Thus, after the first fusion of two spherical 200 nm endosomes, the resulting endosome should contain only an external tubule. After the second round of fusion the invagination should appear, and so on. Finally, when the function of the flippases is significantly affected at low temperatures and the exchange of membranes is not possible, the formation of varicosites should induce immediate formation of constrictions, and in the case of their subsequent breakdown into vesicles, TAA of the membrane should not change.
2.1. Role of the Trans-Membrane Area Asymmetry (TAA) in Golgi Reorganization
To test the first prediction, we inhibited the SNARE machinery by microinjecting HeLa cells with a dominant-negative (L294A) His6-a-SNAP mutant (aSNAPmu). Analysis at the EM level 30 min after this microinjection showed enhanced vesiculation of the GA and the formation of invaginated cisternae (
Figure 2A–C, arrows). To study earlier events after blocking membrane fusion, HeLa cells were treated on ice with NEM. Under these conditions, NEM inactivates the fusion factor NSF without affecting cell viability [
28]. By 30 s after the wash-out of NEM and with re-warming of the cells, the number of COPI vesicles had increased (
Figure 2D, striated bars). This vesiculation was accompanied by narrowing of the cisternal pores and smoothing of the cisternal rims (
Figure 2F-yellow cisterna,
Figure 2E-white bars) whereas after incubation with brefeldin A (BFA), the sizes of the cisternal pores increased (
Figure 2F-orange,
Figure 2E-black bars) in comparison with the control cells (
Figure 2F-blue;
Figure 2E, white bars).
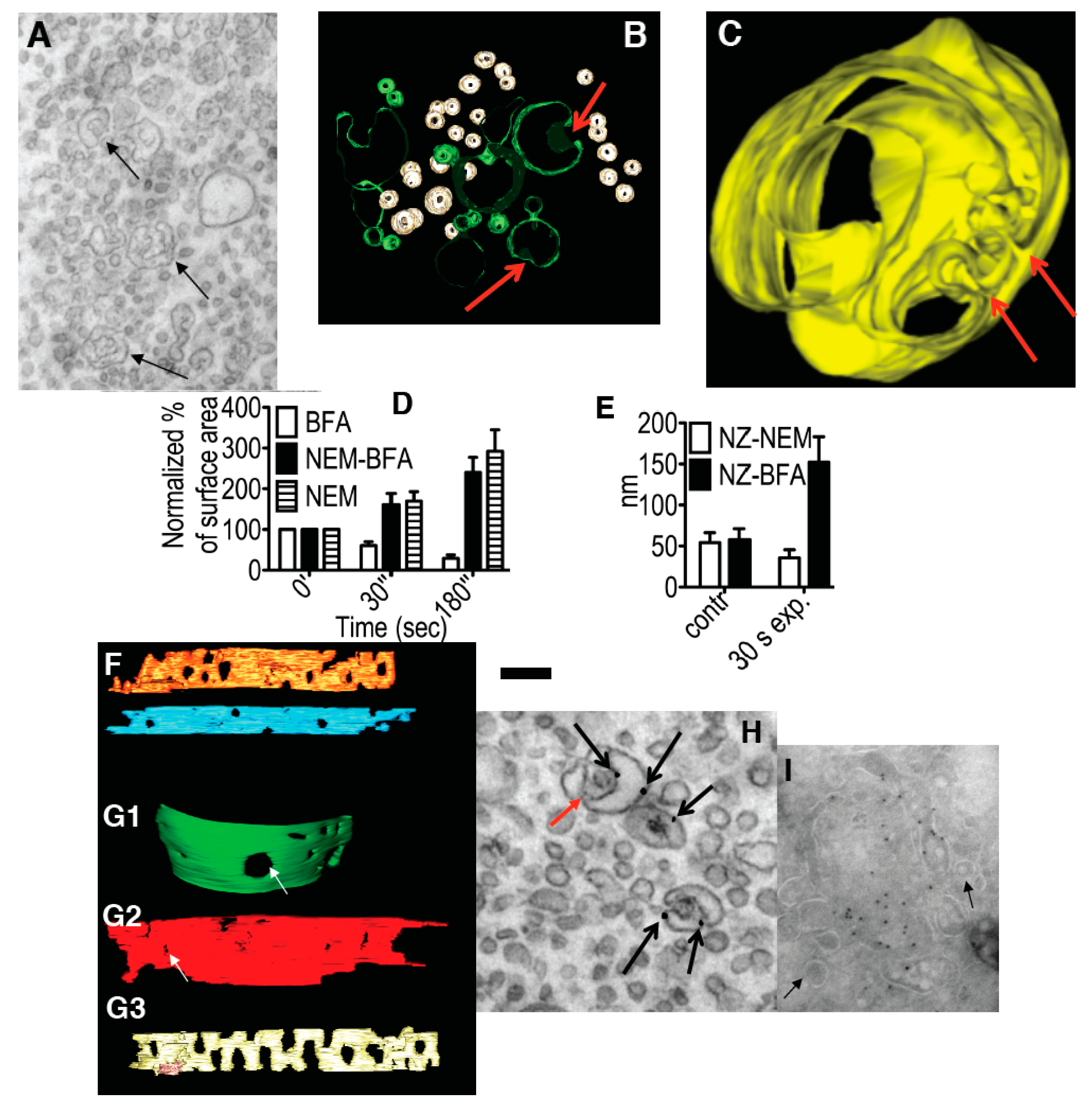
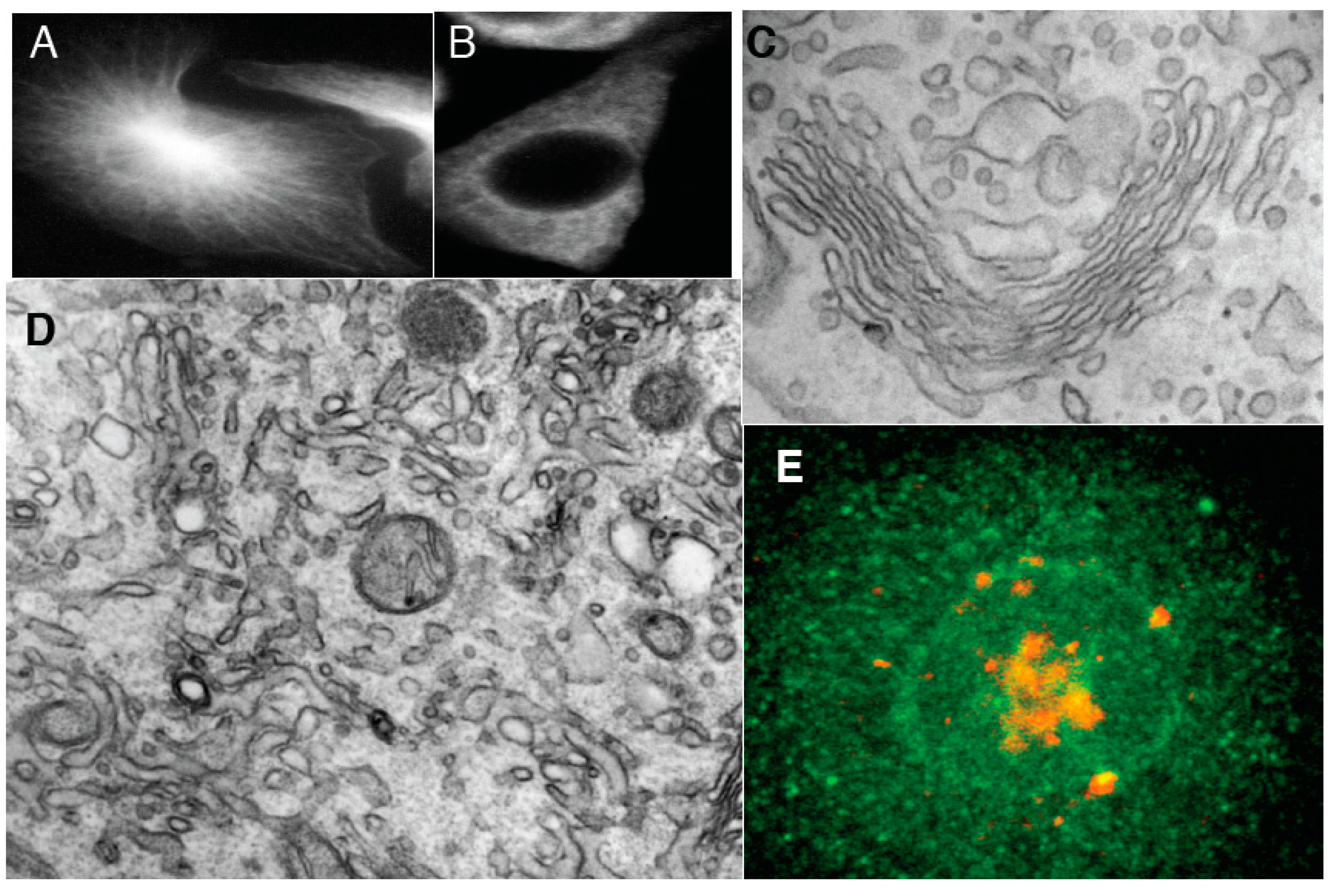
Figure 2.
