
Monitoring the Spatiotemporal Activities of miRNAs in Small Animal Models Using Molecular Imaging Modalities

Abstract
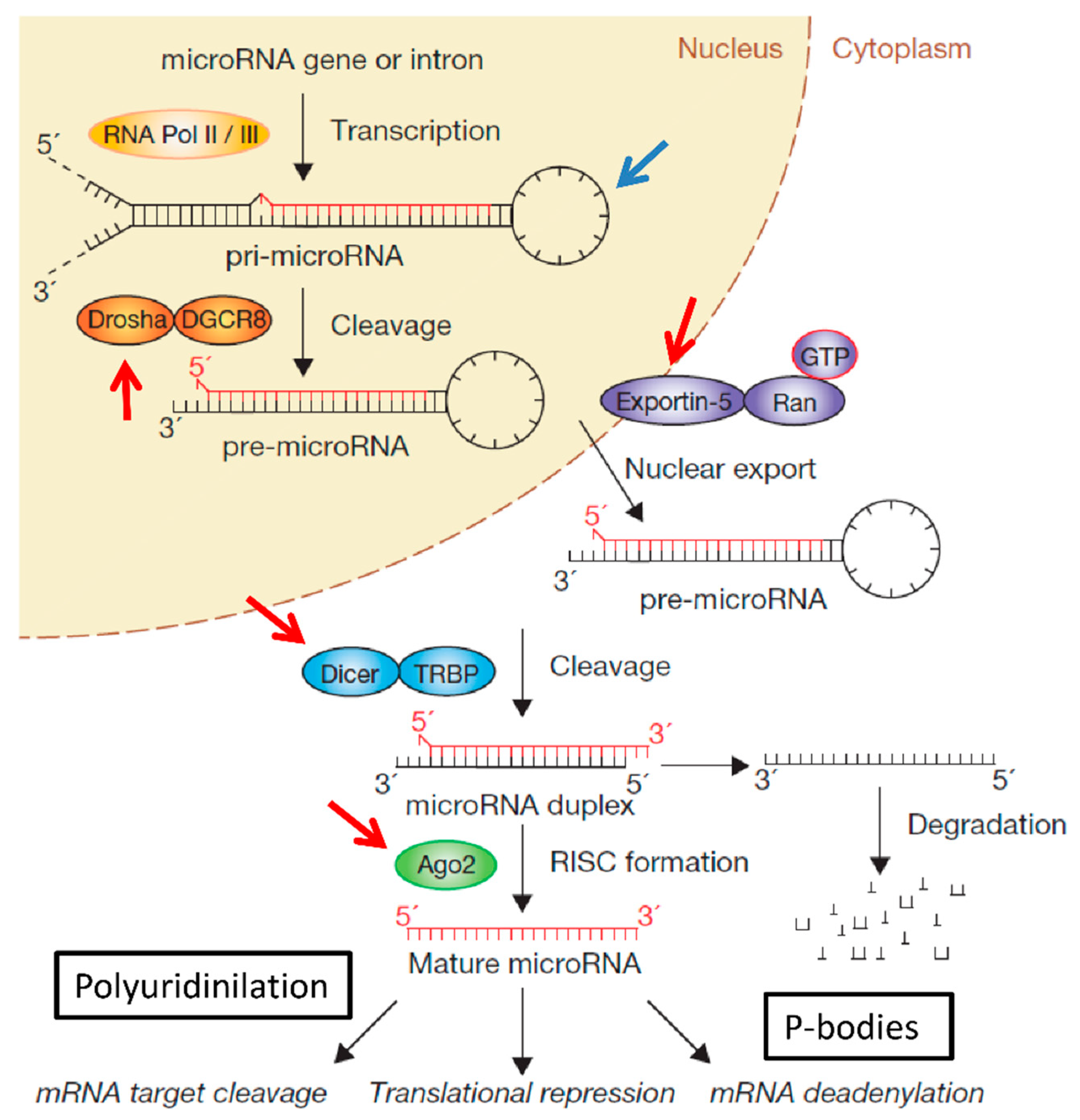
:1. Introduction

2. Conventional Methods for miRNA Detection: PCR, Northern-Blot, Deep Sequencing and in Situ Hybridisation (ISH)
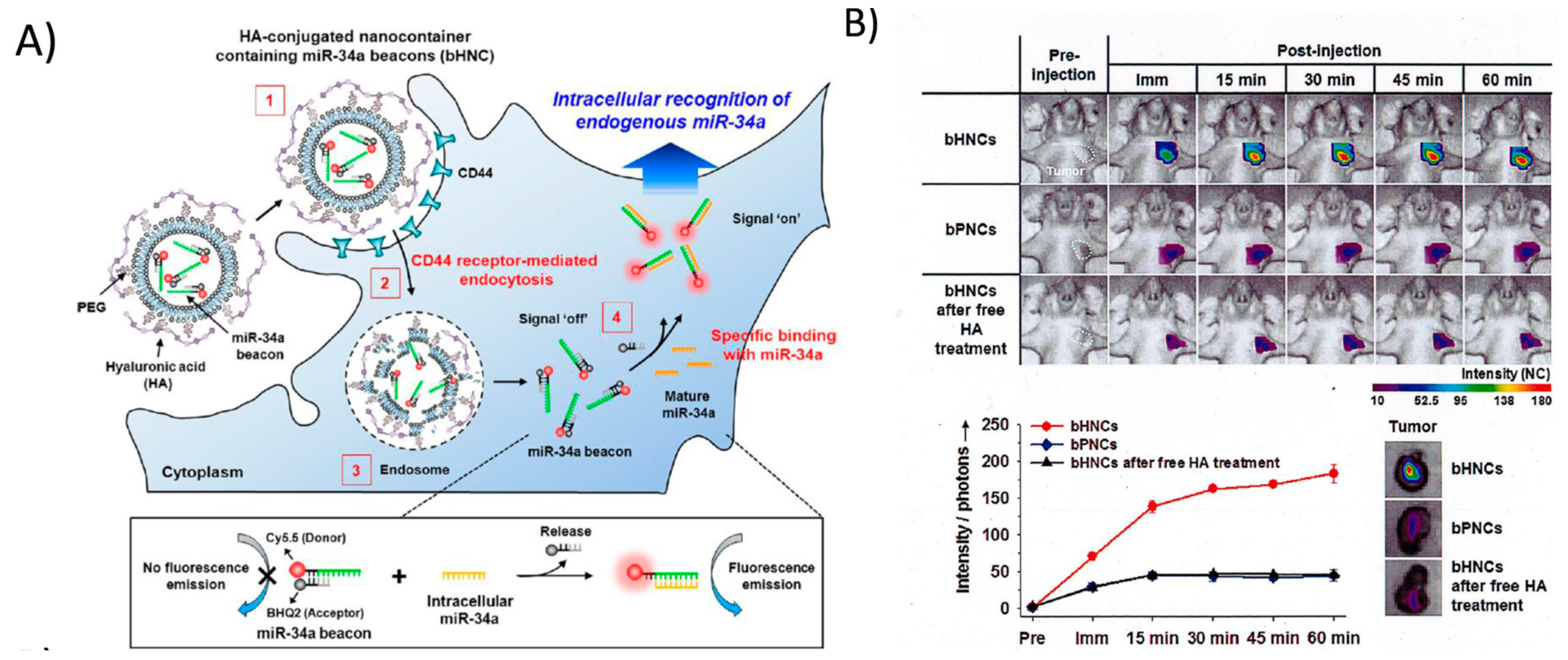
3. Molecular Imaging Using Synthetic Fluorophore-Labelled Probes

4. Molecular Imaging Using Biological Probes: Applications in Basic Research and RNAi-Based-Therapy Including Gene and Cell Therapy
4.1. Negative Read out miRNA Monitoring Systems
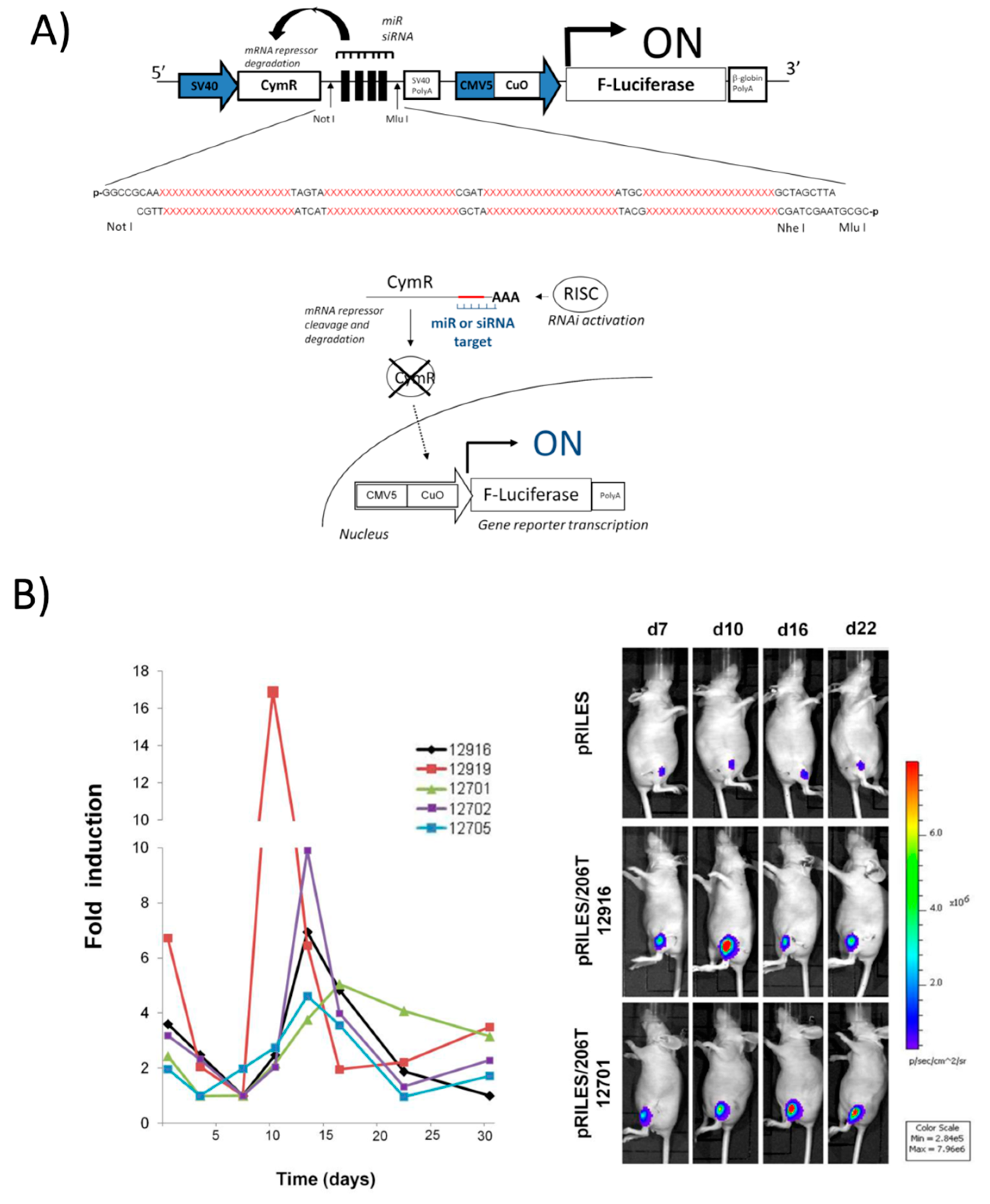
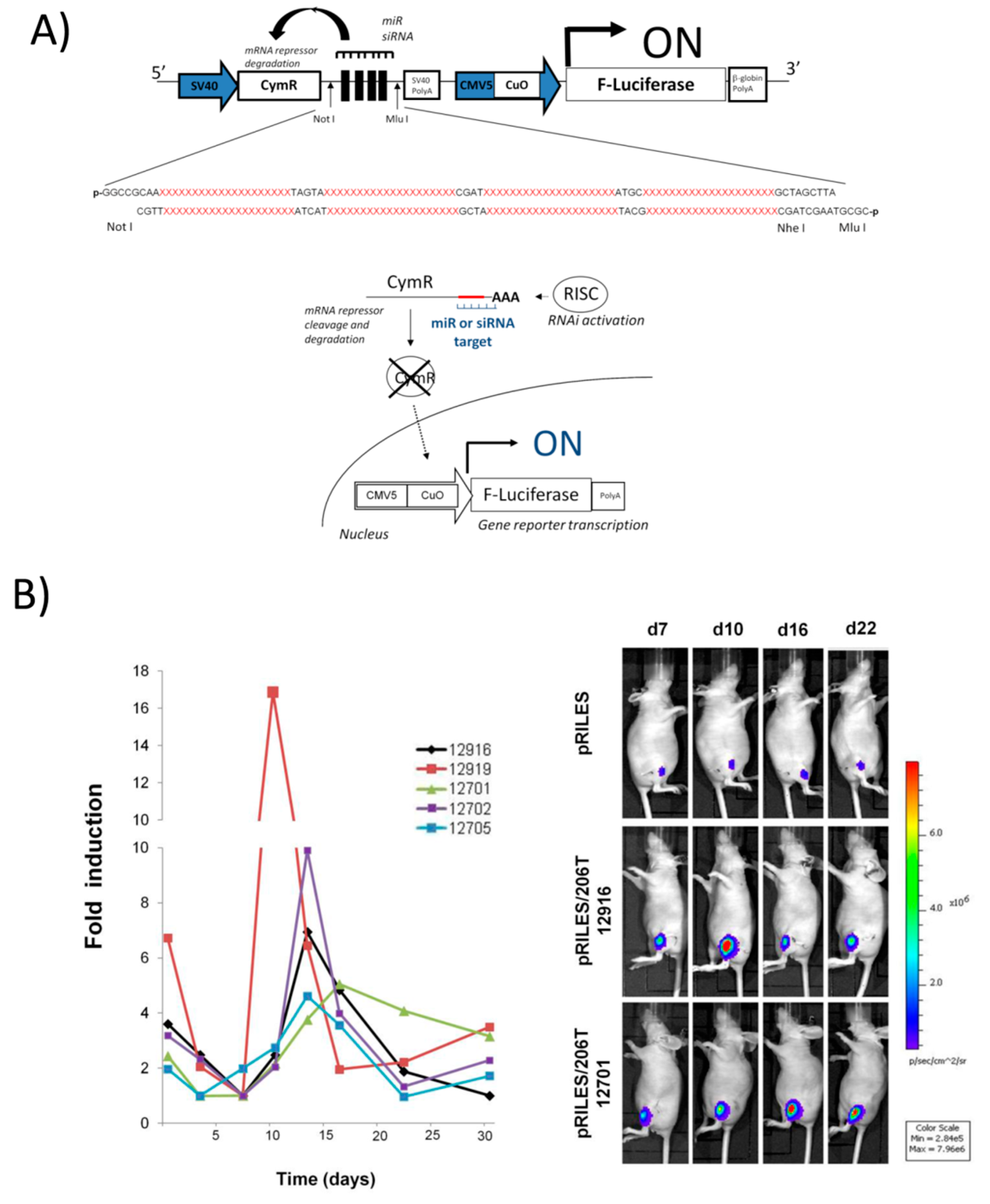
4.2. Positive Read-out miRNA Monitoring Systems

{kind=link}
{kind=link}
{kind=link}
| miRNA Monitoring Methods | Functional Monitoring | Spatio-Temporal Resolution | Advantages | Drawbacks | Ref. |
|---|---|---|---|---|---|
| Invasive Methods | |||||
