
Monitoring of Apoptosis in 3D Cell Cultures by FRET and Light Sheet Fluorescence Microscopy


Abstract
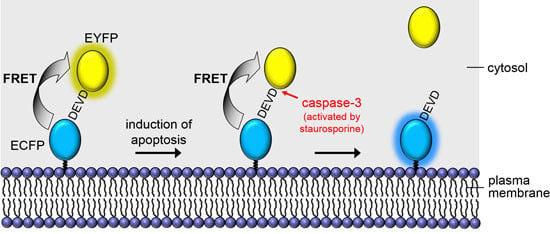
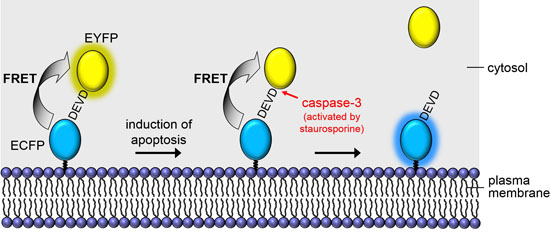
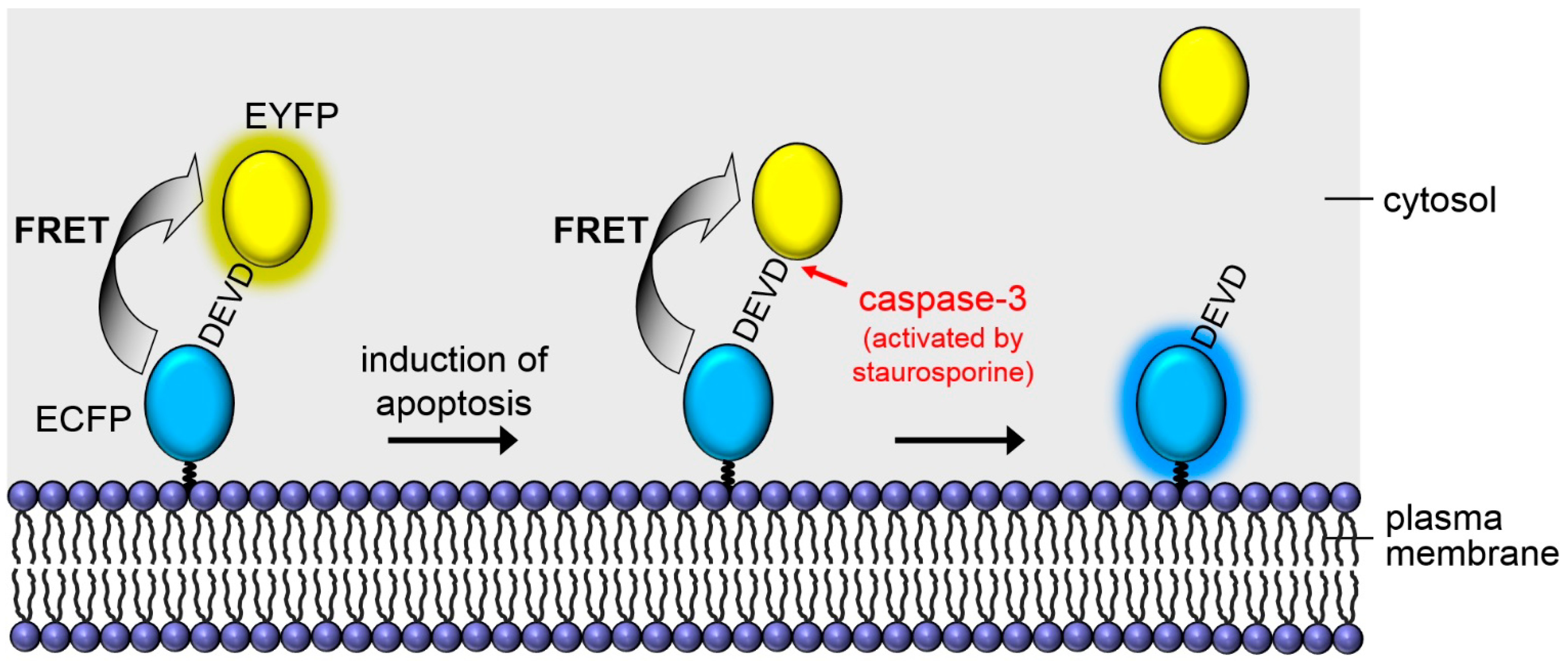
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
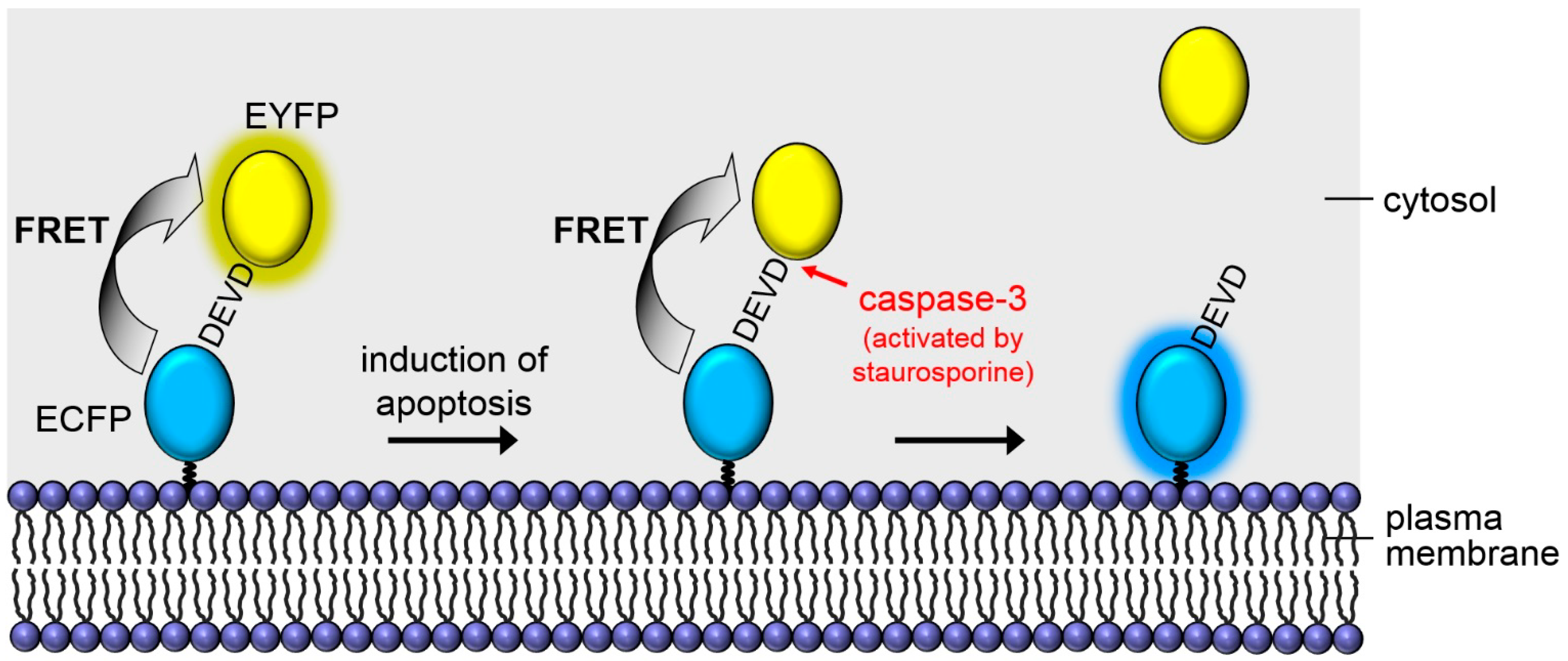
2. Results
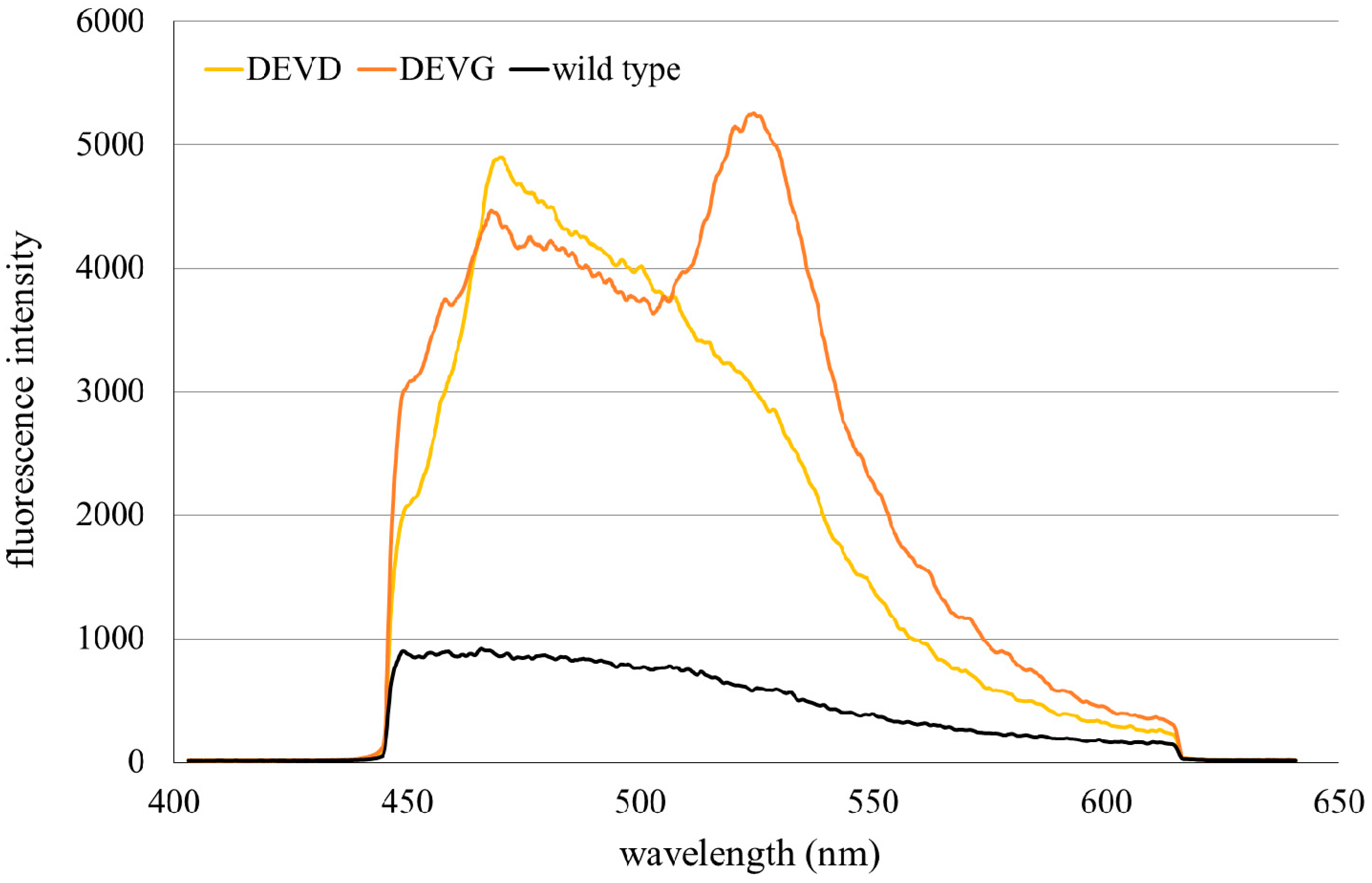
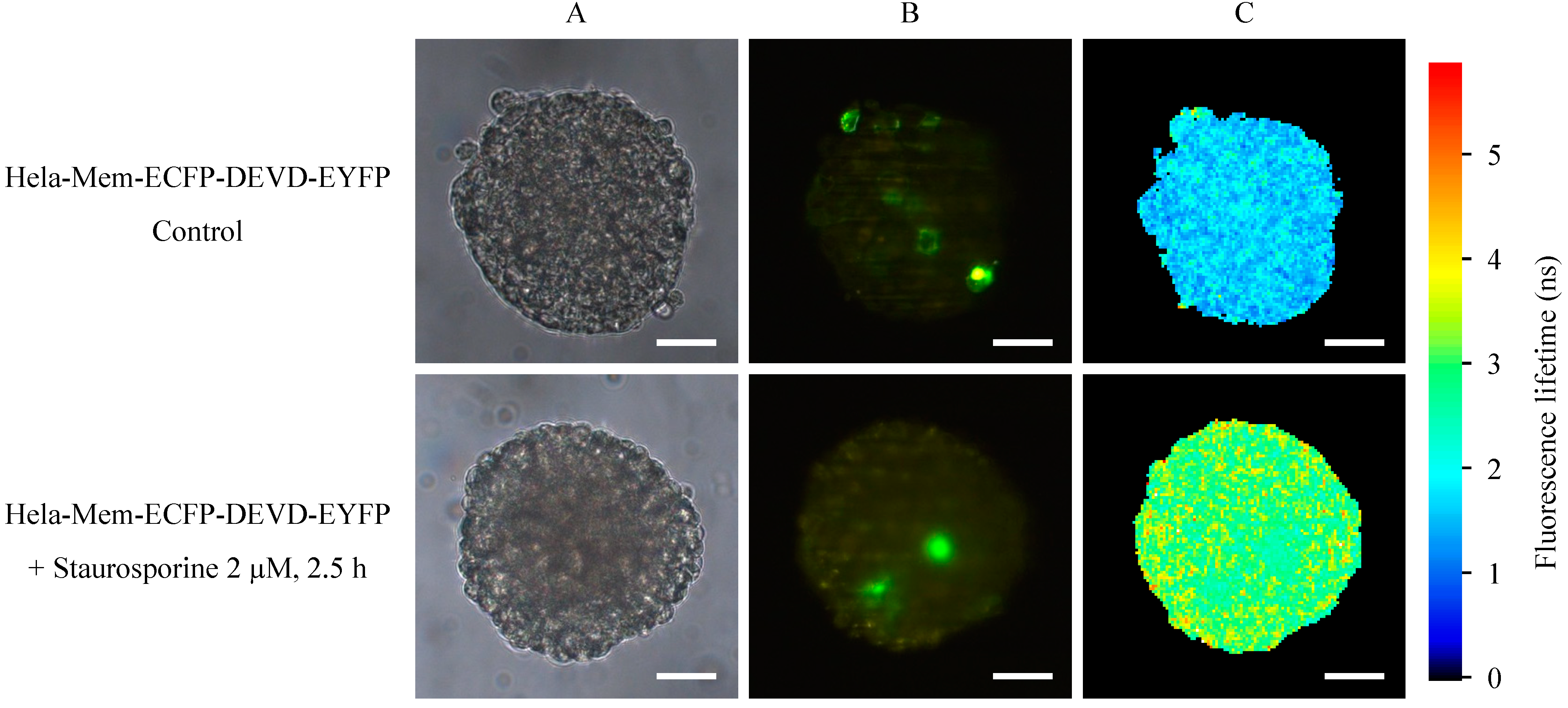
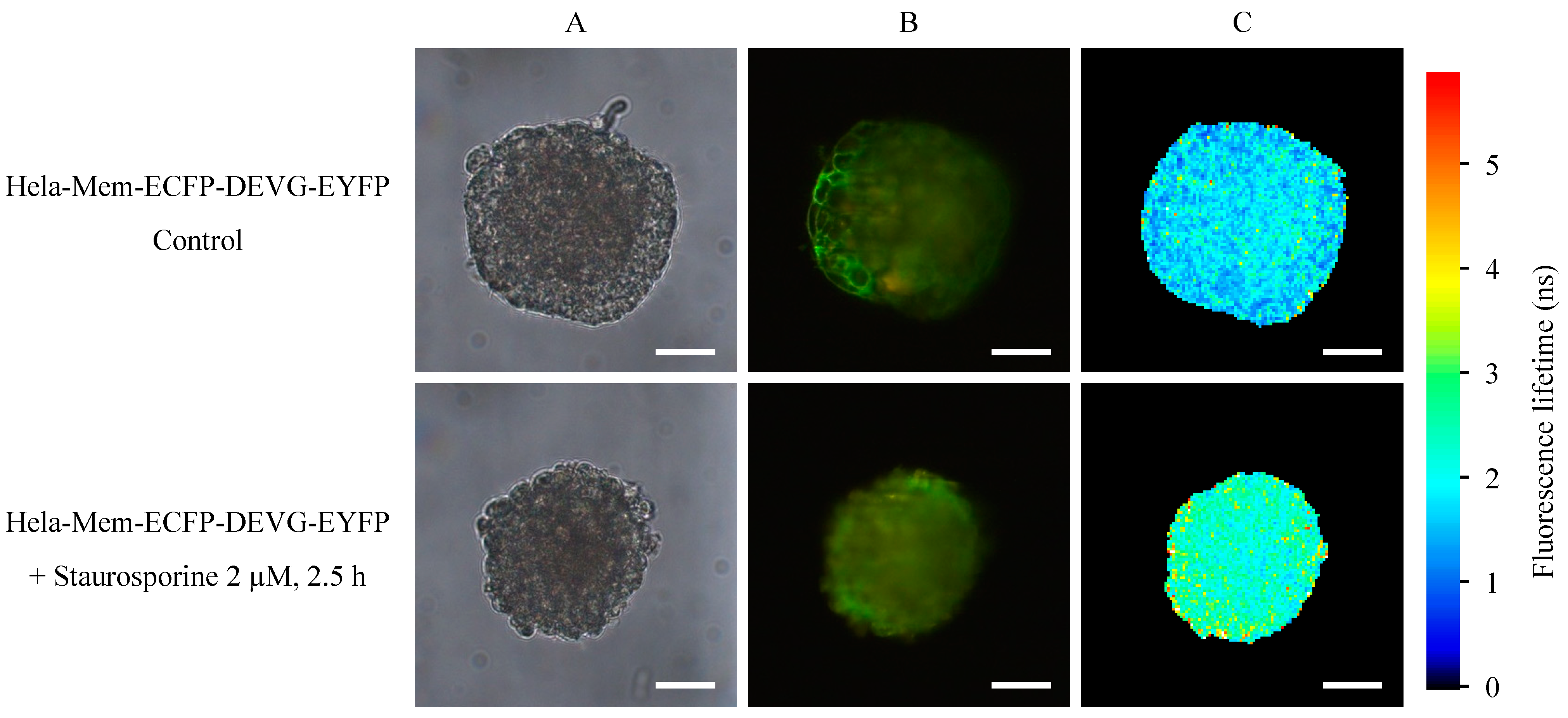
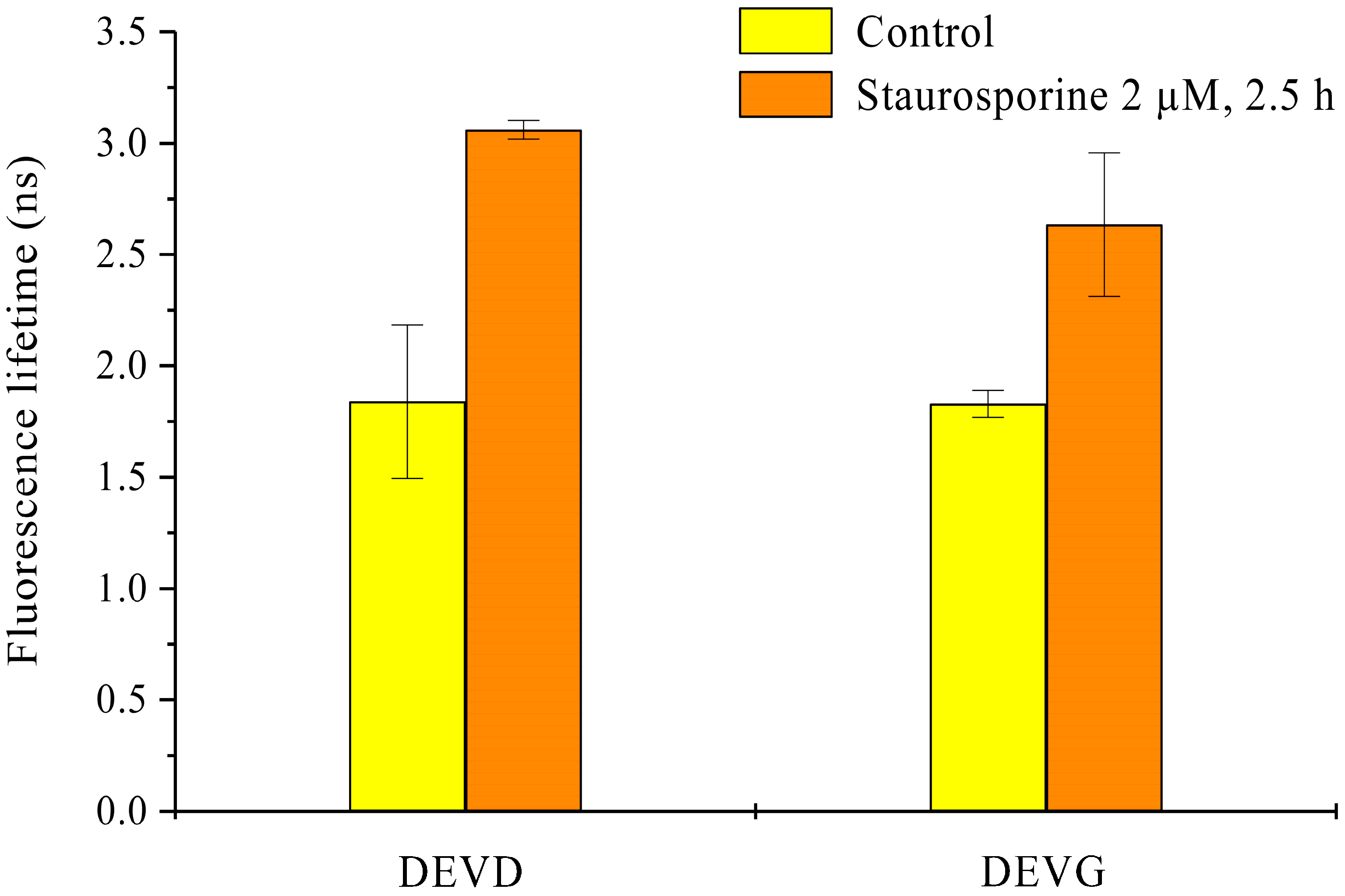
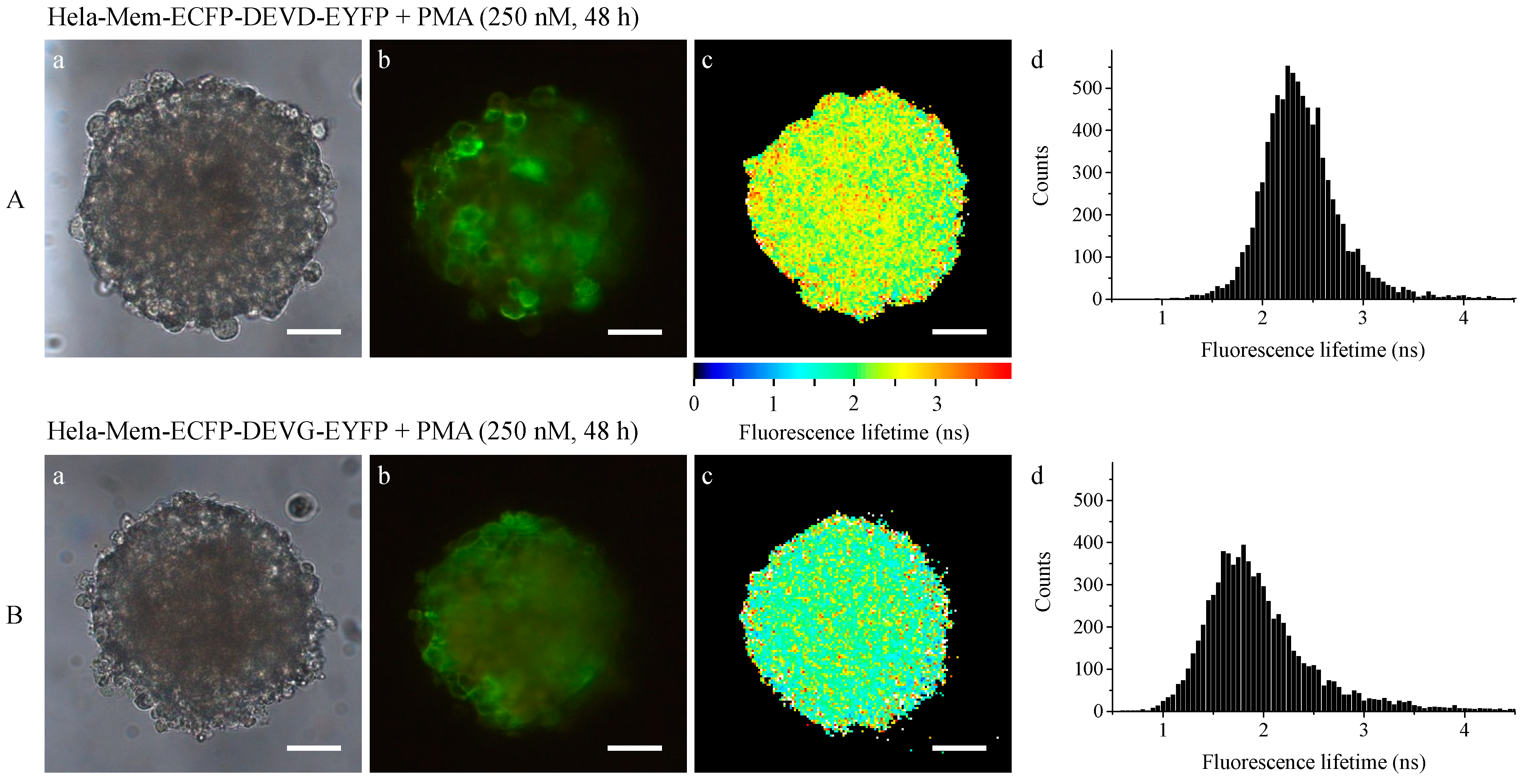
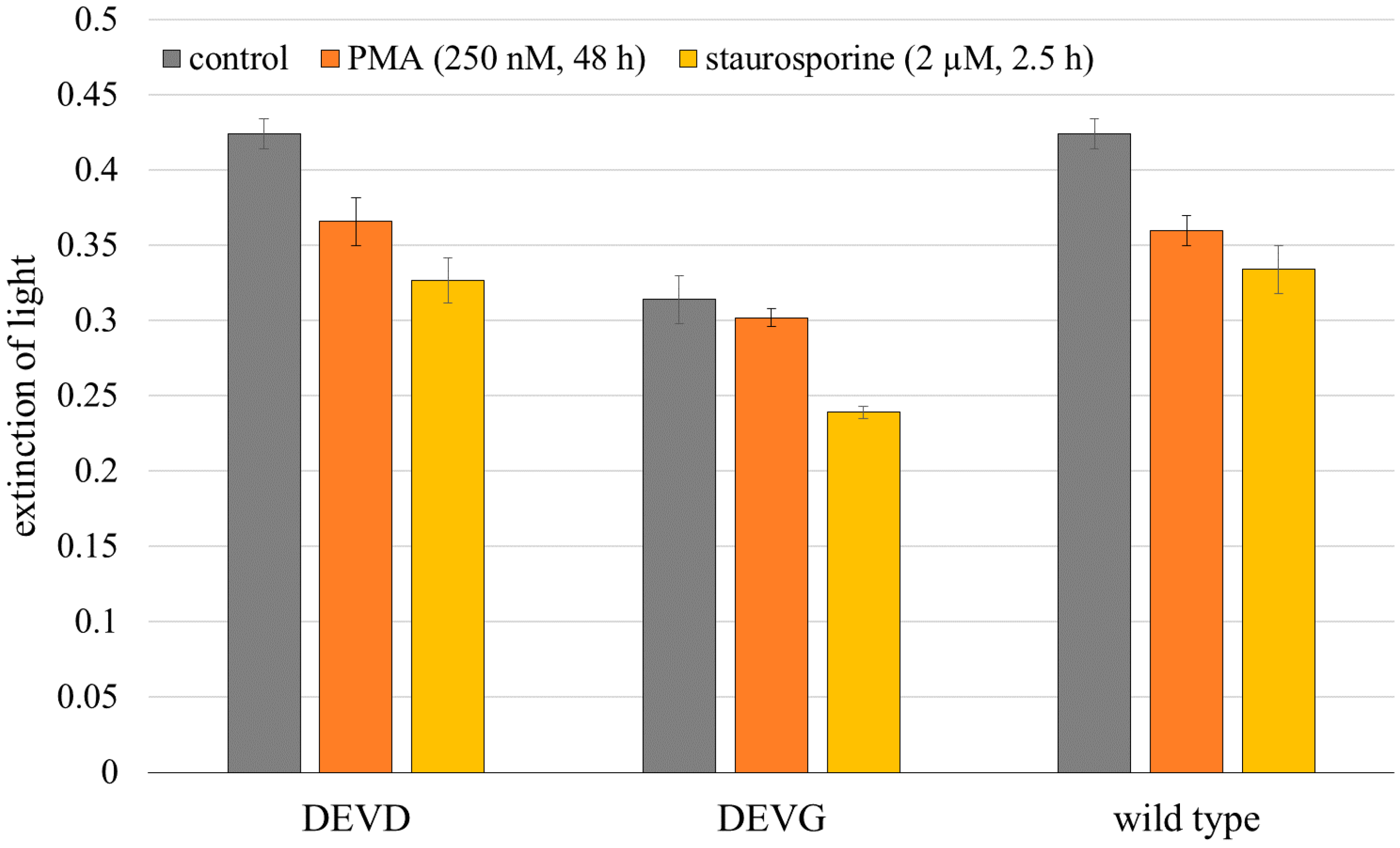
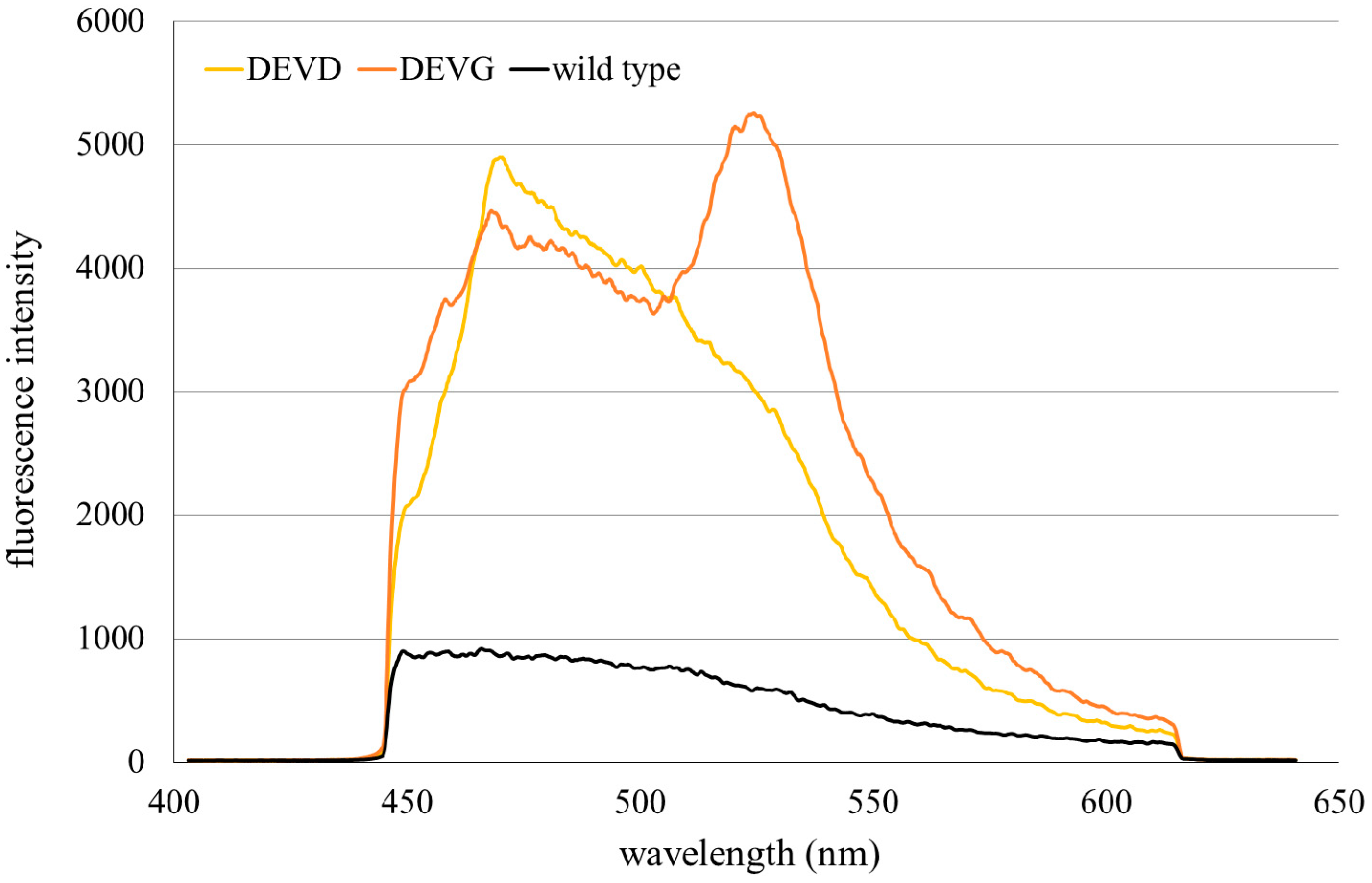
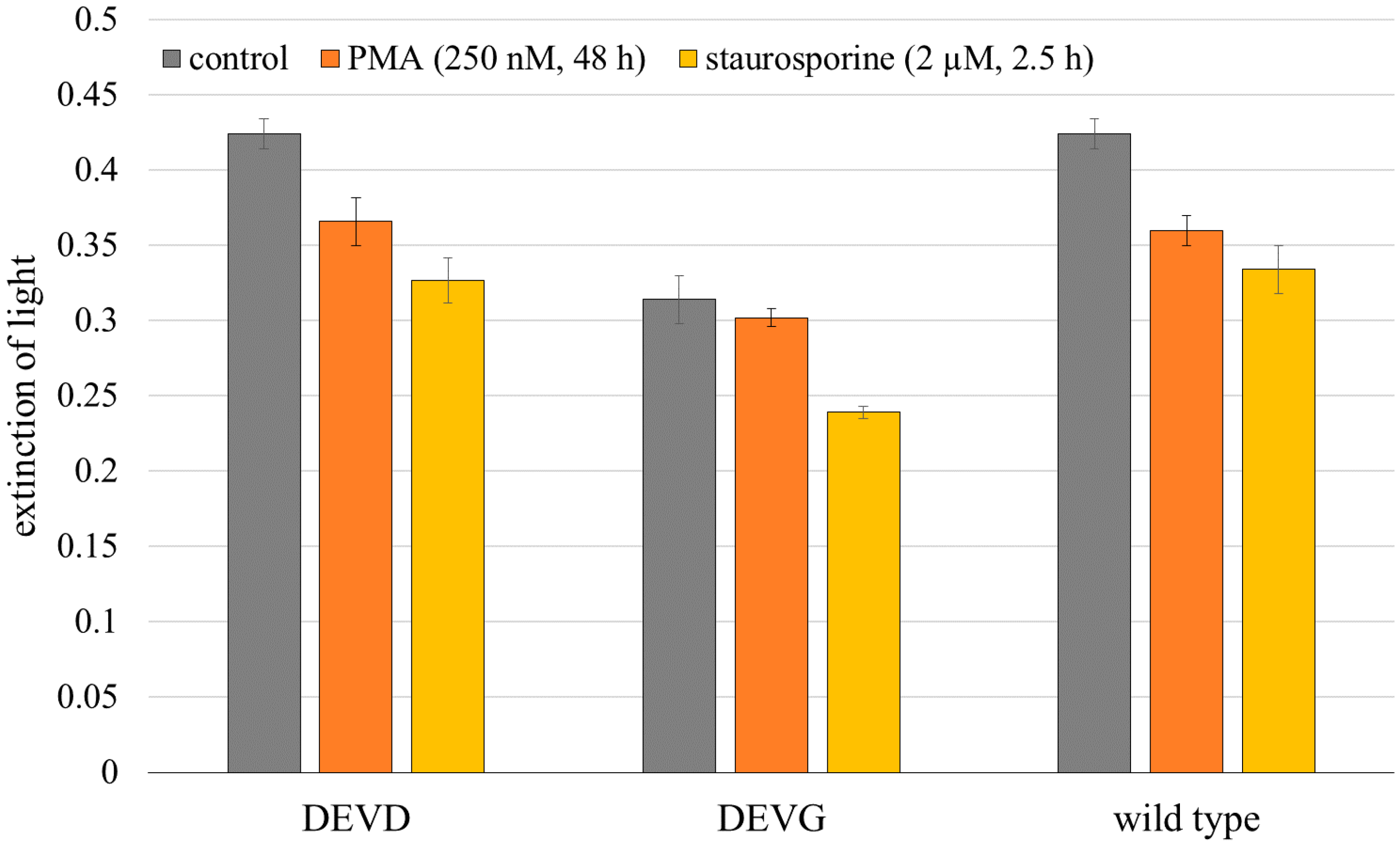
2.1. Microspectral Analysis