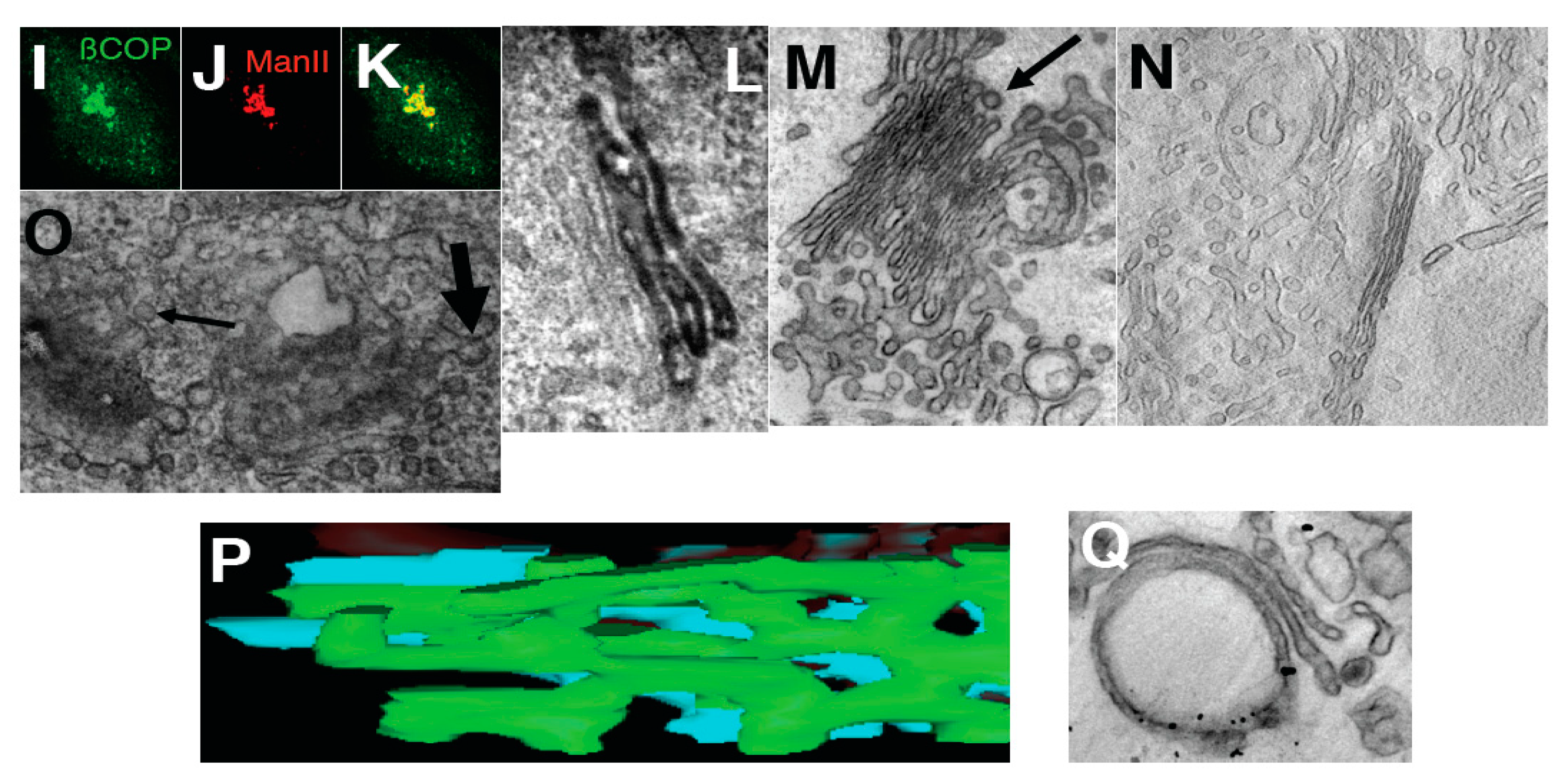
Effects of inhibition of membrane fusion on the Golgi. (A) HeLa cells 30 min after microinjection of αSNAPmu. Arrows, cisternal remnants; (B,C) Three-dimensional models of cisternal remnants after surface rendering. Arrows, invaginations. Cisternal remnants are green (B) and yellow (C). Vesicles in (B) are white; (D) Normalized surface areas of RPs versus Golgi cisternae after treatment with BFA alone (white bars), 30 s after treatment with N-ethylmaleimide (NEM) and then brefeldin A (BFA; black bars), or 30 s after NEM treatment alone (striated bars); (E) Mean diameter of cisternal pores determined using EM tomography. Each experiment was compared with its own control; (F) Cells were treated with 33 µM nocodazole (NZ) or 5 min, and then additionally incubated 30 s with NZ alone (blue cisterna in F), NZ + 5 µg/mL BFA (orange cisterna) or NZ + 1 µM NEM (yellow cisterna). (F,G) Examples of three-dimensional models of control (blue in F) penultimate medial cisternae after inhibition of vesiculation (NZ + BFA, orange in F) or membrane fusion (NZ + NEM, yellow in F) machineries; (G) Isolated Golgi membranes were incubated with 5 mg/mL native cytosol, the ATP restoration system (ARS), GTP: without (G1) or with the mutant of αSNAP (αSNAPmu; G2) for 20 min. Pores are indicated with white arrows. Addition of α-SNAPmu induced narrowing of cisternal pores; (H) HeLa cells treated with NEM and analyzed 3 min after re-warming; mannosidase II (ManII) was labeled by nano-gold (arrows). Red arrow, cisternal invagination; (I) Incubation of isolated Golgi membranes with native cytosol, ARS/GTP (G) and αSNAPmu for 90 min. Cryosection labeled for ManII. Vesicles (arrows) do not contain ManII. Bars. 300 nm (A); 100 nm (B); 50 nm (C); 250 nm (F,G); 120 nm (H,I).
Figure 2.
Effects of inhibition of membrane fusion on the Golgi. (A) HeLa cells 30 min after microinjection of αSNAPmu. Arrows, cisternal remnants; (B,C) Three-dimensional models of cisternal remnants after surface rendering. Arrows, invaginations. Cisternal remnants are green (B) and yellow (C). Vesicles in (B) are white; (D) Normalized surface areas of RPs versus Golgi cisternae after treatment with BFA alone (white bars), 30 s after treatment with N-ethylmaleimide (NEM) and then brefeldin A (BFA; black bars), or 30 s after NEM treatment alone (striated bars); (E) Mean diameter of cisternal pores determined using EM tomography. Each experiment was compared with its own control; (F) Cells were treated with 33 µM nocodazole (NZ) or 5 min, and then additionally incubated 30 s with NZ alone (blue cisterna in F), NZ + 5 µg/mL BFA (orange cisterna) or NZ + 1 µM NEM (yellow cisterna). (F,G) Examples of three-dimensional models of control (blue in F) penultimate medial cisternae after inhibition of vesiculation (NZ + BFA, orange in F) or membrane fusion (NZ + NEM, yellow in F) machineries; (G) Isolated Golgi membranes were incubated with 5 mg/mL native cytosol, the ATP restoration system (ARS), GTP: without (G1) or with the mutant of αSNAP (αSNAPmu; G2) for 20 min. Pores are indicated with white arrows. Addition of α-SNAPmu induced narrowing of cisternal pores; (H) HeLa cells treated with NEM and analyzed 3 min after re-warming; mannosidase II (ManII) was labeled by nano-gold (arrows). Red arrow, cisternal invagination; (I) Incubation of isolated Golgi membranes with native cytosol, ARS/GTP (G) and αSNAPmu for 90 min. Cryosection labeled for ManII. Vesicles (arrows) do not contain ManII. Bars. 300 nm (A); 100 nm (B); 50 nm (C); 250 nm (F,G); 120 nm (H,I).
![Ijms 16 05299 g002]()
By 3 min, the GA was almost completely vesiculated, and invaginated cisternal remnants appeared that labeled for ManII, which indicated that they originated from the Golgi cisternae (
Figure 2H). As an independent approach, we used a well-established
in vitro system based on isolated rat liver Golgi membranes. As shown in
Figure 2G2 (compared with
Figure 2G1 as control), after a 20 min incubation with native cytosol in the presence of an ATP regeneration system, GTP (ARS/GTP) and αSNAPmu, the mean cisternal pore diameter had clearly decreased, and by 90 min the cisternae were transformed into invaginated saccular structures that labeled for ManII and were surrounded by COPI vesicles (
Figure 2I). Thus, inhibition of the SNARE machinery induces Golgi vesiculation, and it is accompanied by a narrowing of the cisternal pores that is followed by the generation of cisternal remnants with invaginations, in agreement with the TAA hypothesis.
To assess the validity of the second prediction, HeLa cells were treated with BFA, an inhibitor of ARFGEFs [
29]. Simultaneously, to prevent the redistribution of the Golgi membranes into the endoplasmic reticulum under the action of BFA, microtubules were depolymerized using a short pretreatment (5 min) with nocodazole (NZ) before the incubation with BFA, for 0.5, 3, 5, or 20 min (in the continued presence of NZ to avoid microtubule re-polymerization). The short pre-treatment with NZ did not induce any visible changes in the GA morphology (
Figure 3), but it clearly prevented BFA-induced redistribution of the Golgi enzyme ManII into the endoplasmic reticulum, although most of the βCOP had dissociated from the Golgi membranes within 5 min of adding BFA (
Figure 4A).
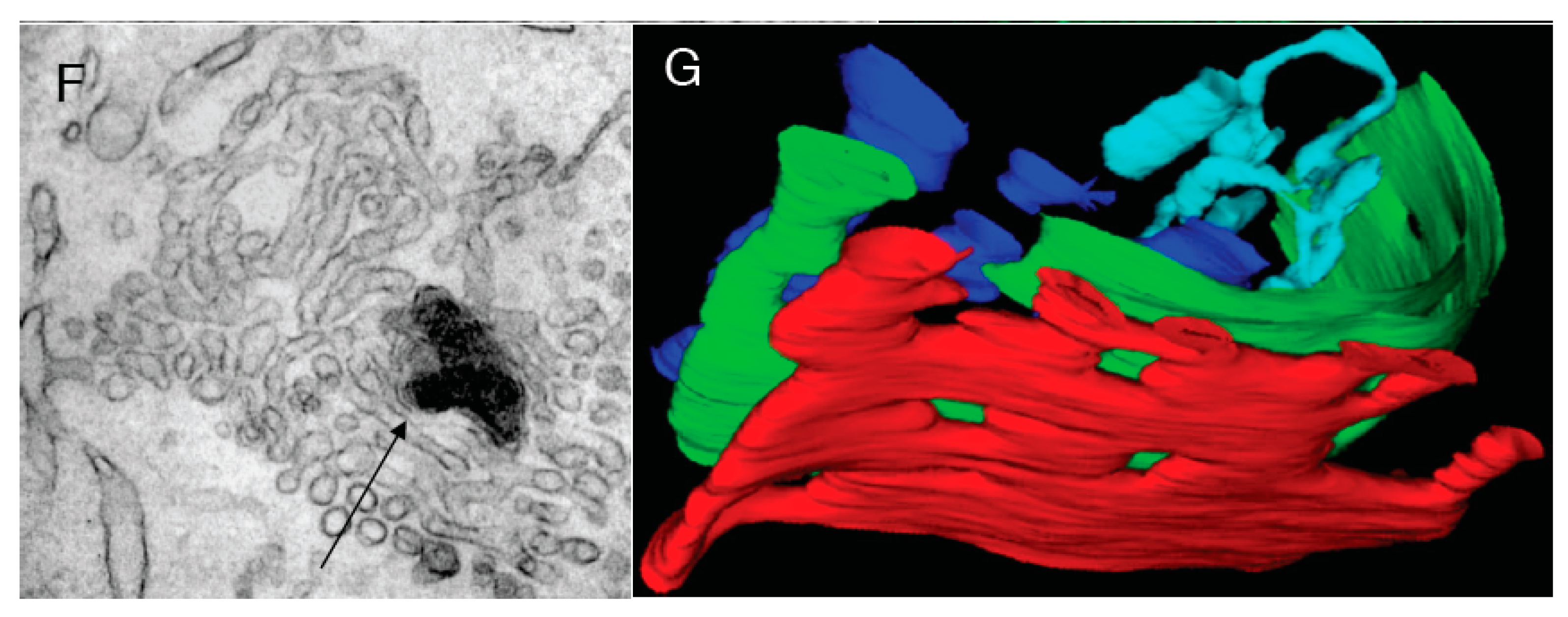
Figure 3.
