
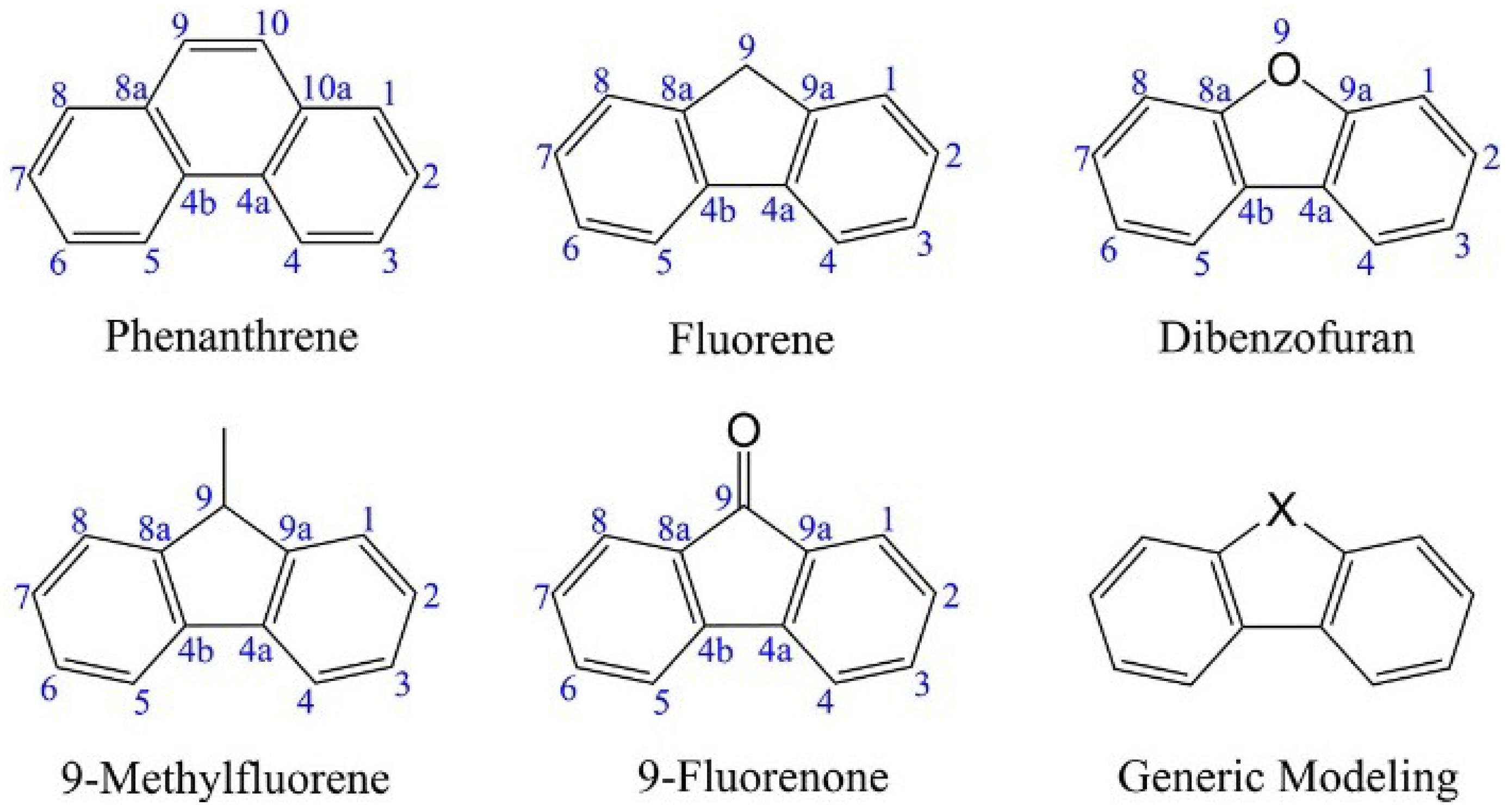
Mechanistic Studies on the Dibenzofuran Formation from Phenanthrene, Fluorene and 9–Fluorenone

Abstract
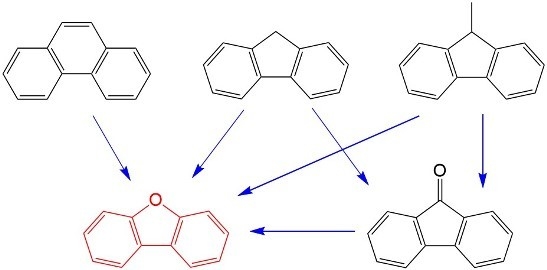
:
1. Introduction
2. Results and Discussion

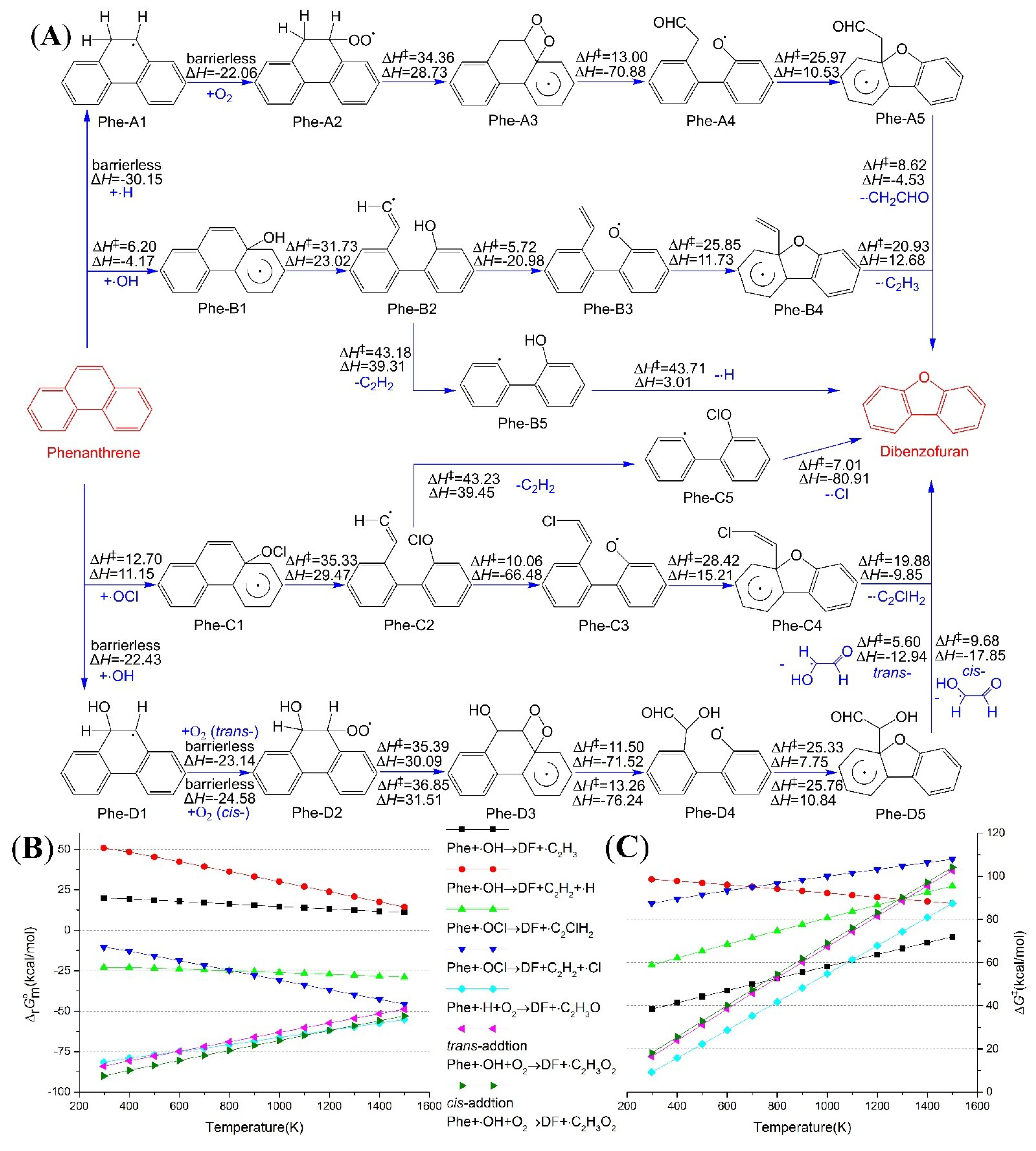
2.1. Reaction from Phenanthrene to Dibenzofuran
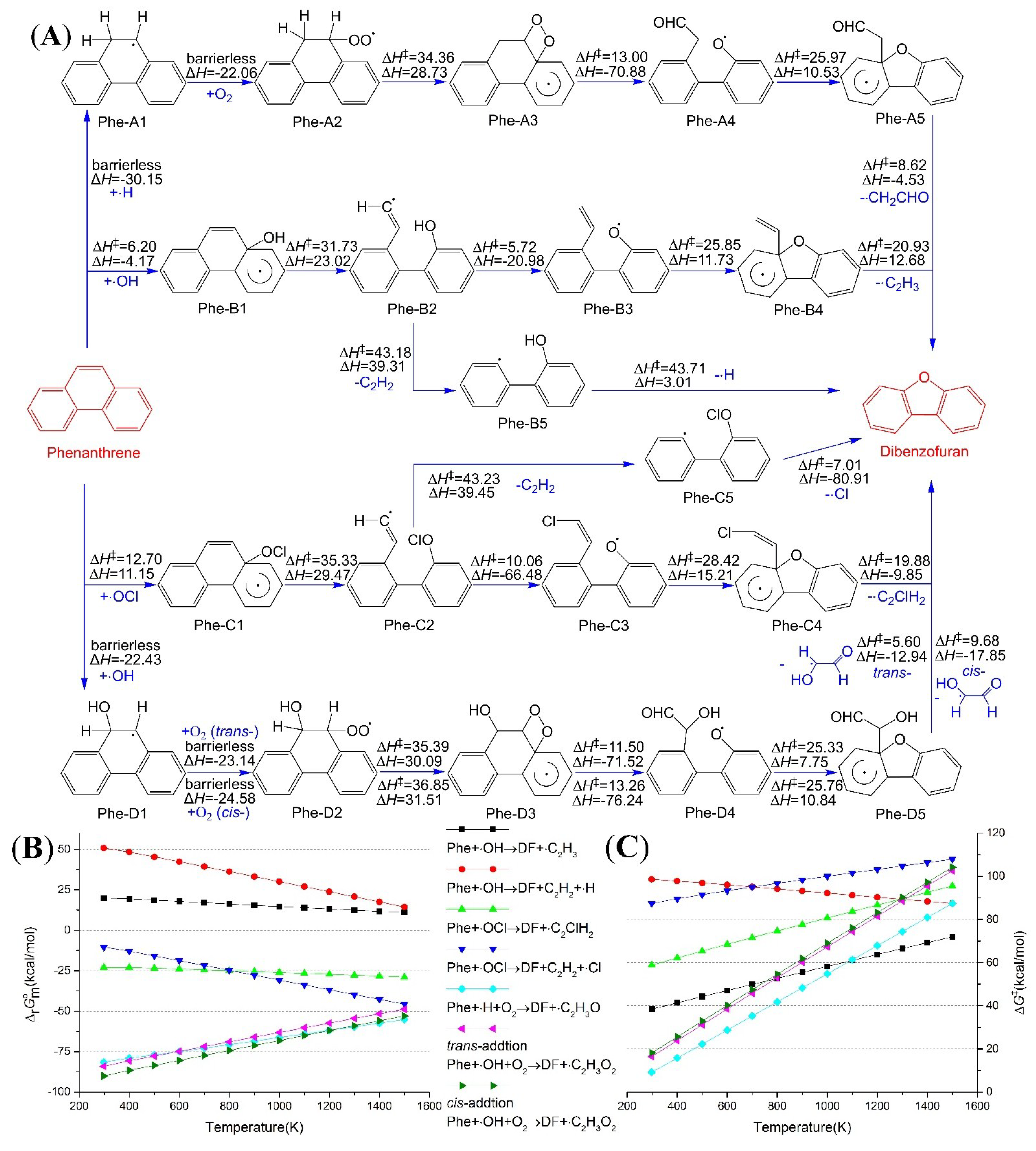
2.2. Reaction from Fluorene to Dibenzofuran and 9-Fluorenone


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H-Abstraction Reactions | |||||
|---|---|---|---|---|---|
| ∙OH | Phenanthrene | 3.65 | −6.22 | 28.79 | −12.50 |
| Fluorene | −0.24 | −35.38 | 24.01 | −38.48 | |
| 9-Methylfluorene | −1.38 | −39.37 | 24.69 | −48.04 | |
| HOO∙ | Phenanthrene | 25.86 | 26.22 | 53.23 | 22.81 |
| Fluorene | 12.12 | −2.93 | 44.63 | −3.67 | |
| 9-Methylfluorene | 10.39 | −6.92 | 41.79 | −12.73 | |
| ∙H | Phenanthrene | 14.96 | 8.58 | 35.54 | 1.97 |
| Fluorene | 4.93 | −20.58 | 28.27 | −24.01 | |
| 9-Methylfluorene | 3.24 | −24.57 | 25.71 | −33.57 | |
| ∙CH3 | Phenanthrene | 17.26 | 6.69 | 45.50 | 6.58 |
| Fluorene | 6.27 | −22.47 | 43.53 | −19.40 | |
| 9-Methylfluorene | 6.75 | −26.46 | 34.63 | −28.96 | |
| O2 | Phenanthrene | 58.34 | 63.55 | 78.94 | 54.91 |
| Fluorene | 36.59 | 34.40 | 62.90 | 28.93 | |
| 9-Methylfluorene | 33.97 | 30.41 | 60.21 | 19.37 | |
2.3. Reaction from 9-Methylfluorene to Dibenzofuran and 9-Fluorenone
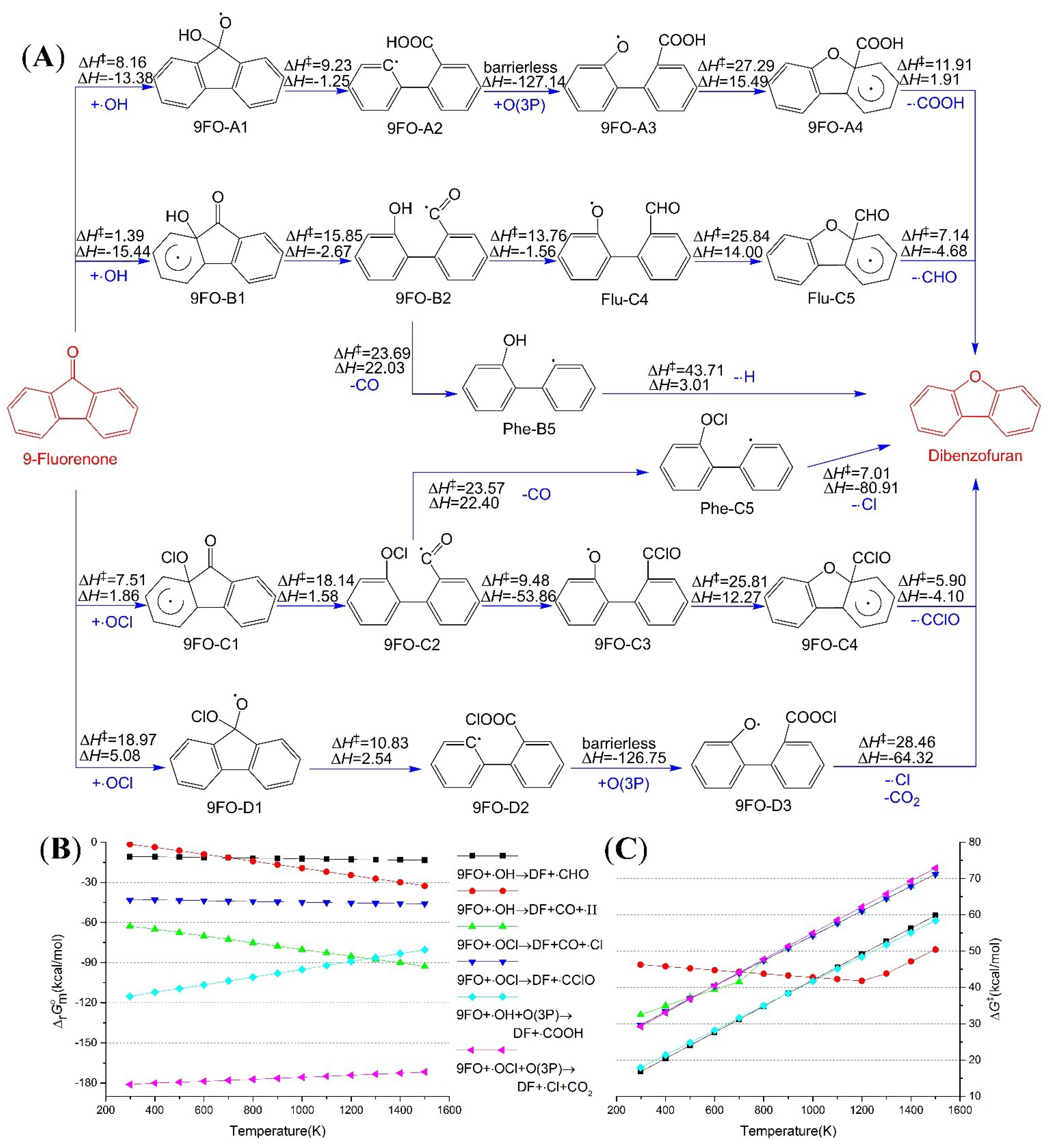
2.4. Reaction from 9-Fluorenone to Dibenzofuran

3. Experimental Section
