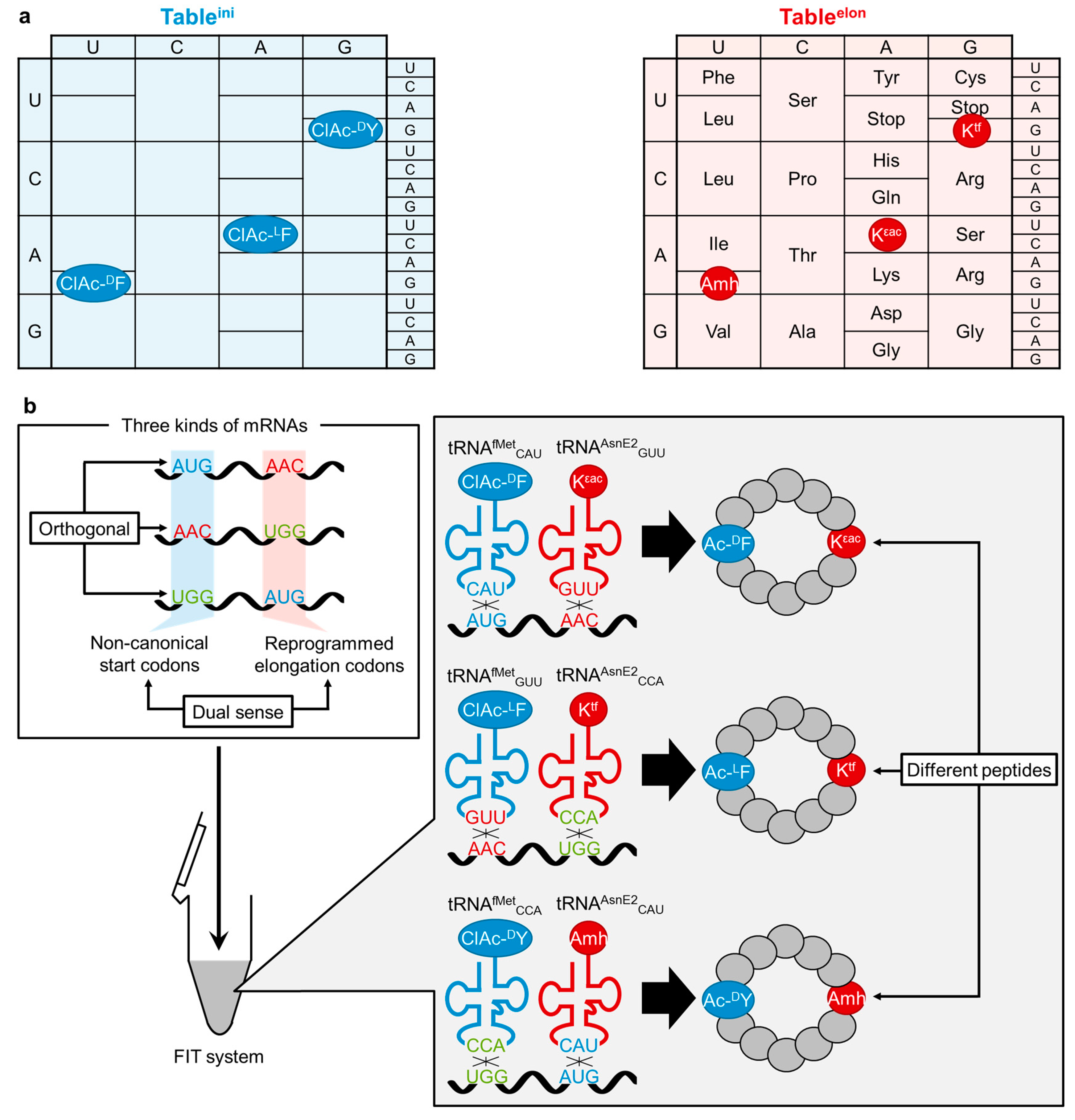{kind=link}
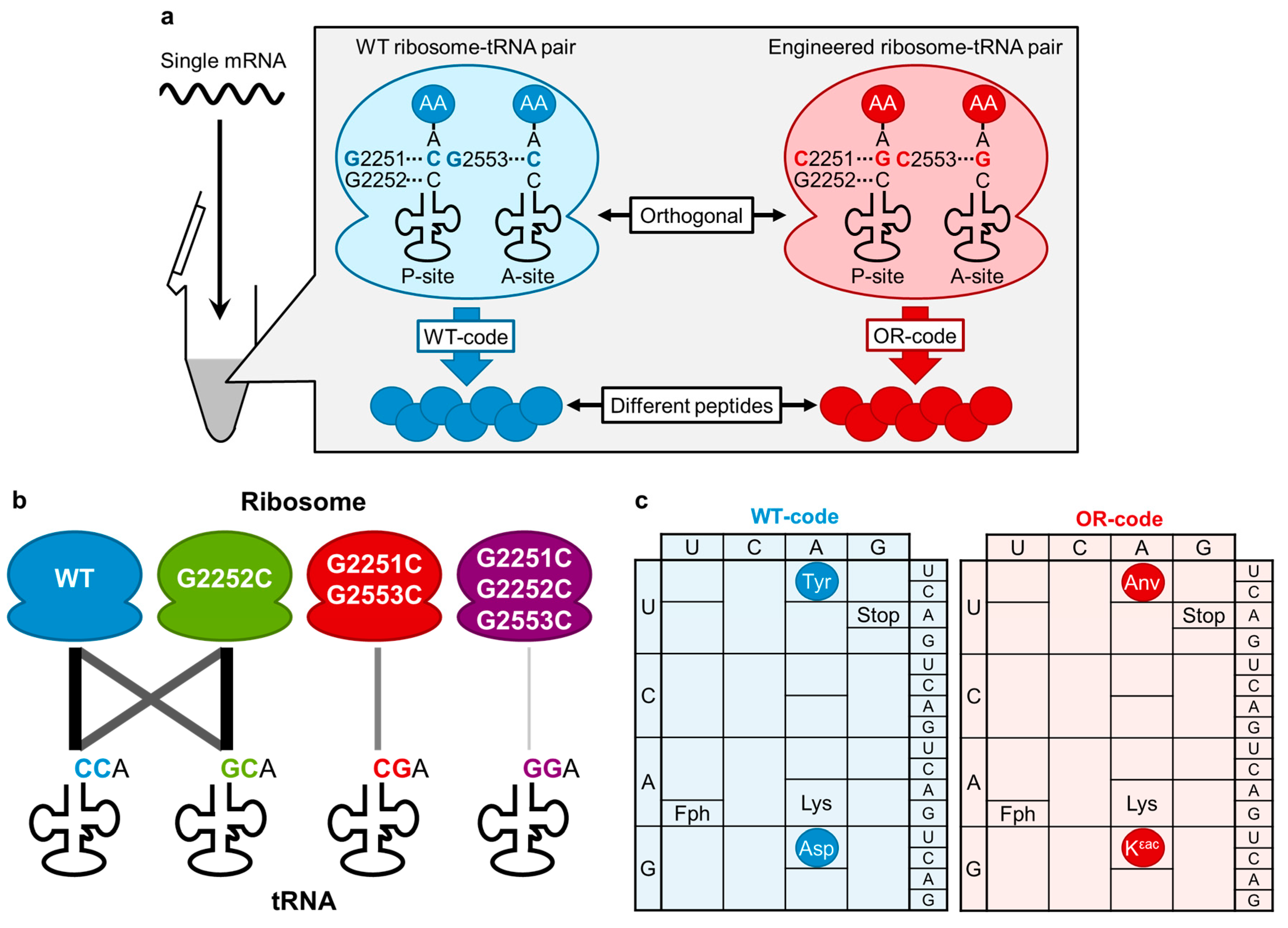
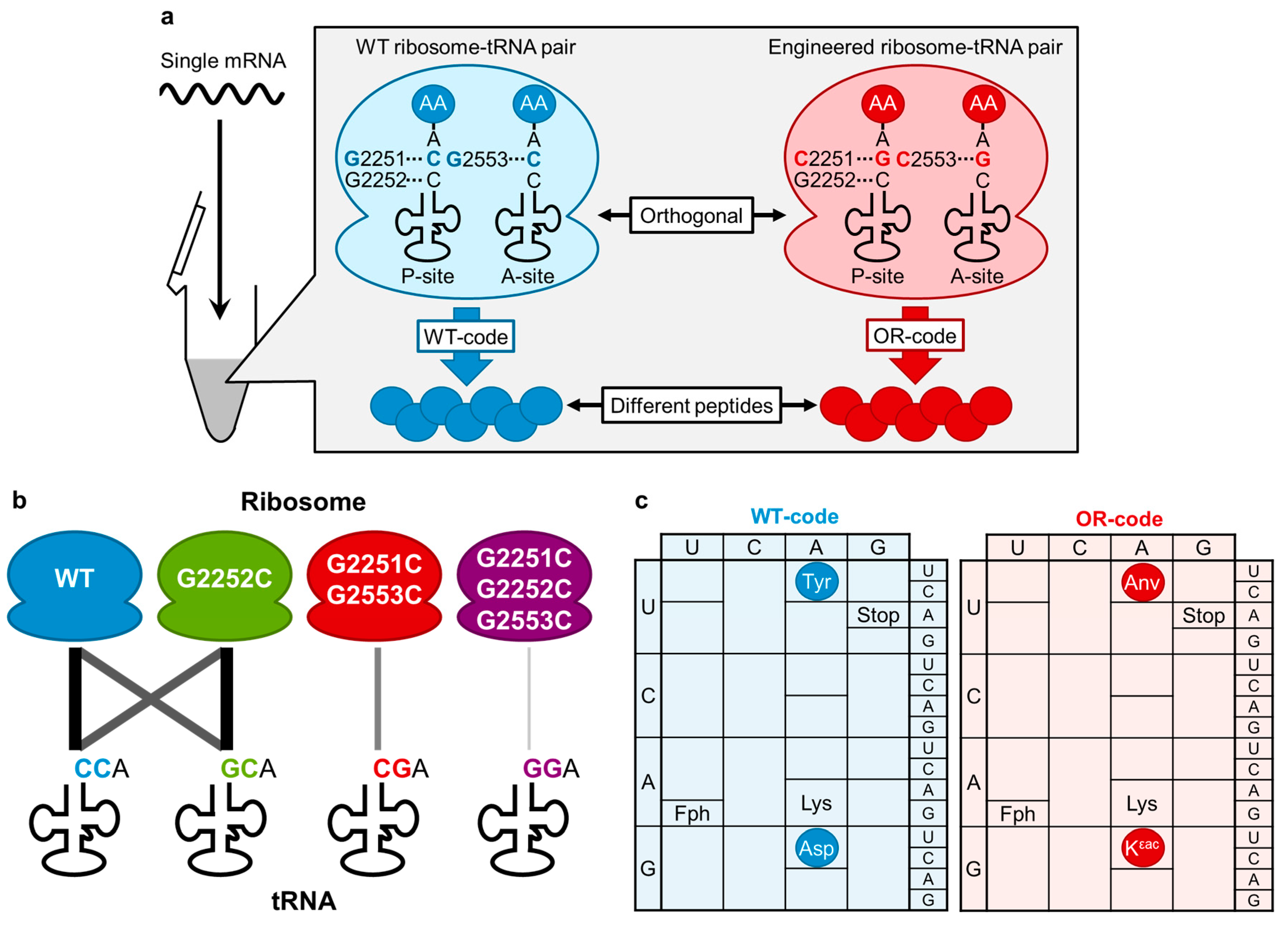{kind=link}
1. Introduction
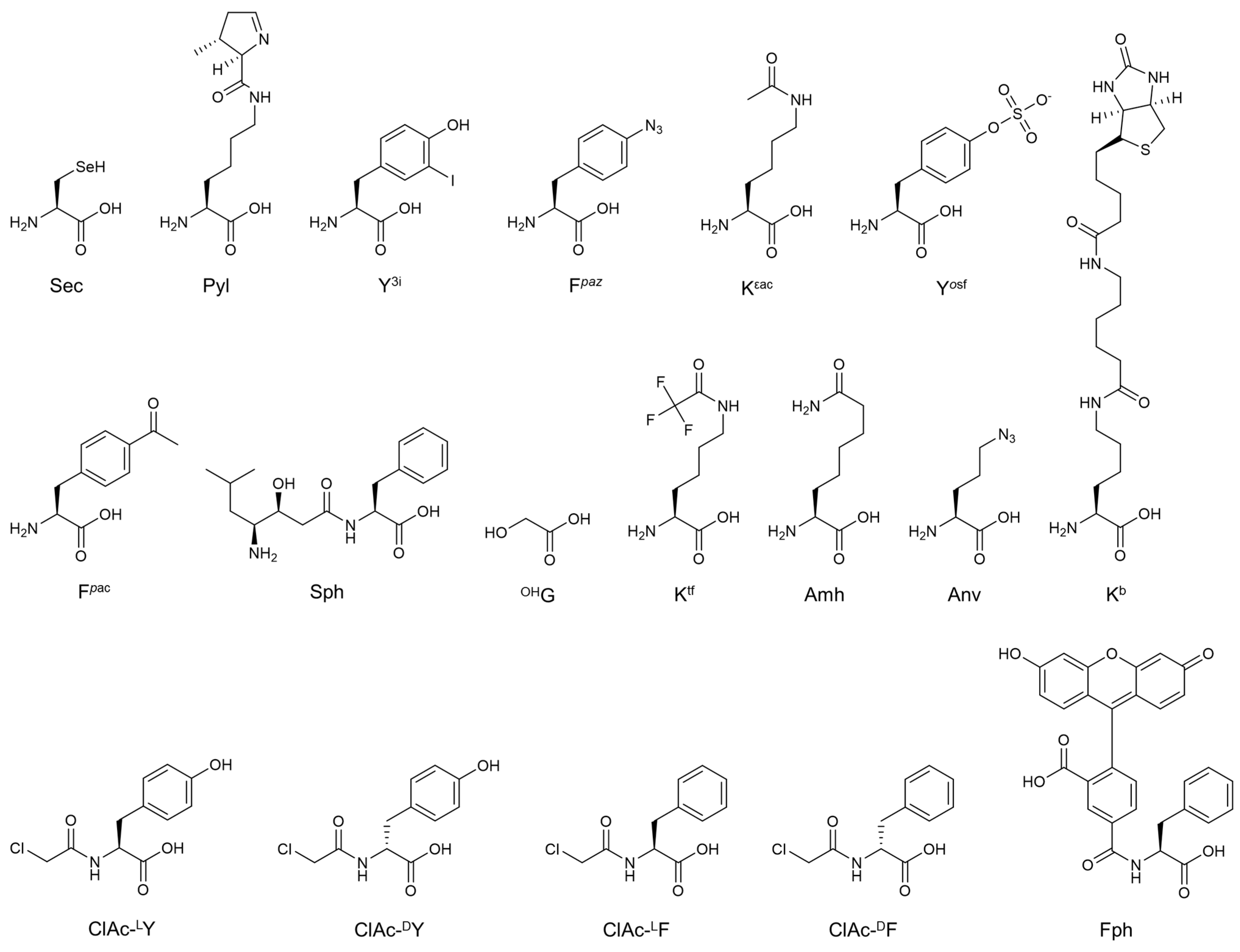

2. Engineering of Translation Components for the Improvement of ncAAs Incorporation Efficiency
3. Engineering of tRNAfMet for Usage of Multiple Initiators and Initiation Codons
4. Engineering of CCA-3' End of tRNA and rRNA for an Orthogonal Translation Machinery
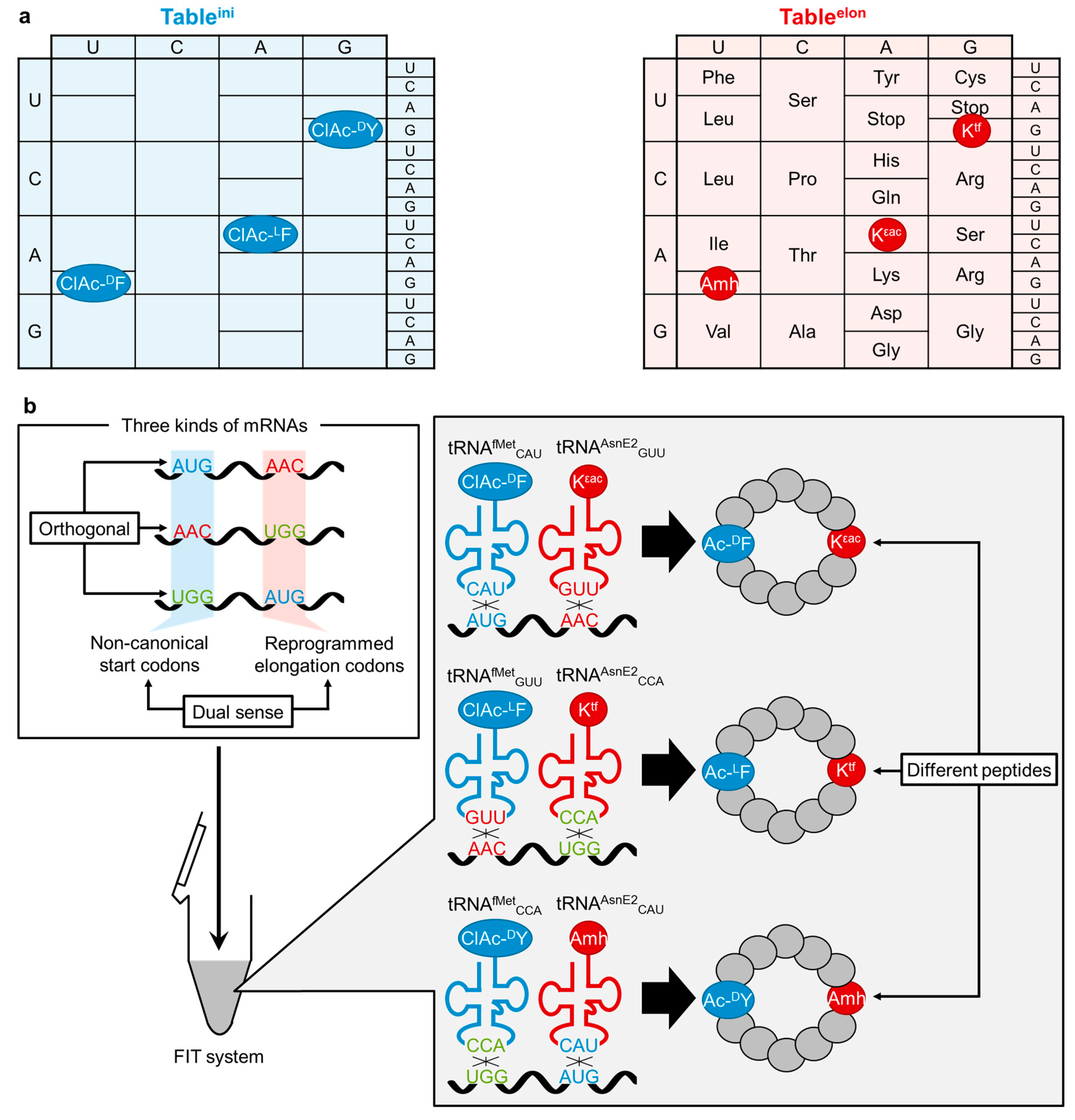
5. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ambrogelly, A.; Palioura, S.; Söll, D. Natural expansion of the genetic code. Nat. Chem. Biol. 2006, 3, 29–35. [Google Scholar] [CrossRef]
- Macino, G.; Coruzzi, G.; Nobrega, F.G. Use of the UGA terminator as a tryptophan codon in yeast mitochondria. Proc. Natl. Acad. Sci. USA 1979, 76, 3784–3785. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, N.N.; Schwientek, P.; Tripp, H.J.; Rinke, C.; Pati, A.; Huntemann, M.; Visel, A.; Woyke, T.; Kyrpides, N.C.; Rubin, E.M. Stop codon reassignments in the wild. Science 2014, 344, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Zinoni, F.; Birkmann, A.; Leinfelder, W.; Böck, A. Cotranslational insertion of selenocysteine into formate dehydrogenase from Escherichia coli directed by a UGA codon. Proc. Natl. Acad. Sci. USA 1987, 84, 3156–3160. [Google Scholar] [CrossRef] [PubMed]
- Hao, B.; Gong, W.M.; Ferguson, T.K.; James, C.M.; Krzycki, J.A.; Chan, M.K. A new UAG-encoded residue in the structure of a methanogen methyltransferase. Science 2002, 296, 1462–1466. [Google Scholar] [CrossRef] [PubMed]
- Gaston, M.A.; Jiang, R.; Krzycki, J.A. Functional context, biosynthesis, and genetic encoding of pyrrolysine. Curr. Opin. Microbiol. 2011, 14, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Schultz, P.G. Adding new chemistries to the genetic code. Annu. Rev. Biochem. 2010, 79, 413–444. [Google Scholar] [CrossRef] [PubMed]
- Magliery, T.J.; Anderson, J.C.; Schultz, P.G. Expanding the genetic code: Selection of efficient suppressors of four-base codons and identification of “shifty” four-base codons with a library approach in Escherichia coli. J. Mol. Biol. 2001, 307, 755–769. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Brock, A.; Herberich, B.; Schultz, P.G. Expanding the genetic code of Escherichia coli. Science 2001, 292, 498–500. [Google Scholar] [CrossRef] [PubMed]
- Forster, A.C.; Tan, Z.P.; Nalam, M.N.L.; Lin, H.N.; Qu, H.; Cornish, V.W.; Blacklow, S.C. Programming peptidomimetic syntheses by translating genetic codes designed de novo. Proc. Natl. Acad. Sci. USA 2003, 100, 6353–6357. [Google Scholar] [CrossRef] [PubMed]