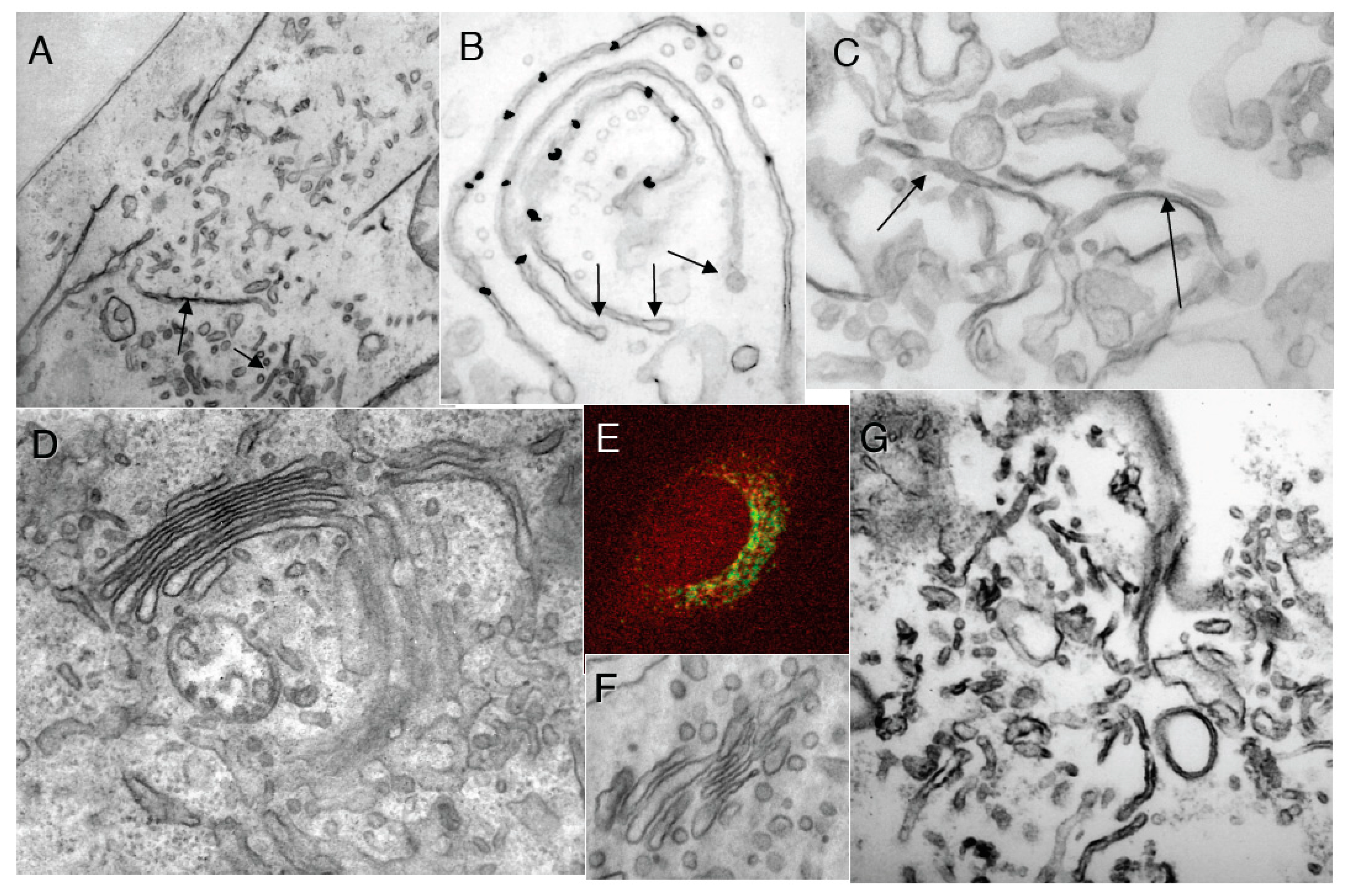
Control experiments. (A) Control HeLe cells labeled with an anti-tubulin antibody; (B) Cells treated with NZ; (C) Preservation of Golgi complex morphology in HeLa cells after 5 min NZ treatment; (D,E) BFA was added for 5 min after an irrelevant IgG (D) or the mutant of αSNAP (αSNAPmu; E) microinjection. Mannosidase II (ManII; red) did not redistribute into the endoplasmic reticulum, although most of the β-subunit of coatomer I (green) detached from the Golgi and diffusely labeled the cytosol; the Golgi stack was preserved; (F) Tubulated Golgi membranes after loading of cells with the WGA lectin conjugated with horse-radish peroxidase. The endosomal TGN is stained with OsO4 (arrow); (G) Three-dimensional tomogram reconstruction of an isolated Golgi stack. Bars: 10 µm (A,B); 150 nm (C,D); 5 µm (E); 100 nm (F,G).
Figure 3.
Control experiments. (A) Control HeLe cells labeled with an anti-tubulin antibody; (B) Cells treated with NZ; (C) Preservation of Golgi complex morphology in HeLa cells after 5 min NZ treatment; (D,E) BFA was added for 5 min after an irrelevant IgG (D) or the mutant of αSNAP (αSNAPmu; E) microinjection. Mannosidase II (ManII; red) did not redistribute into the endoplasmic reticulum, although most of the β-subunit of coatomer I (green) detached from the Golgi and diffusely labeled the cytosol; the Golgi stack was preserved; (F) Tubulated Golgi membranes after loading of cells with the WGA lectin conjugated with horse-radish peroxidase. The endosomal TGN is stained with OsO4 (arrow); (G) Three-dimensional tomogram reconstruction of an isolated Golgi stack. Bars: 10 µm (A,B); 150 nm (C,D); 5 µm (E); 100 nm (F,G).
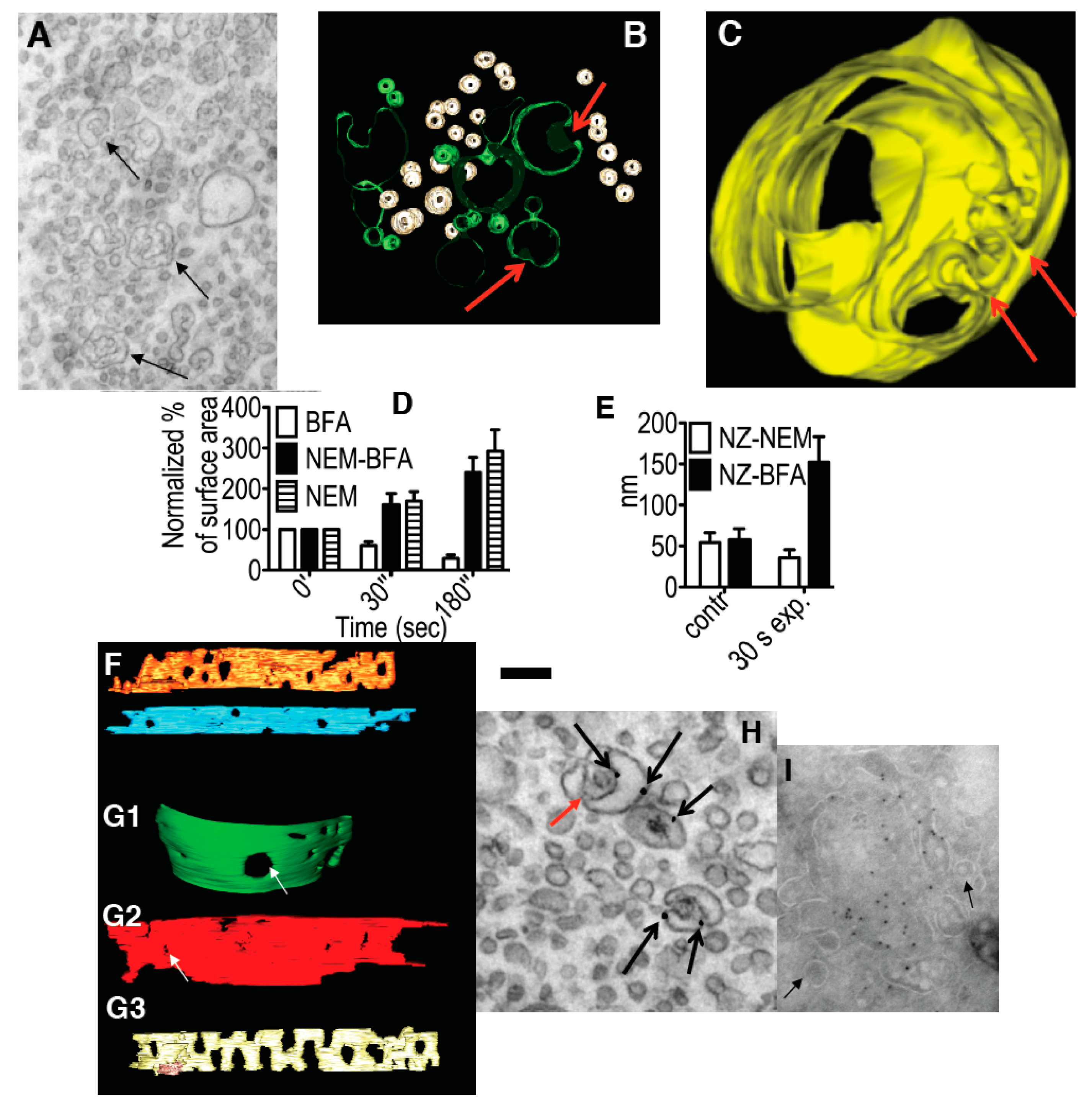
At the EM level, at 0.5 min and, in particular, 3 min after BFA addition, the number of peri-Golgi vesicles had decreased, while the cisternal pores were enlarged (
Figure 2G3,D: white bars). By 5 min, the cisternae were transformed into tubular networks, as seen by EM (
Figure 4B) and electron tomography (
Figure 4C). The tubulated membranes were labeled for the Golgi marker ManII (
Figure 4D). The tubulated GA with WGA-HRP inside the lumen in the endosomal TGN is shown in
Figure 3F. In an independent approach, the ARF1/COPI machinery was inhibited by microinjecting cells deprived of microtubules with anti-βCOP Abs; here the GA remained in the cell center, and again it was tubulated (
Figure 4E). Thus, inhibition of the ARF/COPI machinery induces widening of the cisternal pores that is followed by Golgi tubulation.
To determine whether conversion of the tubulated GA into stacks is ARF/COPI specific using an independent approach that avoided drug treatments, we used mutant ldl F cells. These ldl F cells contain a temperature-sensitive version of ε-COP that is inactivated and completely degraded by an incubation at 40 °C for 6 h [
30]. At the permissive temperature (34 °C), the Golgi stacks in these cells were normal, but after inactivation of εCOP as above, the GA was transformed into tubular networks (
Figure 4F). If the ldl F cells were permeabilized after ε-COP inactivation and incubated with cytosol and ARS/GTP, the tubulated Golgi cisternae (
Figure 4G,H) were converted back into flat and smooth cisternae that were surrounded by 52 nm vesicles (
Figure 4I). This transformation occurred rapidly, and it was complete within 2 to 3 min. COPI-depleted cytosol (
Figure 4J), ARF-depleted cytosol (
Figure 3G), and incubation in the presence of an anti-βCOP antibody (
Figure 4K) did not allow the Golgi stacks to reform. In contrast, after recomplementation of COPI-depleted cytosol with partially purified coatomer (
Figure 4L), or of ARF-depleted cytosol with myristoylated ARF1 (
Figure 4M), the Golgi stacks reformed normally and βCOP was concentrated again in the pericentriolar area (
Figure 4N). Thus, the transformation of tubulated or highly perforated Golgi cisternae into more disk-like structures requires the extraction of membrane curvature into buds, varicose tubules, or vesicles by the ARF/COPI machinery, and this occurs at a striking speed and efficiency upon addition of coatomer I.
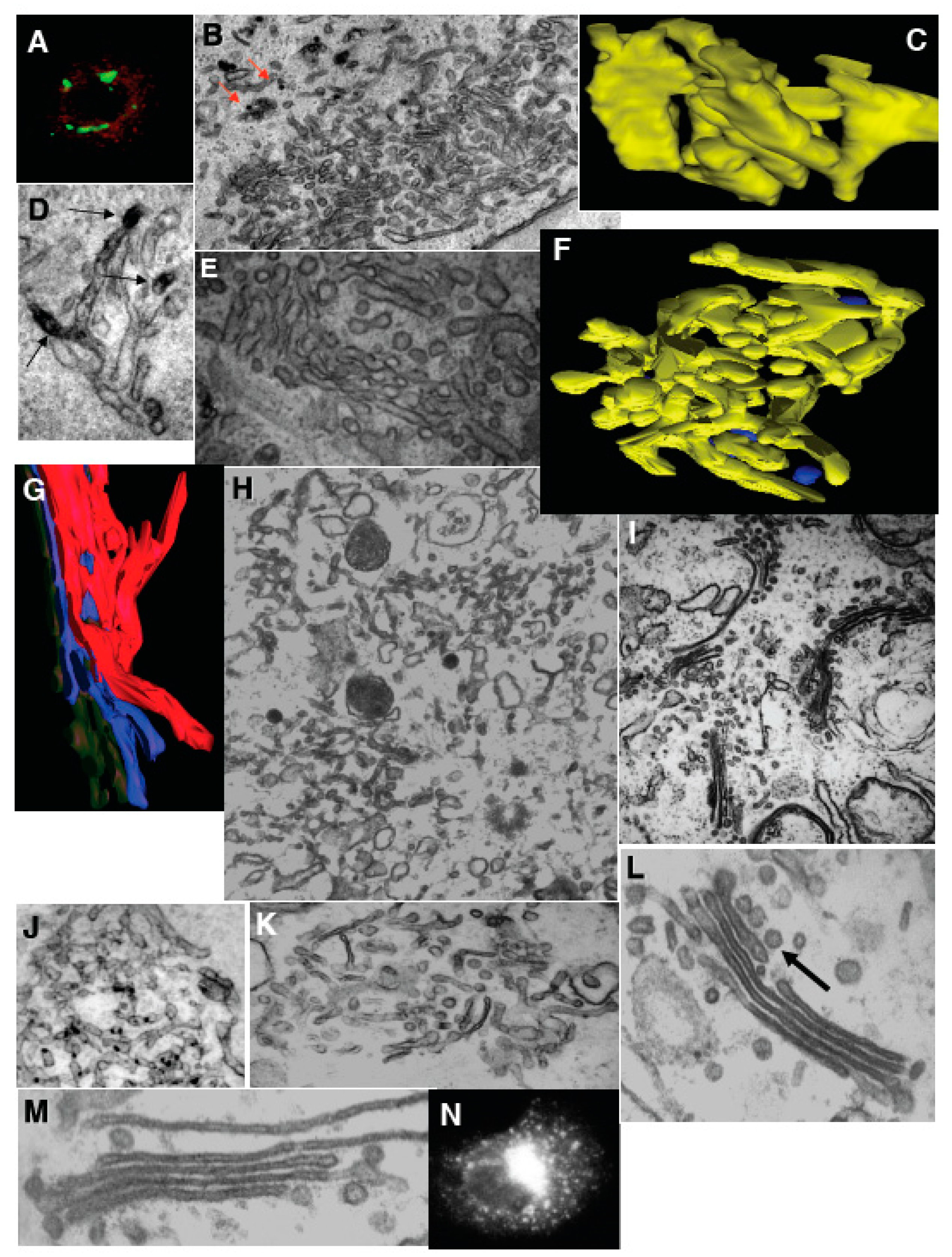
Figure 4.