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Arena, U. Process and technological aspects of municipal solid waste gasification. A review. Waste Manag. 2012, 32, 625–639. [Google Scholar] [CrossRef] [PubMed]
- McKay, G. Dioxin characterisation, formation and minimisation during municipal solid waste (MSW) incineration: Review. Chem. Eng. J. 2002, 86, 343–368. [Google Scholar] [CrossRef]
- Fernandez-Salguero, P.; Pineau, T.; Hilbert, D.M.; McPhail, T.; Lee, S.S.T.; Kimura, S.; Nebert, D.W.; Rudikoff, S.; Ward, J.M.; Gonzalez, F.J. Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor. Science 1995, 268, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, M.; Birnbaum, L.S.; Denison, M.; De Vito, M.; Farland, W.; Feeley, M.; Fiedler, H.; Hakansson, H.; Hanberg, A.; Haws, L.; et al. The 2005 world health organization reevaluation of human and mammalian toxic equivalency factors for dioxins and dioxin-like compounds. Toxicol. Sci. 2006, 93, 223–241. [Google Scholar] [CrossRef] [PubMed]
- Altarawneh, M.; Dlugogorski, B.Z.; Kennedy, E.M.; Mackie, J.C. Mechanisms for formation, chlorination, dechlorination and destruction of polychlorinated dibenzo-p-dioxins and dibenzofurans (PCDD/Fs). Prog. Energ. Combust. 2009, 35, 245–274. [Google Scholar] [CrossRef]
- Ryu, J.Y.; Mulholland, J.A.; Dunn, J.E.; Iino, F.; Gullett, B.K. Potential role of chlorination pathways in PCDD/F formation in a municipal waste incinerator. Environ. Sci. Technol. 2004, 38, 5112–5119. [Google Scholar] [CrossRef] [PubMed]