- Marck, C.; Grosjean, H. tRNomics: Analysis of tRNA genes from 50 genomes of eukarya, archaea, and bacteria reveals anticodon-sparing strategies and domain-specific features. RNA 2002, 8, 1189–1232. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.; Huang, Y.; Wang, Z.; Russell, W.K.; Pai, P.J.; Russell, D.H.; Liu, W.R. A facile system for genetic incorporation of two different noncanonical amino acids into one protein in Escherichia coli. Angew. Chem. Int. Ed. Engl. 2010, 49, 3211–3214. [Google Scholar] [CrossRef] [PubMed]
- Hohsaka, T.; Ashizuka, Y.; Murakami, H.; Sisido, M. Incorporation of nonnatural amino acids into streptavidin through in vitro frame-shift suppression. J. Am. Chem. Soc. 1996, 118, 9778–9779. [Google Scholar] [CrossRef]
- O’Donoghue, P.; Prat, L.; Heinemann, I.U.; Ling, J.; Odoi, K.; Liu, W.R.; Söll, D. Near-cognate suppression of amber, opal and quadruplet codons competes with aminoacyl-tRNA(Pyl) for genetic code expansion. FEBS Lett. 2012, 586, 3931–3937. [Google Scholar] [CrossRef] [PubMed]
- Sando, S.; Kanatani, K.; Sato, N.; Matsumoto, H.; Hohsaka, T.; Aoyama, Y. A small-molecule-based approach to sense codon-templated natural-unnatural hybrid peptides. Selective silencing and reassignment of the sense codon by orthogonal reacylation stalling at the single-codon level. J. Am. Chem. Soc. 2005, 127, 7998–7999. [Google Scholar] [CrossRef] [PubMed]
- Forster, A.C.; Weissbach, H.; Blacklow, S.C. A simplified reconstitution of mRNA-directed peptide synthesis: Activity of the epsilon enhancer and an unnatural amino acid. Anal. Biochem. 2001, 297, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Inoue, A.; Tomari, Y.; Suzuki, T.; Yokogawa, T.; Nishikawa, K.; Ueda, T. Cell-free translation reconstituted with purified components. Nat. Biotechnol. 2001, 19, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Katoh, T.; Suga, H. Flexizymes for genetic code reprogramming. Nat. Protoc. 2011, 6, 779–790. [Google Scholar] [CrossRef] [PubMed]
- Ryden, S.M.; Isaksson, L.A. A temperature-sensitive mutant of Escherichia coli that shows enhanced misreading of UAG/A and increased efficiency for some tRNA nonsense suppressors. Mol. Gen. Genet. 1984, 193, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Mukai, T.; Hayashi, A.; Iraha, F.; Sato, A.; Ohtake, K.; Yokoyama, S.; Sakamoto, K. Codon reassignment in the Escherichia coli genetic code. Nucleic Acids Res. 2010, 38, 8188–8195. [Google Scholar] [CrossRef] [PubMed]
- Mukai, T.; Yanagisawa, T.; Ohtake, K.; Wakamori, M.; Adachi, J.; Hino, N.; Sato, A.; Kobayashi, T.; Hayashi, A.; Shirouzu, M.; et al. Genetic-code evolution for protein synthesis with non-natural amino acids. Biochem. Biophys. Res. Commun. 2011, 411, 757–761. [Google Scholar] [CrossRef] [PubMed]