Transformation of Golgi stacks after inhibition of the ATP restoration system, coatomer I (COPI), and the SNARE-proteins. (A–D) HeLa cells treated with NZ (5 min) and then BFA + NZ (5 min); (E) GA of HeLa cells, 30 min after microinjection of an anti-βCOP antibody. (A) βCOP (red) and ManII (green) labeling; (B) labeling of β-COP (nano-gold-enhancement, red arrows); (C) Three-dimensional model of the GA; (D) immunoperoxidase labeling of ManII (arrows); (E) Routine EM section; (F–N) Inhibition of membrane fusion or ARF/COPI machine impairs GA shape in permeabilized cells. Ldl F cells were warmed to 40 °C for 6 h (F); SLO permeabilized (G,H); and incubated for 90 min at 32 °C with the ATP restoration system (ARS), GTP, plus native (I,K); COPI-depleted (J,K,N) or ARF-depleted (M) cytosol, with an anti-βCOP antibody (K), partially purified COPI cytosol (K,N), or mARF1 (M), and then prepared for EM. (F,G) Three-dimensional tomographic models; (H,I–M) Routine electron microscopic sections. (N) Immunofluorescence. Arrow in (L) shows the section of COPI vesicle. Bars, 10 µm (A,N); 400 nm (B,H,I–K); 70 nm (C,G); 100 (F); 120 nm (D,E,L,M).
Figure 4.
Transformation of Golgi stacks after inhibition of the ATP restoration system, coatomer I (COPI), and the SNARE-proteins. (A–D) HeLa cells treated with NZ (5 min) and then BFA + NZ (5 min); (E) GA of HeLa cells, 30 min after microinjection of an anti-βCOP antibody. (A) βCOP (red) and ManII (green) labeling; (B) labeling of β-COP (nano-gold-enhancement, red arrows); (C) Three-dimensional model of the GA; (D) immunoperoxidase labeling of ManII (arrows); (E) Routine EM section; (F–N) Inhibition of membrane fusion or ARF/COPI machine impairs GA shape in permeabilized cells. Ldl F cells were warmed to 40 °C for 6 h (F); SLO permeabilized (G,H); and incubated for 90 min at 32 °C with the ATP restoration system (ARS), GTP, plus native (I,K); COPI-depleted (J,K,N) or ARF-depleted (M) cytosol, with an anti-βCOP antibody (K), partially purified COPI cytosol (K,N), or mARF1 (M), and then prepared for EM. (F,G) Three-dimensional tomographic models; (H,I–M) Routine electron microscopic sections. (N) Immunofluorescence. Arrow in (L) shows the section of COPI vesicle. Bars, 10 µm (A,N); 400 nm (B,H,I–K); 70 nm (C,G); 100 (F); 120 nm (D,E,L,M).
![Ijms 16 05299 g004]()
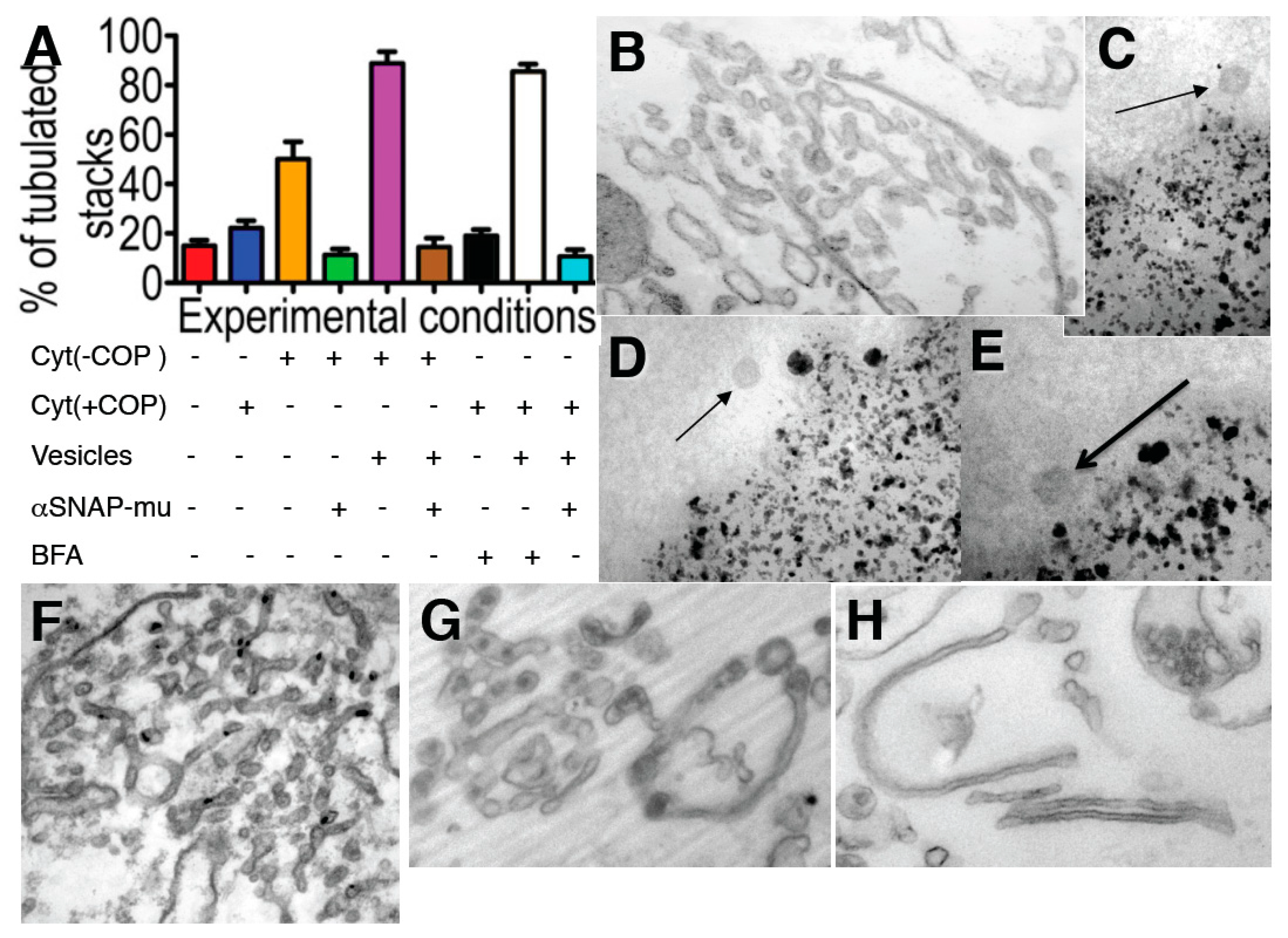
Next, we examined whether the membrane curvature that is stored in COPI-dependent vesicles is actually required for the tubular transformation of the GA. First, when isolated Golgi membranes were incubated with COPI-depleted cytosol in the presence of ARS/GTP for 90 min, the Golgi cisternae were transformed into tubular networks (in agreement with the results of Misteli and Warren [
51]), although only partially (
Figure 5A-orange bar;
Figure 5B). Moreover, tubulation was not seen if αSNAPmu was added to the incubation medium (
Figure 5A-green bar,
Figure 5B). When COPI-dependent vesicles isolated using magnetic beads (Figure 5C–E, see [
31]) (vesicles prepared according to Kweon
et al. [
28] and Yang
et al. [
32] gave identical results; our unpublished observations) were added to the incubation medium, the degree of Golgi tubulation greatly increased (
Figure 5F,
Figure 5A-magenta bar), and this stimulatory effect was blocked by αSNAPmu (
Figure 5A-brown bar). Similarly, BFA (2 µg/mL for 90 min) induced tubulation of isolated Golgi membranes, although only in the presence of externally added COPI-dependent vesicles (
Figure 5G,
Figure 5A-white bar), and not in their absence (
Figure 5A-black bar). This tubulation was also blocked by αSNAPmu (
Figure 5H,
Figure 5A-light blue bar). Thus, BFA-induced Golgi tubulation shows an absolute requirement for the fusion of COPI-dependent vesicles with the Golgi stack. We conclude that the fusion of COPI-dependent vesicles with the Golgi stack provides the curvature necessary for GA tubulation.
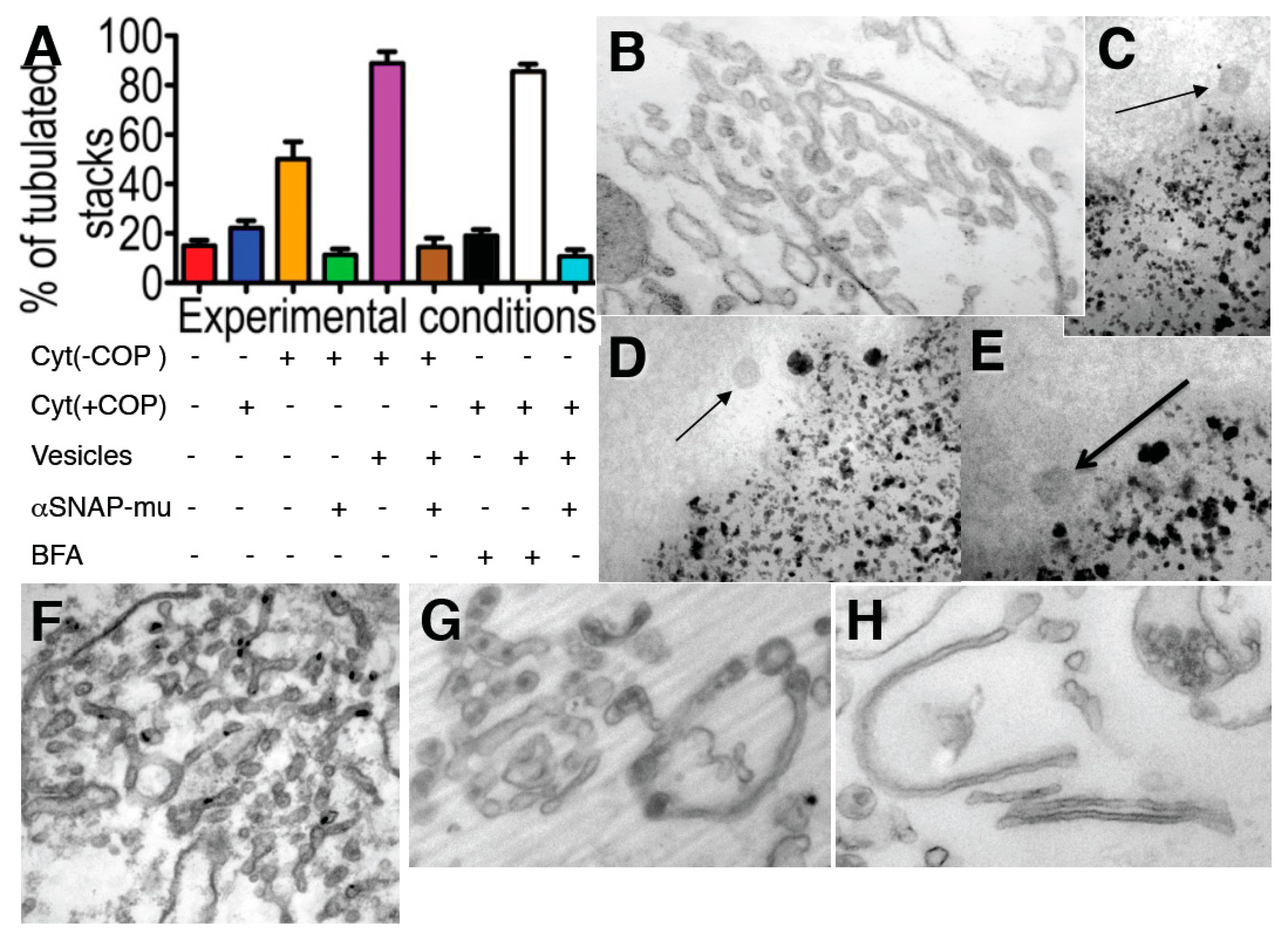
Figure 5.