- Lomnicki, S.; Dellinger, B. A detailed mechanism of the surface-mediated formation of PCDD/F from the oxidation of 2-chlorophenol on a CuO/silica surface. J. Phys. Chem. A 2003, 107, 4387–4395. [Google Scholar] [CrossRef]
- Tuppurainen, K.A.; Ruokojärvi, P.H.; Asikainen, A.H.; Aatamila, M.; Ruuskanen, J. Chlorophenols as precursors of PCDD/Fs in incineration processes: Correlations, PLS modeling, and reaction mechanisms. Environ. Sci. Technol. 2000, 34, 4958–4962. [Google Scholar] [CrossRef]
- Ryu, J.-Y.; Choi, K.-C.; Mulholland, J.A. Polychlorinated dibenzo-p-dioxin (PCDD) and dibenzofurna (PCDF) isomer patterns from municipal waste combustion: Formation mechanism fingerprints. Chemosphere 2006, 65, 1526–1536. [Google Scholar] [CrossRef] [PubMed]
- Altarawneh, M.; Dlugogorski, B.Z.; Kennedy, E.M.; Mackie, J.C. Quantum chemical investigation of formation of polychlorodibenzo-p-dioxins and dibenzofurans from oxidation and pyrolysis of 2‑chlorophenol. J. Phys. Chem. A 2007, 111, 2563–2573. [Google Scholar] [CrossRef] [PubMed]
- Briois, C.; Visez, N.; Baillet, C.; Sawerysyn, J.P. Experimental study on the thermal oxidation of 2-chlorophenol in air over the temperature range 450–900 degrees C. Chemosphere 2006, 62, 1806–1816. [Google Scholar] [CrossRef] [PubMed]
- Altarawneh, M.; Dlugogorski, B.Z.; Kennedy, E.M.; Mackie, J.C. Theoretical study of unimolecular decomposition of catechol. J. Phys. Chem. A 2010, 114, 1060–1067. [Google Scholar] [CrossRef] [PubMed]
- Alsoufi, A.; Altarawneh, M.; Dlugogorski, B.Z.; Kennedy, E.M.; Mackie, J.C. A DFT study on the self-coupling reactions of the three isomeric semiquinone radicals. J. Mol. Struct. 2010, 958, 106–115. [Google Scholar] [CrossRef]