- Lajoie, M.J.; Rovner, A.J.; Goodman, D.B.; Aerni, H.R.; Haimovich, A.D.; Kuznetsov, G.; Mercer, J.A.; Wang, H.H.; Carr, P.A.; Mosberg, J.A.; et al. Genomically recoded organisms expand biological functions. Science 2013, 342, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Xu, J.; Shen, Z.; Takimoto, J.K.; Schultz, M.D.; Schmitz, R.J.; Xiang, Z.; Ecker, J.R.; Briggs, S.P.; Wang, L. RF1 knockout allows ribosomal incorporation of unnatural amino acids at multiple sites. Nat. Chem. Biol. 2011, 7, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Uno, M.; Ito, K.; Nakamura, Y. Functional specificity of amino acid at position 246 in the tRNA mimicry domain of bacterial release factor 2. Biochimie 1996, 78, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Hohn, M.J.; Umehara, T.; Guo, L.T.; Osborne, E.M.; Benner, J.; Noren, C.J.; Rinehart, J.; Soll, D. Expanding the genetic code of Escherichia coli with phosphoserine. Science 2011, 333, 1151–1154. [Google Scholar] [CrossRef] [PubMed]
- Steinfeld, J.B.; Aerni, H.R.; Rogulina, S.; Liu, Y.C.; Rinehart, J. Expanded cellular amino acid pools containing phosphoserine, phosphothreonine, and phosphotyrosine. ACS Chem. Biol. 2014, 9, 1104–1112. [Google Scholar] [CrossRef] [PubMed]
- Wanner, B.L. Gene-regulation by phosphate in enteric bacteria. J. Cell. Biochem. 1993, 51, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Buechter, D.D.; Paolella, D.N.; Leslie, B.S.; Brown, M.S.; Mehos, K.A.; Gruskin, E.A. Co-translational incorporation of trans-4-hydroxyproline into recombinant proteins in bacteria. J. Biol. Chem. 2003, 278, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Fahnesto, S; Rich, A. Ribosome-catalyzed polyester formation. Science 1971, 173, 340–343. [Google Scholar]
- Merryman, C.; Green, R. Transformation of aminoacyl tRNAs for the in vitro selection of “drug-like” molecules. Chem. Biol. 2004, 11, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Hecht, S.M.; Alford, B.L.; Kuroda, Y.; Kitano, S. “Chemical aminoacylation” of tRNA’s. J. Biol. Chem. 1978, 253, 4517–4520. [Google Scholar] [PubMed]
- Lodder, M.; Wang, B.X.; Hecht, S.M. The N-pentenoyl protecting group for aminoacyl-tRNAs. Methods 2005, 36, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Polycarpo, C.R.; Herring, S.; Berube, A.; Wood, J.L.; Soll, D.; Ambrogelly, A. Pyrrolysine analogues as substrates for pyrrolysyl-tRNA synthetase. FEBS Lett. 2006, 580, 6695–6700. [Google Scholar] [CrossRef] [PubMed]
- Neumann, H.; Peak-Chew, S.Y.; Chin, J.W. Genetically encoding N(ε)-acetyllysine in recombinant proteins. Nat. Chem. Biol. 2008, 4, 232–234. [Google Scholar] [CrossRef] [PubMed]
- Mukai, T.; Kobayashi, T.; Hino, N.; Yanagisawa, T.; Sakamoto, K.; Yokoyama, S. Adding l-lysine derivatives to the genetic code of mammalian cells with engineered pyrrolysyl-tRNA synthetases. Biochem. Biophys. Res. Commun. 2008, 371, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, T.; Ishii, R.; Fukunaga, R.; Kobayashi, T.; Sakamoto, K.; Yokoyama, S. Multistep engineering of pyrrolysyl-tRNA synthetase to genetically encode N(ε)-(o-azidobenzyloxycarbonyl) lysine for site-specific protein modification. Chem. Biol. 2008, 15, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Murakami, H.; Kourouklis, D.; Suga, H. Using a solid-phase ribozyme aminoacylation system to reprogram the genetic code. Chem. Biol. 2003, 10, 1077–1084. [Google Scholar] [CrossRef] [PubMed]