The Golgi tubulation induced by brefeldin A (BFA) depends on the availability of coatomer I-dependent (COPI) vesicles. (
A) Incubation of isolated Golgi membranes for 90 min, always in the presence of the ARS, GTP, with the normal cytosol (Cyt (+COPI)), blue) or COPI-depleted cytosol: Cyt (−COPI)), without or with isolated COPI vesicles (Vesicles), the mutant of αSNAP (αSNAPmu) and/or BFA, as indicated below the graph. The degree of tubulation was estimated as indicated in
Supplementary Materials. Red, low level of tubulation before incubation; (
B) Incubation of Golgi membranes with COPI-depleted cytosol in the presence of ARS/GTP for 90 min transforms Golgi cisternae into tubules, although only partially (orange bar in
A). Tubulation was not seen if αSNAPmu was added to the incubation medium (green bar in
A); (
F) Incubation of the Golgi membranes as in (
B), but in the presence of isolated COPI vesicles (magenta bar in
A). Higher degree of tubulation. Addition of αSNAPmu to incubation medium as in (
F) blocked tubulation (brown bar in
A); (
G) BFA (2 µg/mL for 90 min) induced tubulation of isolated Golgi membranes in the presence of externally added COPI-dependent vesicles (white bar in
A), and not in their absence (black bar in
A), or when added together with the vesicles. When αSNAPmu was added, this tubulation was also blocked (
H, light blue bar in
A); (
C–
E) Isolation of COPI vesicles (arrows) isolated with the help of magnetic beads; (
I–
K) Revesibility of the changes in the Golgi shape. (
I–
R) HeLa cells were treated with NZ (5 min) and then with NZ + BFA (5 min), followed by NZ alone (5 min,
B,
F) and then BFA was washed out for 2 min (
I–
K,
N) or 5 min (
L,
M,
O), (
I–
K) Re-appearance of β-COP labeling (green) over ManII-positive structures (red) 2 min after BFA wash out; (
L,
M) Transformation of Golgi tubules back into polarized stacks of cisternae containing coated buds (thick arrow in
O) and surrounded by 50–60 nm round profiles (arrows in
L,
M); (
N) RPs visible 2 min after BFA washout were sections of varicose tubules; (
O) Increase of the number of free 52 nm vesicles and COPI-coated buds; (
P) Re-addition of BFA after reformation of GA (as in
L) induced Golgi tubulation again; (
Q) Re-appearance of stacked cisternae after treatment of isolated Golgi membranes with COPI-depleted cytosol and ARS/GTP for 90 min (see above), re-isolation of the tubulated Golgi membranes and additional treatment of these membrane with normal cytosol and ARS/GTP for a further 90 min. Bars, 200 nm (
B); 100 nm (
C,
D); 80 nm (
E); 200 nm (
F,
M,
N); 150 nm (
G,
H,
L,
Q); 10 µm (
I–
K); 120 nm (
O); 50 nm (
P).
Figure 5.
The Golgi tubulation induced by brefeldin A (BFA) depends on the availability of coatomer I-dependent (COPI) vesicles. (
A) Incubation of isolated Golgi membranes for 90 min, always in the presence of the ARS, GTP, with the normal cytosol (Cyt (+COPI)), blue) or COPI-depleted cytosol: Cyt (−COPI)), without or with isolated COPI vesicles (Vesicles), the mutant of αSNAP (αSNAPmu) and/or BFA, as indicated below the graph. The degree of tubulation was estimated as indicated in
Supplementary Materials. Red, low level of tubulation before incubation; (
B) Incubation of Golgi membranes with COPI-depleted cytosol in the presence of ARS/GTP for 90 min transforms Golgi cisternae into tubules, although only partially (orange bar in
A). Tubulation was not seen if αSNAPmu was added to the incubation medium (green bar in
A); (
F) Incubation of the Golgi membranes as in (
B), but in the presence of isolated COPI vesicles (magenta bar in
A). Higher degree of tubulation. Addition of αSNAPmu to incubation medium as in (
F) blocked tubulation (brown bar in
A); (
G) BFA (2 µg/mL for 90 min) induced tubulation of isolated Golgi membranes in the presence of externally added COPI-dependent vesicles (white bar in
A), and not in their absence (black bar in
A), or when added together with the vesicles. When αSNAPmu was added, this tubulation was also blocked (
H, light blue bar in
A); (
C–
E) Isolation of COPI vesicles (arrows) isolated with the help of magnetic beads; (
I–
K) Revesibility of the changes in the Golgi shape. (
I–
R) HeLa cells were treated with NZ (5 min) and then with NZ + BFA (5 min), followed by NZ alone (5 min,
B,
F) and then BFA was washed out for 2 min (
I–
K,
N) or 5 min (
L,
M,
O), (
I–
K) Re-appearance of β-COP labeling (green) over ManII-positive structures (red) 2 min after BFA wash out; (
L,
M) Transformation of Golgi tubules back into polarized stacks of cisternae containing coated buds (thick arrow in
O) and surrounded by 50–60 nm round profiles (arrows in
L,
M); (
N) RPs visible 2 min after BFA washout were sections of varicose tubules; (
O) Increase of the number of free 52 nm vesicles and COPI-coated buds; (
P) Re-addition of BFA after reformation of GA (as in
L) induced Golgi tubulation again; (
Q) Re-appearance of stacked cisternae after treatment of isolated Golgi membranes with COPI-depleted cytosol and ARS/GTP for 90 min (see above), re-isolation of the tubulated Golgi membranes and additional treatment of these membrane with normal cytosol and ARS/GTP for a further 90 min. Bars, 200 nm (
B); 100 nm (
C,
D); 80 nm (
E); 200 nm (
F,
M,
N); 150 nm (
G,
H,
L,
Q); 10 µm (
I–
K); 120 nm (
O); 50 nm (
P).
![Ijms 16 05299 g005a]()
![Ijms 16 05299 g005b]()
Importantly, during the initial period, the tubular transformation of the GA was fully reversible. If the cells were washed after 5-min BFA treatment (in the continued presence of NZ), the pericentriolar staining for βCOP re-appeared within 2 to 5 min (
Figure 5I–K). Simultaneously, the Golgi tubules were transformed back into polarized stacks of cisternae (
Figure 5L) that contained ManII (DAB precipitate) and coated buds, and were surrounded by 50–60 nm sized round profiles (RPs;
Figure 5M). In the first 2 min after BFA washout, most of these RPs were derived from sections of varicose tubules (
Figure 5N), but after 5 min, the number of free 52 nm vesicles increased three-fold (
Figure 5O). If BFA was re-added to the medium 5 min after its washout, β-COP was once more released from the GA, and the GA became tubulated (
Figure 5P). In a complementary approach, we incubated isolated Golgi membranes with COPI-depleted cytosol and ARS/GTP for 90 min (see above), re-isolated the tubulated Golgi membranes, and then incubated these with normal cytosol under the same conditions for a further 90 min. This incubation led to the re-appearance of stacked cisternae (
Figure 5G). Thus, Golgi tubulation and stack formation are rapidly reversible via COPI-dependent vesicle formation and SNARE-dependent vesicle fusion.