- Chen, K.; Wojtalewicz, D.; Altarawneh, M.; Mackie, J.C.; Kennedy, E.M.; Dlugogorski, B.Z. Formation of polychlorinated dibenzo-p-dioxins and dibenzofurans (PCDD/F) in oxidation of captan pesticide. Proc. Combust. Inst. 2011, 33, 701–708. [Google Scholar] [CrossRef]
- Cosentino, U.; Pitea, D.; Moro, G. Computational modelling of de novo synthesis of dibenzofuran: Oxidative pathways of pyrene and benzodibenzofuran. Theor. Chem. Acc. 2012, 131, 1–12. [Google Scholar] [CrossRef]
- Zimmermann, R.; Blumenstock, M.; Heger, H.J.; Schramm, K.W.; Kettrup, A. Emission of nonchlorinated and chlorinated aromatics in the flue gas of incineration plants during and after transient disturbances of combustion conditions: Delayed emission effects. Environ. Sci. Technol. 2001, 35, 1019–1030. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Austrui, J.; Borrajo, M.A.; Martinez, K.; Adrados, M.A.; Abalos, M.; Van Bavel, B.; Rivera, J.; Abad, E. Assessment of polychlorinated dibenzo-p-dioxin and dibenzofuran emissions from a hazardous waste incineration plant using long-term sampling equipment. Chemosphere 2011, 82, 1343–1349. [Google Scholar] [CrossRef] [PubMed]
- Onofrio, M.; Spataro, R.; Botta, S. The role of a steel plant in north-west Italy to the local air concentrations of PCDD/Fs. Chemosphere 2011, 82, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Hell, K.; Stieglitz, L.; Dinjus, E. Mechanistic aspects of the de-novo synthesis of PCDD/PCDF on model mixtures and MSWI fly ashes using amorphous 12C- and 13C-labeled carbon. Environ. Sci. Technol. 