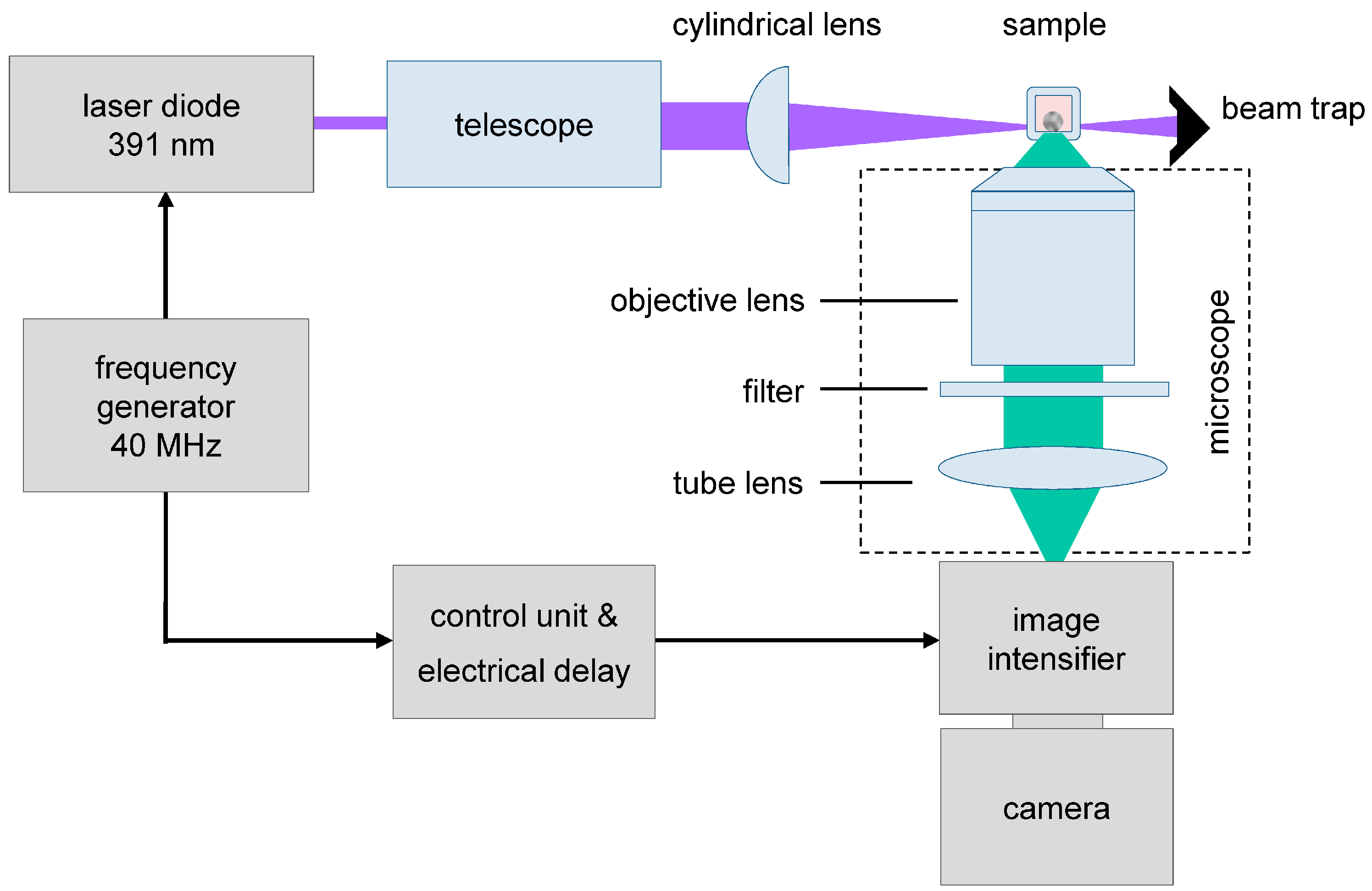
2.2. Light Sheet Based Fluorescence Microscopy and Lifetime Imaging (FLIM)
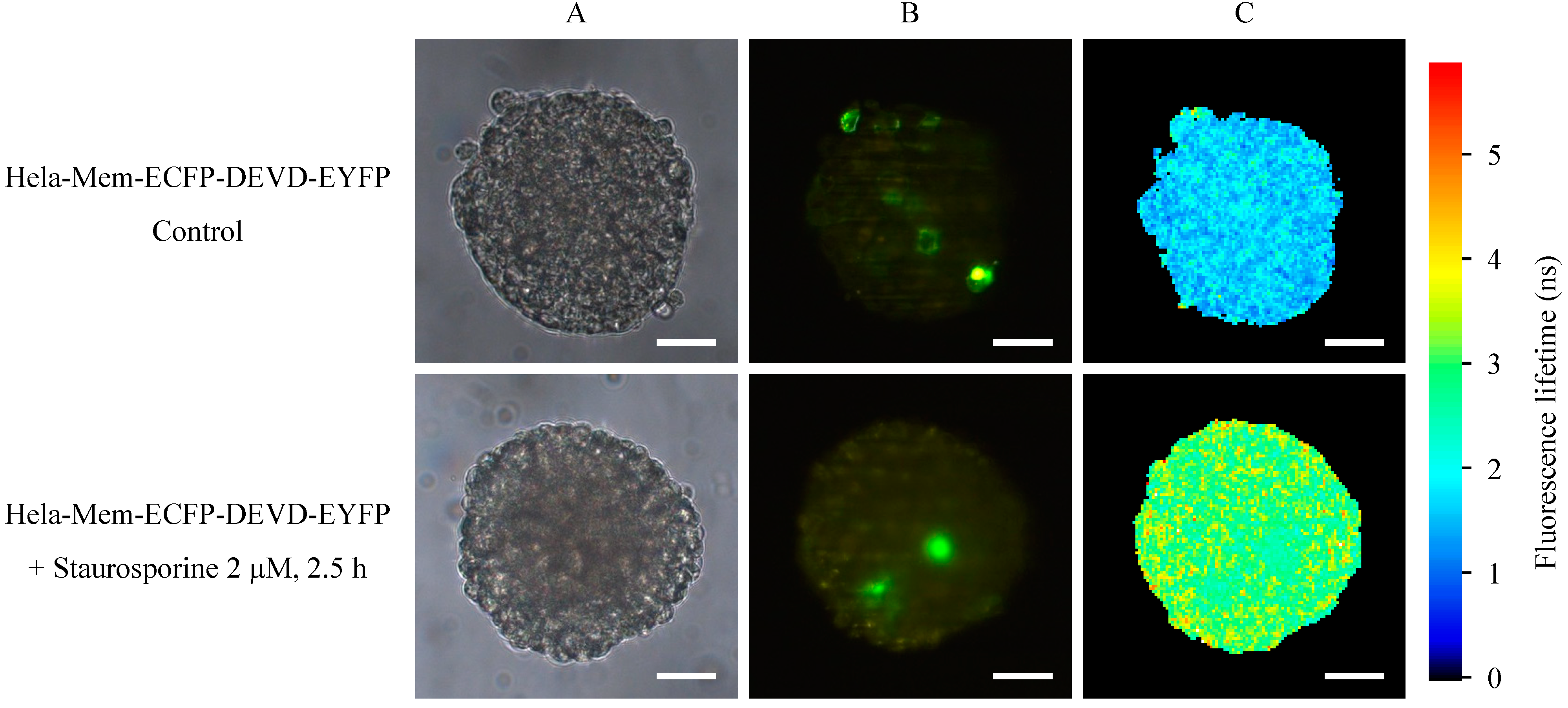

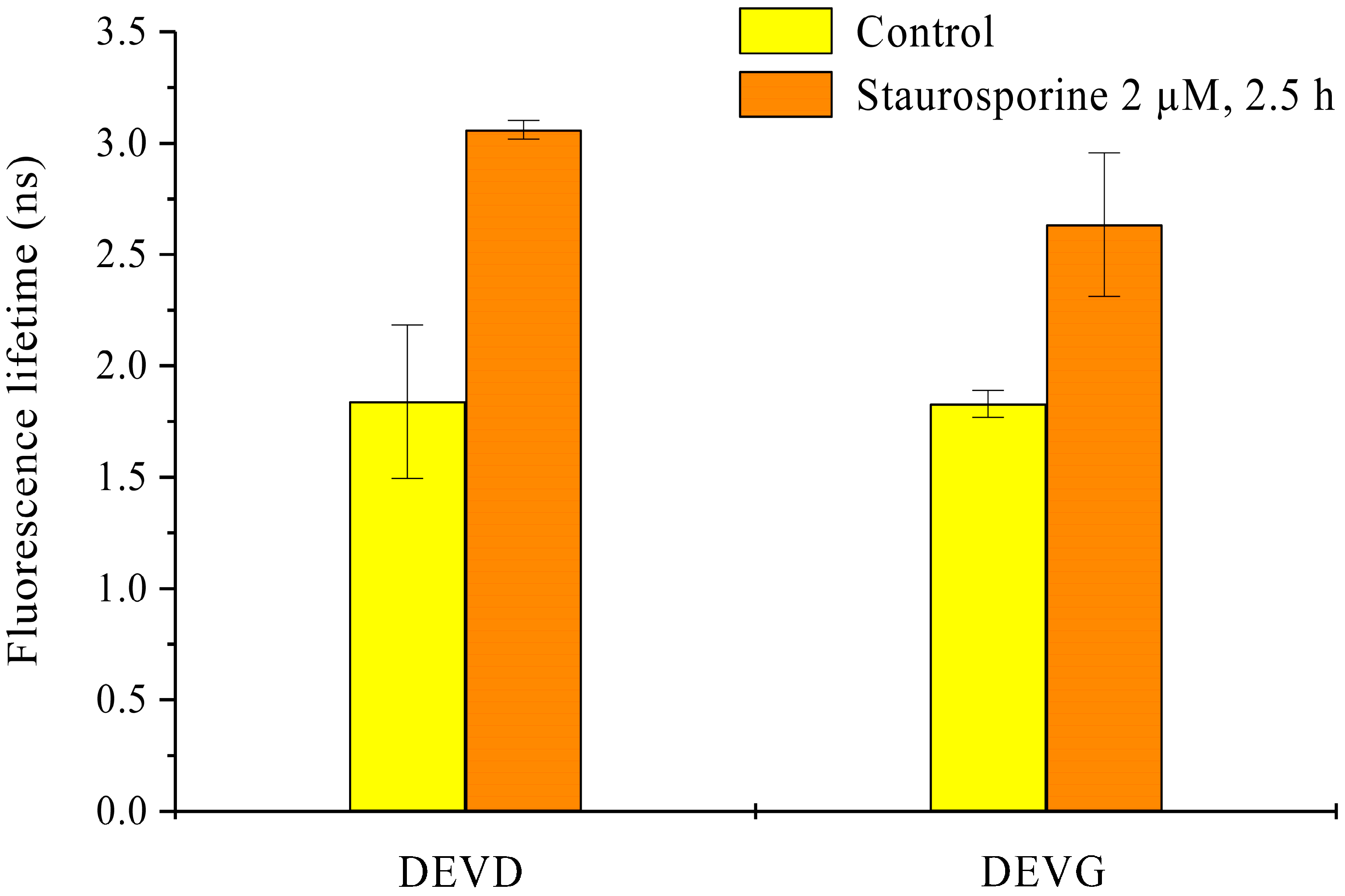
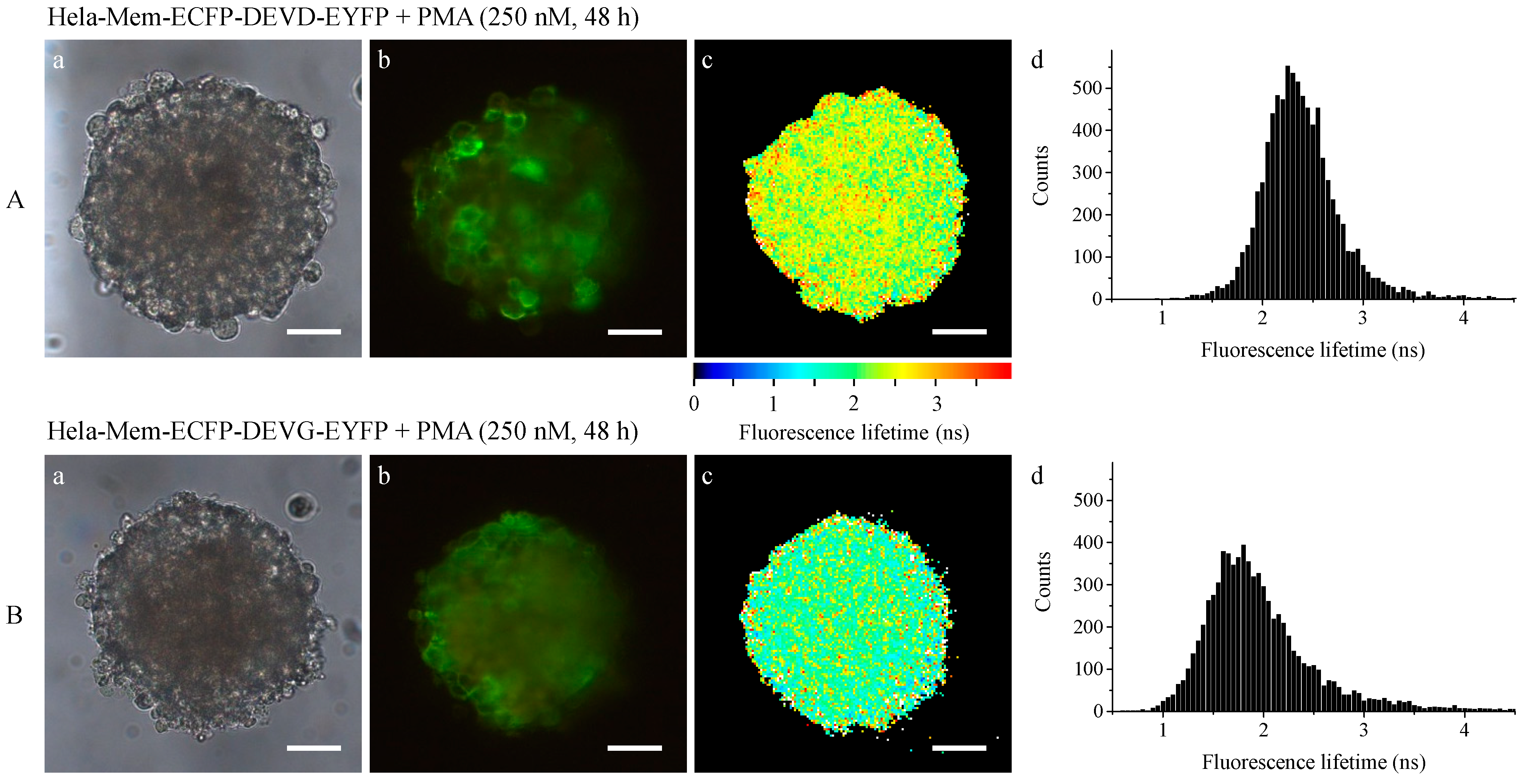
3. Discussion
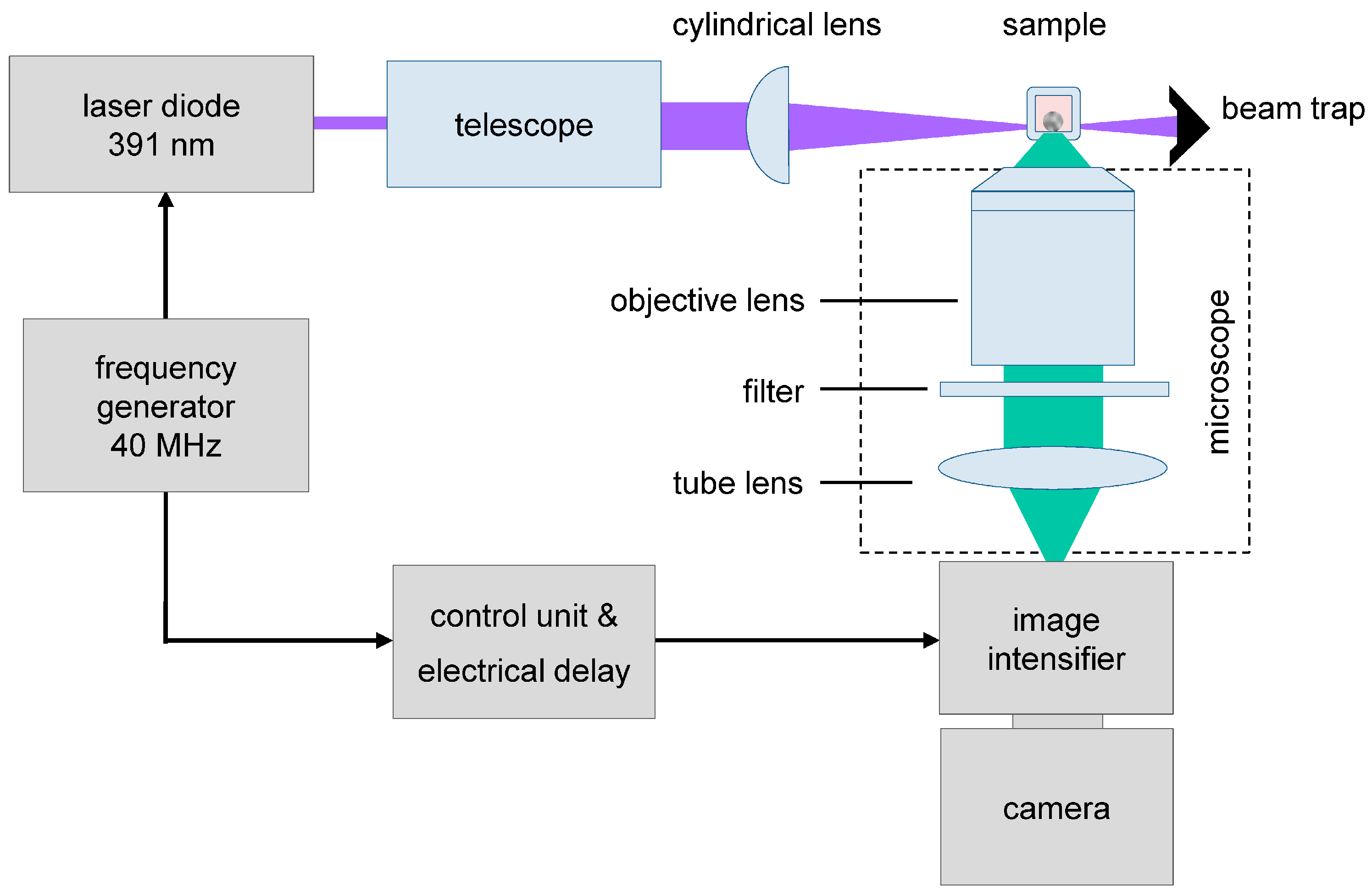
4. Experimental Section
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Angres, B.; Steuer, H.; Weber, P.; Wagner, M.; Schneckenburger, H. A membrane-bound FRET-based caspase sensor for detection of apoptosis using fluorescence lifetime and total internal reflection microscopy. Cytometry 2009, 75, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Förster, T. Zwischenmolekulare energiewanderung und fluoreszenz. Ann. Phys. 1948, 437, 55–75. [Google Scholar] [CrossRef]