- Murakami, H.; Ohta, A.; Ashigai, H.; Suga, H. A highly flexible tRNA acylation method for non-natural polypeptide synthesis. Nat. Methods 2006, 3, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Niwa, N.; Yamagishi, Y.; Murakami, H.; Suga, H. A flexizyme that selectively charges amino acids activated by a water-friendly leaving group. Bioorg. Med. Chem. Lett. 2009, 19, 3892–3894. [Google Scholar] [CrossRef] [PubMed]
- Suga, H.; Hayashi, G.; Terasaka, N. The RNA origin of transfer RNA aminoacylation and beyond. Philos. Trans. R. Soc. Lond. B 2011, 366, 2959–2964. [Google Scholar] [CrossRef]
- Kawakami, T.; Murakami, H.; Suga, H. Messenger RNA-programmed incorporation of multiple N-methyl-amino acids into linear and cyclic peptides. Chem. Biol. 2008, 15, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, T.; Ohta, A.; Ohuchi, M.; Ashigai, H.; Murakami, H.; Suga, H. Diverse backbone-cyclized peptides via codon reprogramming. Nat. Chem. Biol. 2009, 5, 888–890. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, Y.; Shoji, I.; Miyagawa, S.; Kawakami, T.; Katoh, T.; Goto, Y.; Suga, H. Natural product-like macrocyclic N-methyl-peptide inhibitors against a ubiquitin ligase uncovered from a ribosome-expressed de novo library. Chem. Biol. 2011, 18, 1562–1570. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, T.; Murakami, H.; Suga, H. Ribosomal synthesis of polypeptoids and peptoid–peptide hybrids. J. Am. Chem. Soc. 2008, 130, 16861–16863. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, T.; Ishizawa, T.; Murakami, H. Extensive reprogramming of the genetic code for genetically encoded synthesis of highly N-alkylated polycyclic peptidomimetics. J. Am. Chem. Soc. 2013, 135, 12297–12304. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Ohta, A.; Sako, Y.; Yamagishi, Y.; Murakami, H.; Suga, H. Reprogramming the translation initiation for the synthesis of physiologically stable cyclic peptides. ACS Chem. Biol. 2008, 3, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Suga, H. Translation initiation with initiator tRNA charged with exotic peptides. J. Am. Chem. Soc. 2009, 131, 5040–5041. [Google Scholar] [CrossRef] [PubMed]
- Ohta, A.; Murakami, H.; Higashimura, E.; Suga, H. Synthesis of polyester by means of genetic code reprogramming. Chem. Biol. 2007, 14, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Murakami, H.; Suga, H. Initiating translation with d-amino acids. RNA 2008, 14, 1390–1398. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Murakami, H.; Suga, H.; Ferré-D’Amaré, A.R. Structural basis of specific tRNA aminoacylation by a small in vitro selected ribozyme. Nature 2008, 454, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Terasaka, N.; Hayashi, G.; Katoh, T.; Suga, H. An orthogonal ribosome-tRNA pair via engineering of the peptidyl transferase center. Nat. Chem. Biol. 2014, 10, 555–557. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Kourouklis, D.; Suga, H. An in vitro evolved precursor tRNA with aminoacylation activity. EMBO J. 2001, 20, 1797–1806. [Google Scholar] [CrossRef] [PubMed]