2001, 35, 3892–3898. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Li, X.; Zhang, X.; Liu, K.; Yan, J.; Yan, K. Correlation between PAHS and PCDD/Fs in municipal solid waste incinerators. J. Zhejiang Univ. (Eng. Sci.) 2010, 44, 1118–1121. [Google Scholar]
- Johansson, I.; van Bavel, B. Levels and patterns of polycyclic aromatic hydrocarbons in incineration ashes. Sci. Total Environ. 2003, 311, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Weber, R.; Iino, F.; Imagawa, T.; Takeuchi, M.; Sakurai, T.; Sadakata, M. Formation of PCDF, PCDD, PCB, and PCN in de novo synthesis from PAH: Mechanistic aspects and correlation to fluidized bed incinerators. Chemosphere 2001, 44, 1429–1438. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.J.; Liow, M.C.; Tsai, P.J.; Hsieh, L.T. Emission of polycyclic aromatic hydrocarbons from medical waste incinerators. Atmos. Environ. 2002, 36, 781–790. [Google Scholar] [CrossRef]
- Ryu, J.-Y.; Mulholland, J.A.; Chu, B. Chlorination of dibenzofuran and dibenzo-p-dioxin vapor by copper (II) chloride. Chemosphere 2003, 51, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Blumenstock, M.; Zimmermann, R.; Schramm, K.W.; Kettrup, A. Influence of combustion conditions on the PCDD/F-, PCB-, PCBz- and PAH-concentrations in the post-combustion chamber of a waste incineration pilot plant. Chemosphere 2000, 40, 987–993. [Google Scholar] [CrossRef] [PubMed]