- Xu, X.; Gerard, A.L.; Huang, B.C.; Anderson, D.C.; Payan, D.G.; Luo, Y. Detection of programmed cell death using fluorescence energy transfer. Nucleic Acids Res. 1998, 26, 2034–2035. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, N.P.; Harrison-Shostak, D.C.; Michaux, J.; Herman, B. Novel mutant green fluorescent protein protease substrates reveal the activation of specific caspases during apoptosis. Chem. Biol. 1999, 6, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Bruns, T.; Angres, B.; Steuer, H.; Weber, P.; Wagner, M.; Schneckenburger, H. Forster resonance energy transfer-based total internal reflection fluorescence reader for apoptosis. J. Biomed. Opt. 2009, 14, 021003. [Google Scholar] [CrossRef] [PubMed]
- Kunz-Schughart, L.A.; Freyer, J.P.; Hofstaedter, F.; Ebner, R. The use of 3-D cultures for high-throughput screening: The multicellular spheroid model. J. Biomol. Screen. 2004, 9, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Wittig, R.; Richter, V.; Wittig-Blaich, S.; Weber, P.; Strauss, W.S.L.; Bruns, T.; Dick, T.P.; Schneckenburger, H. Biosensor-expressing spheroid cultures for imaging of drug-induced effects in three dimensions. J. Biomol. Screen. 2013, 18, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Seo, K.-S.; Kim, J.