- Ieong, K.W.; Pavlov, M.Y.; Kwiatkowski, M.; Forster, A.C.; Ehrenberg, M. Inefficient delivery but fast peptide bond formation of unnatural l-aminoacyl-tRNAs in translation. J. Am. Chem. Soc. 2012, 134, 17955–17962. [Google Scholar] [CrossRef] [PubMed]
- Doi, Y.; Ohtsuki, T.; Shimizu, Y.; Ueda, T.; Sisido, M. Elongation factor Tu mutants expand amino acid tolerance of protein biosynthesis system. J. Am. Chem. Soc. 2007, 129, 14458–14462. [Google Scholar] [CrossRef] [PubMed]
- Aldag, C.; Bröcker, M.J.; Hohn, M.J.; Prat, L.; Hammond, G.; Plummer, A.; Söll, D. Rewiring translation for elongation factor Tu-dependent selenocysteine incorporation. Angew. Chem. Int. Ed. Engl. 2013, 52, 1441–1445. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Oh, S.; Yang, A.; Kim, J.; Söll, D.; Lee, D.; Park, H.S. A facile strategy for selective incorporation of phosphoserine into histones. Angew. Chem. Int. Ed. Engl. 2013, 52, 5771–5775. [Google Scholar] [CrossRef] [PubMed]
- Dale, T.; Sanderson, L.E.; Uhlenbeck, O.C. The affinity of elongation factor Tu for an aminoacyl-tRNA is modulated by the esterified amino acid. Biochemistry 2004, 43, 6159–6166. [Google Scholar] [CrossRef] [PubMed]
- Ieong, K.W.; Pavlov, M.Y.; Kwiatkowski, M.; Ehrenberg, M.; Forster, A.C. A tRNA body with high affinity for EF-Tu hastens ribosomal incorporation of unnatural amino acids. RNA 2014, 20, 632–643. [Google Scholar] [CrossRef] [PubMed]
- Thyer, R.; Filipovska, A.; Rackham, O. Engineered rRNA enhances the efficiency of selenocysteine incorporation during translation. J. Am. Chem. Soc. 2013, 135, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Neumann, H.; Peak-Chew, S.Y.; Chin, J.W. Evolved orthogonal ribosomes enhance the efficiency of synthetic genetic code expansion. Nat. Biotechnol. 2007, 25, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Neumann, H.; Wang, K.; Davis, L.; Garcia-Alai, M.; Chin, J.W. Encoding multiple unnatural amino acids via evolution of a quadruplet-decoding ribosome. Nature 2010, 464, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Dedkova, L.M.; Fahmi, N.E.; Golovine, S.Y.; Hecht, S.M. Enhanced d-amino acid incorporation into protein by modified ribosomes. J. Am. Chem. Soc. 2003, 125, 6616–6617. [Google Scholar] [CrossRef] [PubMed]
- Dedkova, L.M.; Fahmi, N.E.; Paul, R.; del Rosario, M.; Zhang, L.Q.; Chen, S.X.; Feder, G.; Hecht, S.M. β-Puromycin selection of modified ribosomes for in vitro incorporation of β-amino acids. Biochemistry 2012, 51, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Maini, R.; Nguyen, D.T.; Chen, S.X.; Dedkova, L.M.; Chowdhury, S.R.; Alcala-Torano, R.; Hecht, S.M. Incorporation of β-amino acids into dihydrofolate reductase by ribosomes having modifications in the peptidyltransferase center. Bioorg. Med. Chem. 2013, 21, 1088–1096. [Google Scholar] [CrossRef] [PubMed]