- Tokmakov, I.V.; Kim, G.-S.; Kislov, V.V.; Mebel, A.M.; Lin, M.C. The reaction of phenyl radical with molecular oxygen: A G2M study of the potential energy surface. J. Phys. Chem. A 2005, 109, 6114–6127. [Google Scholar] [CrossRef] [PubMed]
- Sebbar, N.; Bockhorn, H.; Bozzelli, J.W. Thermochemical similarities among three reaction systems: Vinyl + O2–phenyl + O2–dibenzofuranyl + O2. Combust. Sci. Technol. 2008, 180, 959–974. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, L.; Tian, Z.; Yuan, T.; Wang, J.; Yang, B.; Qi, F. Experimental study of a fuel-rich premixed toluene flame at low pressure. Energ. Fuel 2009, 23, 1473–1485. [Google Scholar] [CrossRef]
- Khachatryan, L.; Burcat, A.; Dellinger, B. The role of chlorine atoms and hydroxyl radicals in the formation of PCDDs from the oxidative pyrolysis of 2,4,6-trichlorophenol. Int. J. Chem. Kinet. 2010, 42, 90–97. [Google Scholar] [CrossRef]
- Aizawa, T. Diode-laser wavelength-modulation absorption spectroscopy for quantitative in situ measurements of temperature and OH radical concentration in combustion gases. Appl. Opt. 2001, 40, 4894–4903. [Google Scholar] [CrossRef] [PubMed]
- Yuasa, S.; Yushina, S.; Uchida, T.; Shiraishi, N. Ignition and combustion of ammonium perchlorate in a hydrogen atmosphere. Proc. Combust. Inst. 2000, 28, 863–870. [Google Scholar] [CrossRef]
- Canneaux, S.; Hammaecher, C.; Cours, T.; Louis, F.; Ribaucour, M. Theoretical study of H‑abstraction reactions from CH3Cl and CH3Br molecules by ClO and BrO radicals. J. Phys. Chem. A 2012, 116, 4396–4408. [Google Scholar] [CrossRef] [PubMed]