-S.; Park, J.-H.; Song, K.-S.; Yun, E.-J.; Park, J.-I.; Kweon, G.R.; Yoon, W.-H.; Lim, K.; Hwang, B.-D. PMA synergistically enhances apicularen A-induced cytotoxicity by disrupting microtubule networks in HeLa cells. BMC Cancer 2014, 14, 36. [Google Scholar] [CrossRef] [PubMed]
- Huisken, J.; Swoger, J.; del Bene, F.; Wittbrodt, J.; Stelzer, E.H.K. Optical sectioning deep inside live embryos by selective plane illumination microscopy. Science 2004, 305, 1007–1009. [Google Scholar] [CrossRef] [PubMed]
- Bruns, T.; Schickinger, S.; Wittig, R.; Schneckenburger, H. Preparation strategy and illumination of three-dimensional cell cultures in light sheet-based fluorescence microscopy. J. Biomed. Opt. 2012, 17, 101518. [Google Scholar] [CrossRef] [PubMed]
- Pawley, J.B. Handbook of Biological Confocal Microscopy, 3rd ed.; Springer: New York, NY, USA, 2006. [Google Scholar]
- Webb, R.H. Confocal optical microscopy. Rep. Prog. Phys. 1996, 59, 427–471. [Google Scholar] [CrossRef]
- Denk, W.; Strickler, J.H.; Webb, W.W. Two-photon laser scanning fluorescence microscopy. Science 1990, 248, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Neil, M.A.; Juskaitis, R.; Wilson, T. Method of obtaining optical sectioning by using structured light in a conventional microscope. Opt. Lett. 1997, 22, 1905–1907. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, M.G.L.; Shao, L.; Carlton, P.M.; Wang, C.J.R.; Golubovskaya, I.N.; Cande, W.Z.; Agard, D.A.; Sedat, J.W. Three-dimensional resolution doubling in wide-field fluorescence microscopy by structured illumination. Biophys. J. 2008, 94, 4957–4970. [Google Scholar] [CrossRef] [PubMed]
- Schneckenburger, H.; Weber, P.; Wagner, M.; Schickinger, S.; Richter, V.; Bruns, T.; Strauss, W.S.L.; Wittig, R. Light exposure and cell viability in fluorescence microscopy. J. Microsc. 2012, 245, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weber, P.; Schickinger, S.; Wagner, M.; Angres, B.; Bruns, T.; Schneckenburger, H. Monitoring of Apoptosis in 3D Cell Cultures by FRET and Light Sheet Fluorescence Microscopy. Int. J. Mol. Sci. 2015, 16, 5375-5385. https://doi.org/10.3390/ijms16035375
Weber P, Schickinger S, Wagner M, Angres B, Bruns T, Schneckenburger H. Monitoring of Apoptosis in 3D Cell Cultures by FRET and Light Sheet Fluorescence Microscopy. International Journal of Molecular Sciences. 2015; 16(3):5375-5385. https://doi.org/10.3390/ijms16035375
Chicago/Turabian StyleWeber, Petra, Sarah Schickinger, Michael Wagner, Brigitte Angres, Thomas Bruns, and Herbert Schneckenburger. 2015. "Monitoring of Apoptosis in 3D Cell Cultures by FRET and Light Sheet Fluorescence Microscopy" International Journal of Molecular Sciences 16, no. 3: 5375-5385. https://doi.org/10.3390/ijms16035375