- Fujino, T.; Goto, Y.; Suga, H.; Murakami, H. Reevaluation of the d-amino acid compatibility with the elongation event in translation. J. Am. Chem. Soc. 2013, 135, 1830–1837. [Google Scholar] [CrossRef] [PubMed]
- Kozak, M. Comparison of initiation of protein synthesis in procaryotes, eucaryotes, and organelles. Microbiol. Rev. 1983, 47, 1–45. [Google Scholar] [PubMed]
- Spurio, R.; Brandi, L.; Caserta, E.; Pon, C.L.; Gualerzi, C.O.; Misselwitz, R.; Krafft, C.; Welfle, K.; Welfle, H. The C-terminal subdomain (IF2 C-2) contains the entire fMet-tRNA binding site of initiation factor IF2. J. Biol. Chem. 2000, 275, 2447–2454. [Google Scholar] [CrossRef] [PubMed]
- Hansen, P.K.; Wikman, F.; Clark, B.F.C.; Hershey, J.W.B.; Petersen, H.U. Interaction between initiator Met-tRNA(fMet) and elongation factor EF-Tu from Escherichia coli. Biochimie 1986, 68, 697–703. [Google Scholar] [CrossRef] [PubMed]
- RajBhandary, U.L. Initiator transfer RNAs. J. Bacteriol. 1994, 176, 547–552. [Google Scholar] [PubMed]
- Rasmussen, L.C.V.; Laursen, B.S.; Mortensen, K.K.; Sperling-Petersen, H.U. Initiator tRNAs in bacteria and eukaryotes. eLS 2009. [Google Scholar] [CrossRef]
- Gualerzi, C.O.; Pon, C.L. Initiation of mRNA translation in prokaryotes. Biochemistry 1990, 29, 5881–5889. [Google Scholar] [CrossRef] [PubMed]
- Varshney, U.; RajBhandary, U.L. Initiation of protein synthesis from a termination codon. Proc. Natl. Acad. Sci. USA 1990, 87, 1586–1590. [Google Scholar] [CrossRef] [PubMed]
- Chattapadhyay, R.; Pelka, H.; Schulman, L.H. Initiation of in vivo protein synthesis with non-methionine amino acids. Biochemistry 1990, 29, 4263–4268. [Google Scholar] [CrossRef] [PubMed]
- Mamaev, S.; Olejnik, J.; Olejnik, E.K.; Rothschild, K.J. Cell-free N-terminal protein labeling using initiator suppressor tRNA. Anal. Biochem. 2004, 326, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Olejnik, J.; Gite, S.; Mamaev, S.; Rothschild, K.J. N-Terminal labeling of proteins using initiator tRNA. Methods 2005, 36, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Muranaka, N.; Miura, M.; Taira, H.; Hohsaka, T. Incorporation of unnatural non-α-amino acids into the N-terminus of proteins in a cell-free translation system. Chembiochem 2007, 8, 1650–1653. [Google Scholar] [CrossRef] [PubMed]
- Ohshiro, Y.; Nakajima, E.; Goto, Y.; Fuse, S.; Takahashi, T.; Doi, T.; Suga, H. Ribosomal synthesis of backbone-macrocyclic peptides containing γ-amino acids. Chembiochem 2011, 12, 1183–1187. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Iseki, M.; Hitomi, A.; Murakami, H.; Suga, H. Nonstandard peptide expression under the genetic code consisting of reprogrammed dual sense codons. ACS Chem. Biol. 2013, 8, 2630–2634. [Google Scholar] [CrossRef] [PubMed]
- Crick, F.H. On protein synthesis. Symp. Soc. Exp. Biol. 1958, 12, 138–163. [Google Scholar] [PubMed]