- Johnson III, R.D. NIST computational chemistry comparison and benchmark database. Release 16 August 2013. Available online: http://cccbdb.nist.gov/ (accessed on 20 December 2014).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Goerigk, L. How do DFT-DCP, DFT-NL, and DFT-D3 compare for the description of London-dispersion effects in conformers and general thermochemistry? J. Chem. Theory Comput. 2014, 10, 968–980. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Steinmann, S.N.; Corminboeuf, C. Comprehensive benchmarking of a density-dependent dispersion correction. J. Chem. Theory Comput. 2011, 7, 3567–3577. [Google Scholar] [CrossRef]
- Goerigk, L.; Grimme, S. A thorough benchmark of density functional methods for general main group thermochemistry, kinetics, and noncovalent interactions. Phys. Chem. Chem. Phys. 2011, 13, 6670–6688. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.; Zhang, Q. Mechanistic Studies on the Dibenzofuran Formation from Phenanthrene, Fluorene and 9–Fluorenone. Int. J. Mol. Sci. 2015, 16, 5271-5284. https://doi.org/10.3390/ijms16035271
Li S, Zhang Q. Mechanistic Studies on the Dibenzofuran Formation from Phenanthrene, Fluorene and 9–Fluorenone. International Journal of Molecular Sciences. 2015; 16(3):5271-5284. https://doi.org/10.3390/ijms16035271
Chicago/Turabian StyleLi, Shanqing, and Qingzhu Zhang. 2015. "Mechanistic Studies on the Dibenzofuran Formation from Phenanthrene, Fluorene and 9–Fluorenone" International Journal of Molecular Sciences 16, no. 3: 5271-5284. https://doi.org/10.3390/ijms16035271