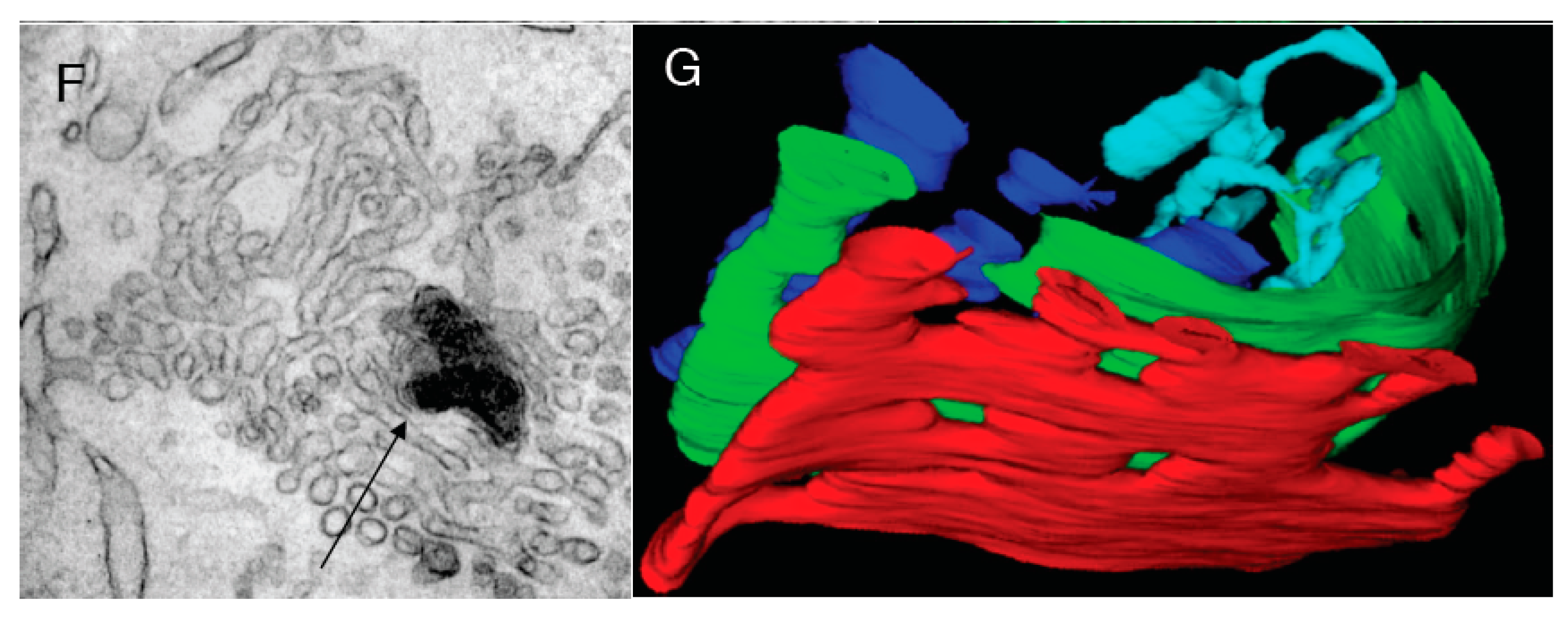
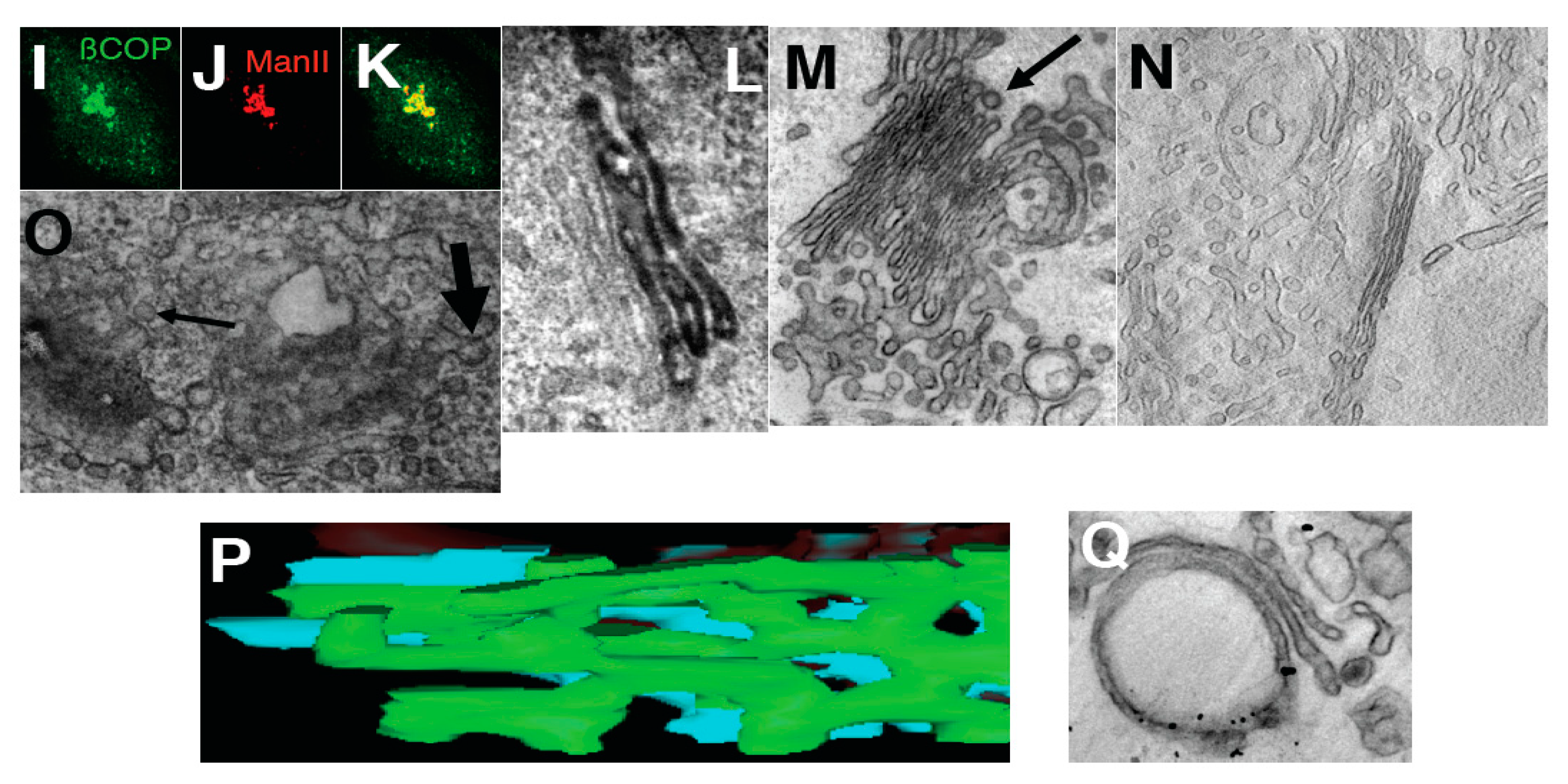
To examine the possibility that cisterna stacking contributes to the tubule-to-cisterna conversion, permeabilized ldl F cells (
Figure 6A,B) with a pre-tubulated GA, or isolated Golgi membranes (tubulated by COPI-depleted cytosol and then re-isolated;
Figure 6C), were incubated with cytosol and ARS/GTP in the presence of a mixture of antibodies directed against GRASP-55 and GRASP-65 that have been reported to almost completely inhibit Golgi stacking [
34,
35]. Under these conditions, the Golgi stacking was indeed strongly inhibited, but the efficiency with which tubules were converted into (isolated) cisternae, and the number of 50–60 nm buds/vesicles was not affected (
Figure 6A–C). Collectively, these results show that the conversion of tubulated into smooth cisternal Golgi elements by the ARF/COPI machinery does not depend on the stacking of the Golgi cisternae.
Figure 6.
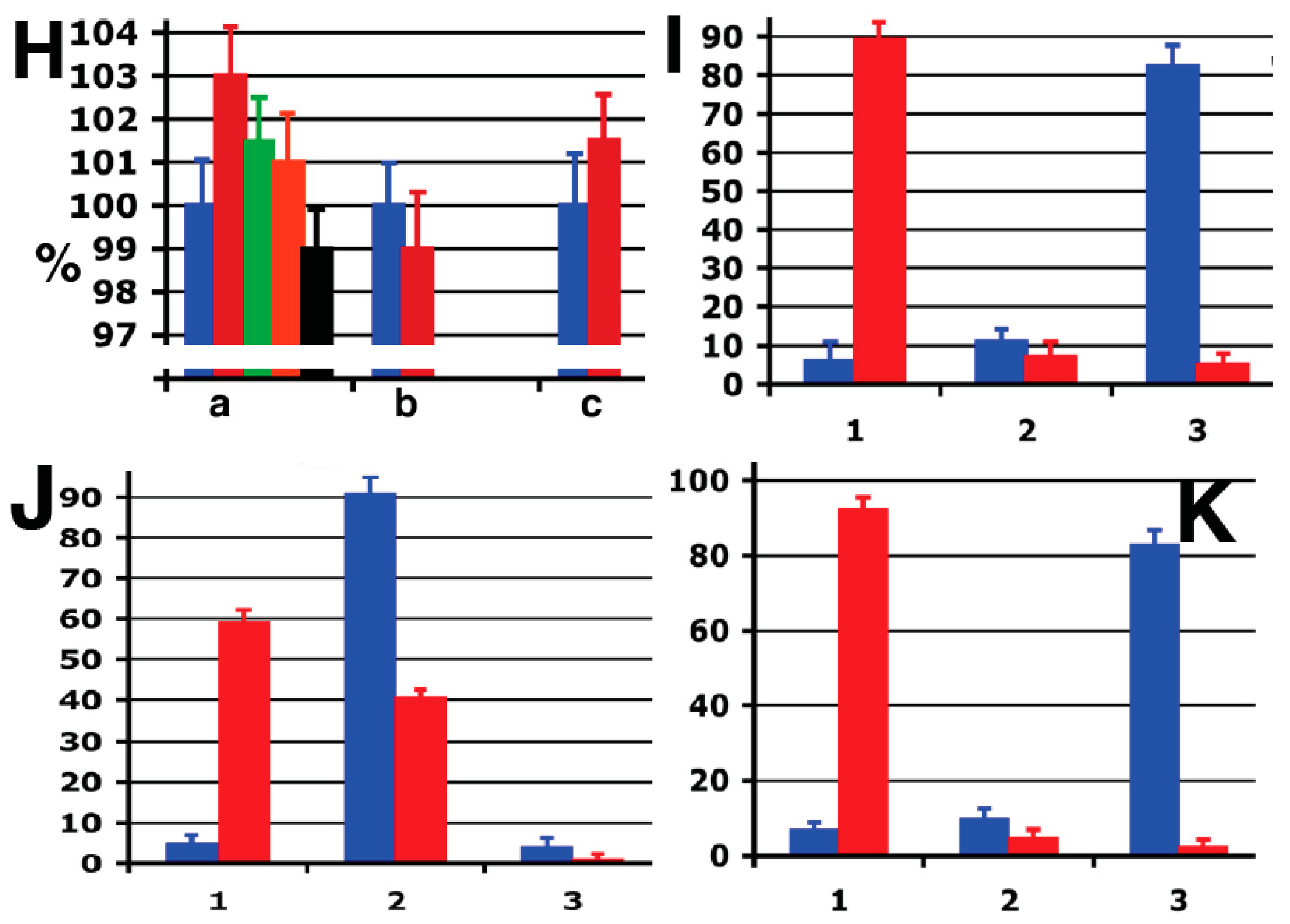
Protein machineries that are involved in GA shaping. (
A,
B) The ldl F cells were treated at 40 °C for 6 h, permeabilized with streptolysis O (SLO), then incubated with native cytosol, the ATP restoration system (ARS), GTP and a mixture of anti-GRASP55 and GRASP65 antibodies for 90 min, and prepared for electron microscopy (EM;
A) or immuno-nano-gold-EM labeling for ManII (
B). Arrows show non-stacked Golgi cisternae (
A) or COPI-coated buds (
B); (
C) Isolated Golgi membranes were incubated with COPI-depleted cytosol and ARS/GTP and then re-isolated. Arrows show individual cisternae; (
D,
E) HeLa cells were microinjected with the mutants of αSNAP protein (αSNAPmu), then placed on ice for 10 min, treated with BFA for 30 min, and then prepared for EM (
D) or immunofluorescence (
E; anti-βCOP, red; anti-mannosidase II (ManII, green); (
F) HeLa cells were microinjected with a mixture of αSNAPmu and an anti-β-COP antibody, and after 30 min prepared for EM; (
G) Ldl F cells were treated at 40 °C for 6 h and permeabilized with SLO, and then incubated with COPI-depleted cytosol and ARS/GTP, plus αSNAPmu, for 90 min; (
H–
K) Quantification of mean Golgi membrane curvature; (
H) Ratio between the cytosolic and luminal surface areas (a); integrated mean curvature (b); and tomographic estimation (c). Data were normalized with respect to the curvature 5 min after NZ treatment. Note: the
Y-axis in
H is shown with a break; (
I–
K) Distribution of surface area (blue bars) and TAA (red bars) between 50–60 nm RPs (1 in
H–
K), elongated profiles (2 in
B–
D) and cisternae (3 in
H–
K) after treatment of HeLa cells with NZ (5 min,
I); NZ (5 min) and then NZ + BFA (5 min,
J); or NZ (5 min), NZ + BFA (5 min) followed by BFA washout in the presence of NZ (5 min,
K). For details on curvature quantification, see
Supplementary Materials. Bars, 500 nm (
A); 300 nm (
B); 200 nm (
C,
D,
F); 5 µm (
E); 400 nm (
G).
Figure 6.