- Ibba, M.; Becker, H.D.; Stathopoulos, C.; Tumbula, D.L.; Söll, D. The adaptor hypothesis revisited. Trends Biochem. Sci. 2000, 25, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Terasaka, N.; Suga, H. Flexizymes-facilitated genetic code reprogramming leading to the discovery of drug-like peptides. Chem. Lett. 2014, 43, 11–19. [Google Scholar] [CrossRef]
- Morimoto, J.; Hayashi, Y.; Suga, H. Discovery of macrocyclic peptides armed with a mechanism-based warhead: Isoform-selective inhibition of human deacetylase SIRT2. Angew. Chem. Int. Ed. 2012, 51, 3423–3427. [Google Scholar] [CrossRef]
- Bashan, A.; Agmon, I.; Zarivach, R.; Schluenzen, F.; Harms, J.; Berisio, R.; Bartels, H.; Franceschi, F.; Auerbach, T.; Hansen, H.A.; et al. Structural basis of the ribosomal machinery for peptide bond formation, translocation, and nascent chain progression. Mol. Cell 2003, 11, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.L.; Schmeing, T.M.; Moore, P.B.; Steitz, T.A. Structural insights into peptide bond formation. Proc. Natl. Acad. Sci. USA 2002, 99, 11670–11675. [Google Scholar] [CrossRef] [PubMed]
- Nissen, P.; Hansen, J.; Ban, N.; Moore, P.B.; Steitz, T.A. The structural basis of ribosome activity in peptide bond synthesis. Science 2000, 289, 920–930. [Google Scholar] [CrossRef] [PubMed]
- Steitz, T.A. A structural understanding of the dynamic ribosome machine. Nat. Rev. Mol. Cell Biol. 2008, 9, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Moazed, D.; Noller, H.F. Sites of interaction of the CCA end of peptidyl-tRNA with 23S rRNA. Proc. Natl. Acad. Sci. USA 1991, 88, 3725–3728. [Google Scholar] [CrossRef] [PubMed]
- Samaha, R.R.; Green, R.; Noller, H.F. A base pair between tRNA and 23S rRNA in the peptidyl transferase centre of the ribosome. Nature 1995, 377, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Lescoute, A.; Westhof, E. The interaction networks of structured RNAs. Nucleic Acids Res. 2006, 34, 6587–6604. [Google Scholar] [CrossRef] [PubMed]
- Green, R.; Switzer, C.; Noller, H.F. Ribosome-catalyzed peptide-bond formation with an A-site substrate covalently linked to 23S ribosomal RNA. Science 1998, 280, 286–289. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.F.; Green, R. Base-pairing between 23S rRNA and tRNA in the ribosomal a site. Mol. Cell 1999, 4, 859–864. [Google Scholar] [CrossRef] [PubMed]
- Cavarelli, J.; Moras, D. Recognition of tRNAs by aminoacyl-tRNA synthetases. FASEB J. 1993, 7, 79–86. [Google Scholar] [PubMed]
- Ruff, M.; Krishnaswamy, S.; Boeglin, M.; Poterszman, A.; Mitschler, A.; Podjarny, A.; Rees, B.; Thierry, J.C.; Moras, D. Class II aminoacyl transfer RNA synthetases: Crystal structure of yeast aspartyl-tRNA synthetase complexed with tRNA(Asp). Science 1991, 252, 1682–1689. [Google Scholar] [CrossRef] [PubMed]