Protein machineries that are involved in GA shaping. (
A,
B) The ldl F cells were treated at 40 °C for 6 h, permeabilized with streptolysis O (SLO), then incubated with native cytosol, the ATP restoration system (ARS), GTP and a mixture of anti-GRASP55 and GRASP65 antibodies for 90 min, and prepared for electron microscopy (EM;
A) or immuno-nano-gold-EM labeling for ManII (
B). Arrows show non-stacked Golgi cisternae (
A) or COPI-coated buds (
B); (
C) Isolated Golgi membranes were incubated with COPI-depleted cytosol and ARS/GTP and then re-isolated. Arrows show individual cisternae; (
D,
E) HeLa cells were microinjected with the mutants of αSNAP protein (αSNAPmu), then placed on ice for 10 min, treated with BFA for 30 min, and then prepared for EM (
D) or immunofluorescence (
E; anti-βCOP, red; anti-mannosidase II (ManII, green); (
F) HeLa cells were microinjected with a mixture of αSNAPmu and an anti-β-COP antibody, and after 30 min prepared for EM; (
G) Ldl F cells were treated at 40 °C for 6 h and permeabilized with SLO, and then incubated with COPI-depleted cytosol and ARS/GTP, plus αSNAPmu, for 90 min; (
H–
K) Quantification of mean Golgi membrane curvature; (
H) Ratio between the cytosolic and luminal surface areas (a); integrated mean curvature (b); and tomographic estimation (c). Data were normalized with respect to the curvature 5 min after NZ treatment. Note: the
Y-axis in
H is shown with a break; (
I–
K) Distribution of surface area (blue bars) and TAA (red bars) between 50–60 nm RPs (1 in
H–
K), elongated profiles (2 in
B–
D) and cisternae (3 in
H–
K) after treatment of HeLa cells with NZ (5 min,
I); NZ (5 min) and then NZ + BFA (5 min,
J); or NZ (5 min), NZ + BFA (5 min) followed by BFA washout in the presence of NZ (5 min,
K). For details on curvature quantification, see
Supplementary Materials. Bars, 500 nm (
A); 300 nm (
B); 200 nm (
C,
D,
F); 5 µm (
E); 400 nm (
G).
![Ijms 16 05299 g006a]()
![Ijms 16 05299 g006b]()
To test the third prediction of the TAA redistribution model, we treated cells with BFA after microinjection of αSNAPmu to allow spreading of the mutant (
Figure 6D), or with NEM. Under these conditions, the GA morphology was preserved (
Figure 6D) and the Golgi enzymes did not redistribute into the endoplasmic reticulum, despite the normal inhibition of the ARF/COPI machinery by BFA (as seen by the diffuse cytosolic staining of βCOP;
Figure 6E). When the cells were first treated with NEM, and BFA was added 20 s after re-warming, the increase in the number of RPs near the GA was halted, and they remained constant in number upon further incubation (3 min), while in control cells that were not treated with BFA, the number of RPs continued to increase (
Figure 2D, striated bars).
Similarly, microinjection of a mixture of an anti-β-COP antibody and αSNAPmu did not affect Golgi morphology for at least 40 min (
Figure 6F), whereas when HeLa cells were microinjected with the anti-βCOP antibody alone, the Golgi cisternae were converted into a highly perforated and tubulated state (see
Figure 4E). In addition, incubation of isolated Golgi membranes with a COPI-depleted cytosol in the presence of ARS/GTP and αSNAPmu did not result in GA tubulation or vesiculation (see
Figure 5A-green bar). Finally, incubation of permeabilized ldl F cells (after heat-induced GA tubulation) with COPI-depleted cytosol in the presence of αSNAPmu did not affect the tubular state of the GA (
Figure 6G). Thus, simultaneous inhibition of both the ARF/COPI and SNARE machineries stabilizes the GA morphology in any given state, in agreement with the TAA redistribution model.
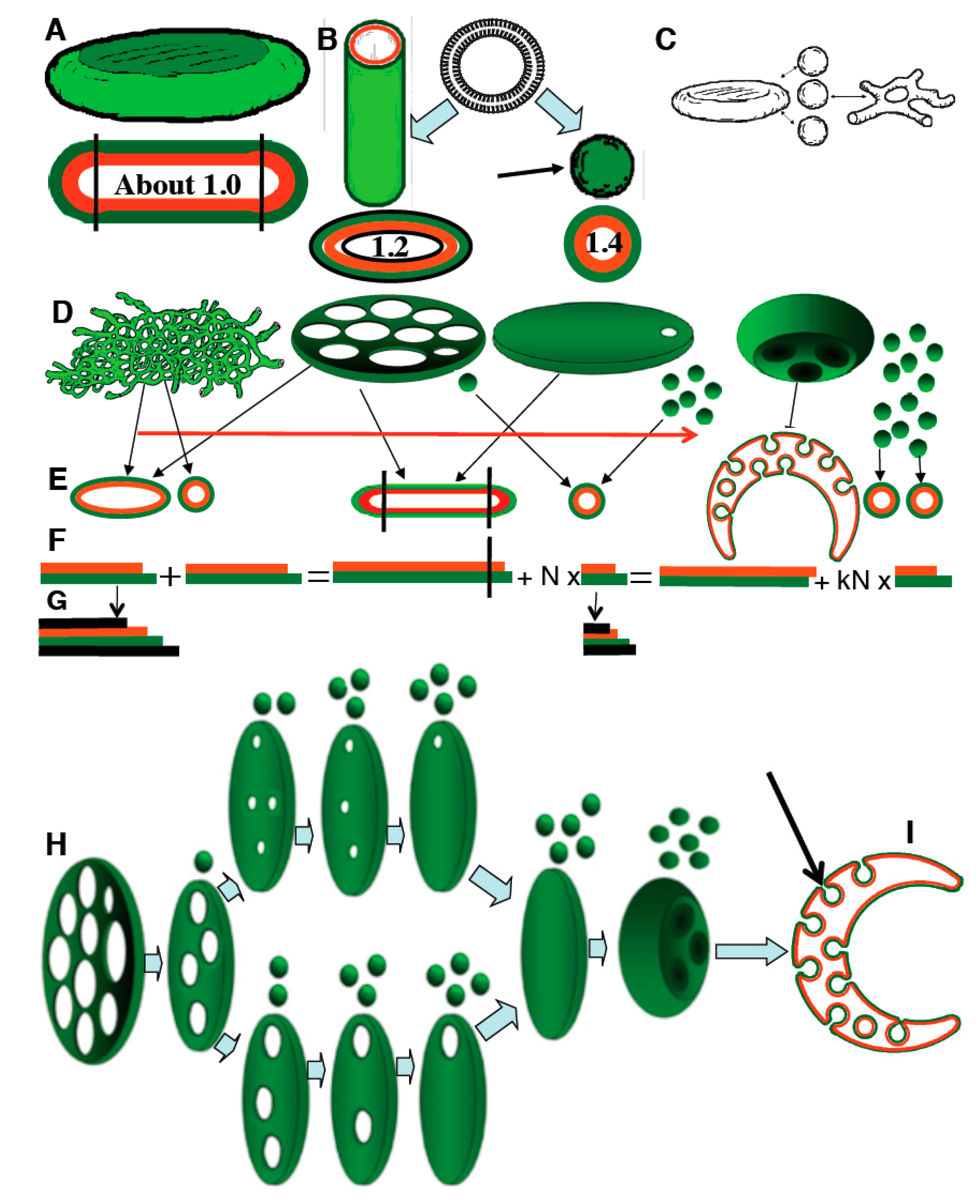
The TAA redistribution model assumes that the total TAA and total volume of all of the structural elements of the GA are constant. To test this assumption, three different methods were used to estimate Golgi membrane curvature and volume. The following results were obtained for HeLa cells treated with NZ for 5 min: the ratio between the surface area of the cytosolic and the lum
inal leaflets, was 1.08 ± 0.01; EM tomography gave the ratio 1.09 ± 0.015; and the integral mean curvature was 331 ± 5 µm/µm
3. The relative contribution of each structural element of the GA to the differences in surface area between the cytoplasmic and luminal membrane leaflets of the GA as a whole is given as the percentage TAA. The measured total TAA (see
Table 1) of NZ-treated cells (mostly cisternae plus vesicles) was 1.08; the TAA for RPs of <60 nm diameter was 2.20 ± 0.02, for elongated profiles, 1.12 ± 0.03, and for cisternae, 1.01 ± 0.03. All of the other data were normalized with respect to the curvature 5 min after NZ treatment. All three of these methods showed that the total surface area of the Golgi membranes, the total GA volume, and the total (or mean) TAA did not change during GA tubulation or GA stack re-formation (
Figure 6H–K).
Table 1.
Distribution of the trans-membrane area asymmetry (TAA) between the structural elements of the Golgi.
Table 1.
Distribution of the trans-membrane area asymmetry (TAA) between the structural elements of the Golgi.
| Experimental Design | Percentage Intersections | Percentage TAA |
|---|
| 50–60 nm Round Profiles | Elongated Profiles | Cisternae | 50–60 nm Round Profiles | Elongated Profiles | Cisternae |
|---|
| 5 min; nocodazole (NZ) | 7 | 10 | 83 | 92.5 | 5 | 2.5 |
| 5 min; brefeldin A (BFA) | 5 | 91 | 4 | 59.2 | 40.7 | 0.1 |
| 5 min BFA, 5 min w/o BFA | 6 | 11 | 82 | 89 | 7 | 5 |
Importantly, the distribution of TAA among the different structural elements of the GA (buds/vesicles, tubules, cisternae) changed dramatically during the shape transitions of the GA. Starting from a flat cisternal state with an accumulation of TAA in round profiles (buds/vesicles), BFA treatment resulted in a tubular state with more TAA in elongated profiles (tubules) and less in round profiles. BFA washout restored the flat cisternal state, with almost all of the TAA again concentrated in round profiles (
Figure 6H–K).
In summary, we found that: (i) inhibition of the SNARE machinery alone reduced TAA of the Golgi cisternae and induced narrowing of the cisternal perforations, followed by invagination of cisternal membranes; (ii) inhibition of the ARF/COPI machinery alone increased TAA of the Golgi cisternae and induced widening of the cisternal perforations followed by GA tubulation; (iii) inhibition of both machineries did not change the GA shape significantly; and (iv) in all of these cases, TAA did not change significantly during the transformation of the GA shape, which suggested that the rapid interference with the ARF/COPI or SNARE machineries that results in dramatic changes in the morphology of the GA was not because of a change in overall TAA, but because of a redistribution of TAA among the structural elements of the GA.
2.2. Role of the Trans-Membrane Area Asymmetry in Reorganization of Microvilli of the Apical Plasma Membrane and Endosomes
To demonstrate that the situation with the Golgi is not
ad hoc, we performed additional experiments. A long time ago, the phenomenon of the breakdown of cylindrical microvilli of brush bodies of kidney epithelial cells into vesicles during the preservation of kidneys in an intracellular (
i.e., VNIIKIEX) solution was described [
26]. The TAA hypothesis predicts that during this fragmentation of microvilli, TAA should remain the same. To test whether the TAA hypothesis is valid also for the conditions when the function of flippases is blocked by low temperature, we used our previous procedure for preservation of the kidney in the intercellular isotonic VNIIKIEX solution (see [
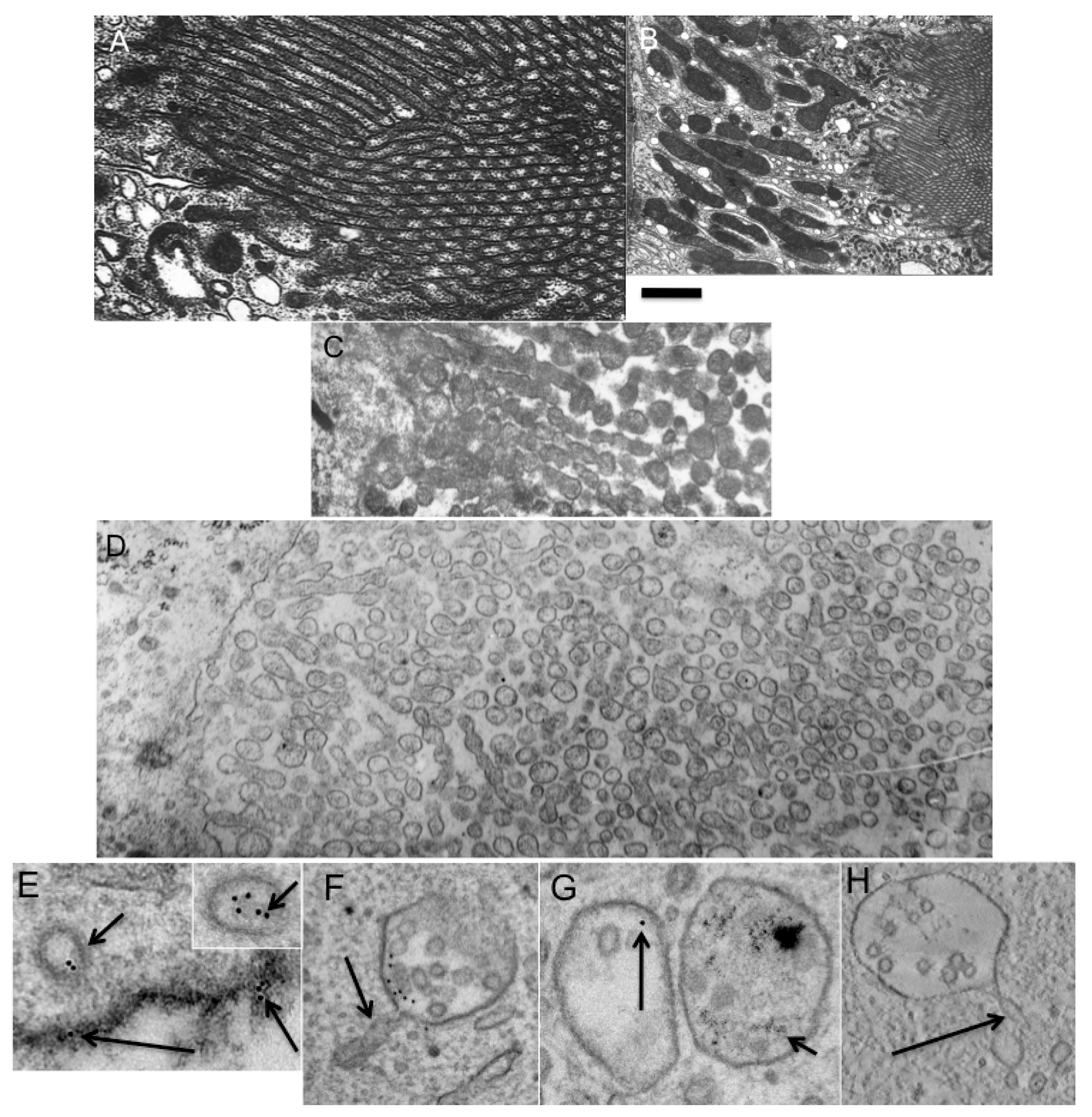
26]). To this end, rat kidneys were removed from the bodies, perfused with the cold (0 °C) VNIIKIEX solution in 30 s, and examined under EM at 8 and 24 h after perfusion. In the control samples, the microvilli were normal just after the perfusion (
Figure 7A,B). By 8 h, the microvilli were transformed into varicose tubules, while at 24 h, only vesicles were visible. This breakdown of the microvilli into spherical vesicles produced vesicles with a diameter of 198 ± 7 nm (
Figure 7C,D). Measurement of TAA revealed that it was 1.13 immediately after the perfusion, and 1.18 after the breakdown of the microvilli into vesicles. If we take into consideration that during the treatment of the sample with OsO
4 the thickness of the membrane increases from 4 to 8 nm, the real TAA was 1.083 and 1.085, respectively. Our calculations based on serial sections revealed that the surface area of the microvilli immediately after perfusion was equal to the surface area of the vesicles that formed from the microvilli. The ratio between the surface area and the volume decreased. The vesicles were almost perfectly spherical. This means that additional volume was delivered from the cellular bodies along the cytosolic continuity of the osmotic pressure into the vesicles, which increased in size when water went into them. Thus, the prediction of TAA claiming that under these conditions TAA would not change is confirmed.
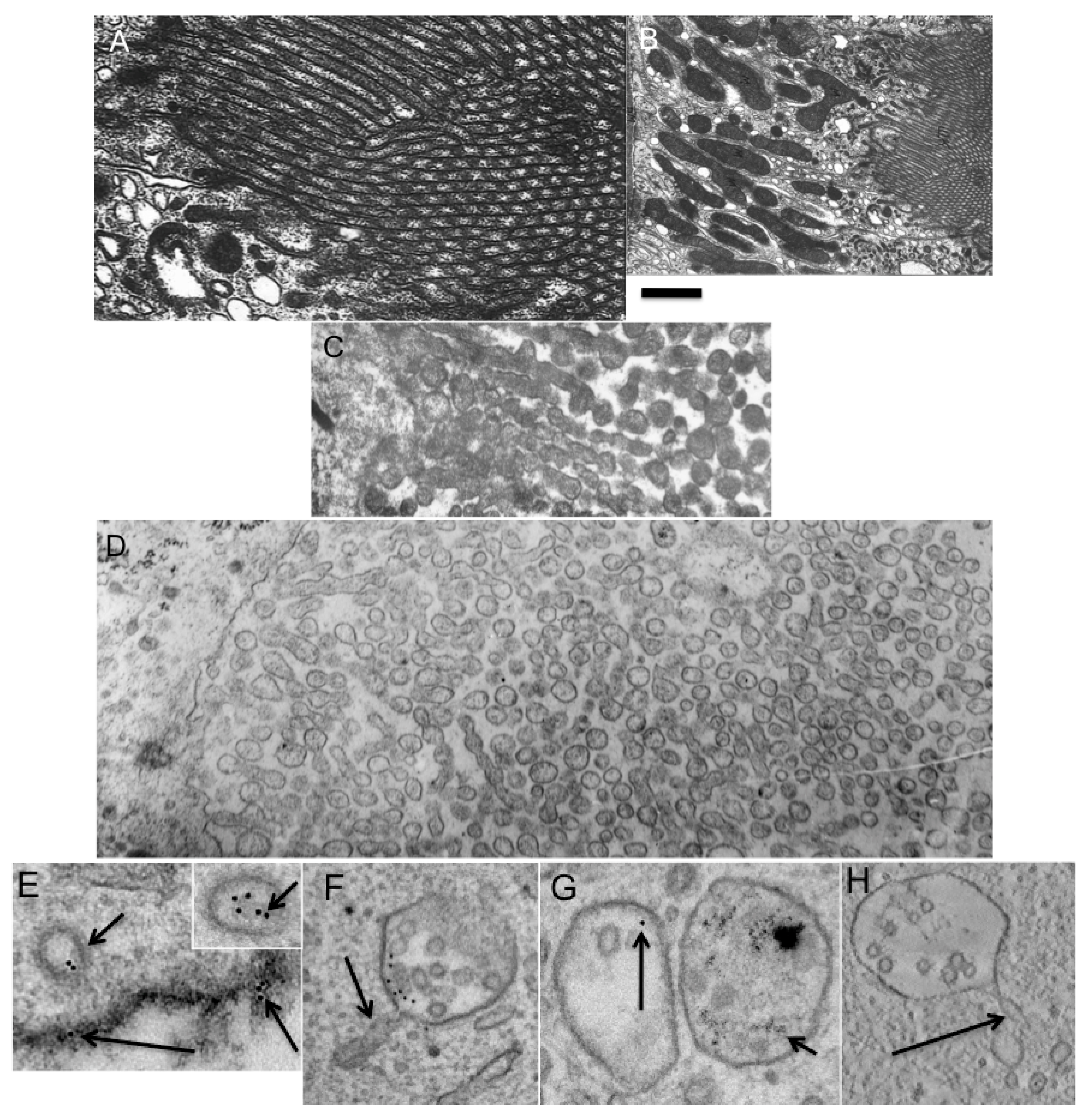
Figure 7.
Breakdown of epithelial microvilli of kidney proximal tubule into membrane spheres. (A). Normal brush border consists of cylindrical protrusion of the apical plasma membrane; (B) General view of the brush border; (C) Formation of varicosite cylinders from cylindrical microvilli; (D) Transformation of varicose cylinders into small membrane spheres; (E) Formation of the endocytic clathrin-coated vesicles (short arrows) with a diameter of 90 and 128 nm (insertion on the right). The large vesicle contains more gold particles but no intraluminal vesicles. Long arrow shows gold particcles embedded into oxidized 3,3'–diaminobenzidine (dark precipitate); (F–H) Formation of multivesicular bodies containing tubular protrusions with a diameter of 40 nm (arrows in F,H). (G) Two multivesicular bodies: one filled with 5-nm gold particles (on the right, short arrow) and another filled with 10-nm gold particles (on the left, long arrow). Bars. 230 nm (A–D); 100 nm (E–H).
Figure 7.
Breakdown of epithelial microvilli of kidney proximal tubule into membrane spheres. (A). Normal brush border consists of cylindrical protrusion of the apical plasma membrane; (B) General view of the brush border; (C) Formation of varicosite cylinders from cylindrical microvilli; (D) Transformation of varicose cylinders into small membrane spheres; (E) Formation of the endocytic clathrin-coated vesicles (short arrows) with a diameter of 90 and 128 nm (insertion on the right). The large vesicle contains more gold particles but no intraluminal vesicles. Long arrow shows gold particcles embedded into oxidized 3,3'–diaminobenzidine (dark precipitate); (F–H) Formation of multivesicular bodies containing tubular protrusions with a diameter of 40 nm (arrows in F,H). (G) Two multivesicular bodies: one filled with 5-nm gold particles (on the right, short arrow) and another filled with 10-nm gold particles (on the left, long arrow). Bars. 230 nm (A–D); 100 nm (E–H).
To test whether the transformation of endosome shape during their fusion and maturation occurs in agreement of our mathematical model (see Appendix;
Table S3), we prepared cells where endosomes were labeled with 5 nm and 10 nm gold (see Experimental Section). After such treatment, we observed three populations of endosomes inside the cells: newly formed; previously formed; and a small proportion of newly formed endosomes that fused with previously formed endosomes. The old endosomes contained only 5 nm gold, the newly formed endosomes contained only 10 nm gold (
Figure 7E), while the endosomes that were newly formed by fusion with old endosomes contained both types of gold particles. After this, the cells were incubated at 37 °C for 5 min, and examined under EM (
Figure 7F). As a control, we used samples that had been incubated with NEM, to block the fusion of endomembranes. We followed the fate of the endosomes, which were formed from the plasma membrane. These endosomes were labeled only with 10 nm gold. Their diameter was in the range of 100 to 130 nm. These endosomes could fuse with MVBs formed before the addition of 10 nm gold and labeled with 5 nm gold. In this case, we would see both 10 nm gold and 5 nm gold (
Figure 7G). Thus, if an endomembrane structure contained 10 nm gold and 5 nm gold it was not examined. Only endosomes that contained only 10 nm gold particles were examined. These endosomes could not fuse with the plasma membrane, which was stabilized with oxidized DAB (
Figure 7E) and a dilute concentration of 10 nm gold. In experimental cells, the number of gold particles and the mean diameter of the vacuoles that contained gold particles increased 3.2 ± 0.4-fold. Additionally many of these contained tubules that extended from the external membrane, and internal vacuoles and invaginations. Three-dimensional reconstruction revealed that the MVBs always contained not only intraluminal vesicles, but also external tubules with a diameter of 40 nm (
Figure 7F,H). The mean diameter of vacuoles with gold was 240 ± 31 nm, whereas in cells that were not treated with NEM, the diameter was 160 ± 24 nm. The number of internal vesicles was about 2 per vacuole. Intraluminal vesicles were not found in MVBs with diameters <230 nm. In our samples, the ratio between the external (intraluminal) surface area and that internal (inside a vesicle) was 1.53. Endomembranes with a size >150 nm that contained only 10 nm gold had intraluminal vesicles and external tubules. TAA of MVBs with 10 nm gold revealed that it was equal to TAA of the 90–100 nm endosomes that contained 10 nm gold particles observed immediately before fusion. There was a positive correlation between the number of intraluminal vesicles and the size of the MVBs (
r = 0.67). A similar correlation was seen between the number of tubular extensions and the number of intraluminal vesicles (
r = 0.59). In contrast, after incubation in the presence of NEM, the mean number of gold particles inside vacuoles and the mean diameter of these vacuoles were not changed. Thus, fusion of spherical endosomes occurred according to the predictions derived from the TAA hypothesis.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}