- Schulman, L.H.; Pelka, H. Structural requirements for aminoacylation of Escherichia coli formylmethionine transfer RNA. Biochemistry 1977, 16, 4256–4265. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Horowitz, J. Functional transfer RNAs with modifications in the 3'-CCA end: Differential effects on aminoacylation and polypeptide synthesis. Proc. Natl. Acad. Sci. USA 1994, 91, 10389–10393. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.L.; Du, D.H.; Tan, M.; Lei, H.Y.; Ruan, L.L.; Eriani, G.; Wang, E.D. Role of tRNA amino acid-accepting end in aminoacylation and its quality control. Nucleic Acids Res. 2011, 39, 8857–8868. [Google Scholar] [CrossRef] [PubMed]
- Hipolito, C.J.; Bashiruddin, N.K.; Suga, H. Protein cocrystallization molecules originating from in vitro selected macrocyclic peptides. Curr. Opin. Struct. Biol. 2014, 26C, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Ishizawa, T.; Kawakami, T.; Reid, P.C.; Murakami, H. Trap display: A high-speed selection method for the generation of functional polypeptides. J. Am. Chem. Soc. 2013, 135, 5433–5440. [Google Scholar] [CrossRef] [PubMed]
- Hipolito, C.J.; Suga, H. Ribosomal production and in vitro selection of natural product-like peptidomimetics: The FIT and RaPID systems. Curr. Opin. Chem. Biol. 2012, 16, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Coin, I.; Katritch, V.; Sun, T.T.; Xiang, Z.; Siu, F.Y.; Beyermann, M.; Stevens, R.C.; Wang, L. Genetically encoded chemical probes in cells reveal the binding path of Urocortin-I to CRF class B GPCR. Cell 2013, 155, 1258–1269. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, T.; Felsovalyi, K.; Chen, C. Quantitative analysis of T cell receptor complex interaction sites using genetically encoded photo-crosslinkers. ACS Chem. Biol. 2014, 9, 2165–2172. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xiang, Z.; Hu, Y.S.; Lacey, V.; Cang, H. Genetically encoding an electrophilic amino acid for protein stapling and covalent binding to native receptors. ACS Chem. Biol. 2014, 9, 1956–1961. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terasaka, N.; Iwane, Y.; Geiermann, A.-S.; Goto, Y.; Suga, H. Recent Developments of Engineered Translational Machineries for the Incorporation of Non-Canonical Amino Acids into Polypeptides. Int. J. Mol. Sci. 2015, 16, 6513-6531. https://doi.org/10.3390/ijms16036513
Terasaka N, Iwane Y, Geiermann A-S, Goto Y, Suga H. Recent Developments of Engineered Translational Machineries for the Incorporation of Non-Canonical Amino Acids into Polypeptides. International Journal of Molecular Sciences. 2015; 16(3):6513-6531. https://doi.org/10.3390/ijms16036513
Chicago/Turabian StyleTerasaka, Naohiro, Yoshihiko Iwane, Anna-Skrollan Geiermann, Yuki Goto, and Hiroaki Suga. 2015. "Recent Developments of Engineered Translational Machineries for the Incorporation of Non-Canonical Amino Acids into Polypeptides" International Journal of Molecular Sciences 16, no. 3: 6513-6531. https://doi.org/10.3390/ijms16036513