| Northern-blot | No * | Difficult * | Qualitative, no amplification procedure, sensitive | Lysis, large amount of starting material, long procedure | [41,42] |
| Microarray | No * | Difficult * | High-throughput screening, qualitative, biomarkers | Lysis, amplification procedure, false hybridization, biased interpretations | [43,44] |
| Real-time PCR | No * | Difficult * | Qualitative, quantitative, fast and accessible procedure | Lysis, amplification procedure, false hybridization, biased interpretations | [46,47] |
| Deep sequencing | No * | Difficult * | High resolution, qualitative, quantitative, sensitive and specific | Lysis, long procedure, complex bioinformatic analysis | [49,50] |
| ISH | No * | Spatial | Spatial resolution, no amplification procedure, colorimetric method | Low sensitivity, long and complex procedure | [52,53] |
| Non-Invasive Methods | |||||
| Molecular beacons | No * | Yes | Positive monitoring, short fellow-up, fast procedure, theranostic probes | Low signal-to-noise ratio, low resolution | [60,62] |
| Negative read-out | Yes | Yes | Functional monitoring, qualitative and quantitative | Negative monitoring | [72,77] |
| Positive Read-out | |||||
| RILES | Yes | Yes | Positive monitoring, qualitative and quantitative, temporal resolution | Leakiness, immunogenicity, potentially interfering | [85] |
| TetKrab | Yes | Yes | Positive monitoring, qualitative and quantitative, temporal resolution | Leakiness, immunogenicity, potentially interfering | [86] |
| miR-ON | Yes | Yes | Positive monitoring, qualitative and quantitative, temporal resolution | Leakiness, immunogenicity, potentially interfering | [87] |
5. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 1993, 75, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.; Jung, S.; Keller, S.; Gregory, R.I.; Diederichs, S. Many roads to maturity: MicroRNA biogenesis pathways and their regulation. Nat. Cell Biol. 2009, 11, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, E.; Kim, S.Y.; Carmell, M.A.; Murchison, E.P.; Alcorn, H.; Li, M.Z.; Mills, A.A.; Elledge, S.J.; Anderson, K.V.; Hannon, G.J. Dicer is essential for mouse development. Nat. Genet. 2003, 35, 215–217. [Google Scholar] [CrossRef] [PubMed]
- Wienholds, E.; Koudijs, M.J.; van Eeden, F.J.; Cuppen, E.; Plasterk, R.H. The microRNA-producing enzyme Dicer1 is essential for zebrafish development. Nat. Genet. 2003, 35, 217–218. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Kole, R.; Krainer, A.R.; Altman, S. RNA therapeutics: Beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug Discov. 2012, 11, 125–140. [Google Scholar] [PubMed]
- Garzon, R.; Marcucci, G.; Croce, C.M. Targeting microRNAs in cancer: Rationale, strategies and challenges. Nat. Rev. Drug Discov. 2010, 9, 775–789. [Google Scholar] [CrossRef] [PubMed]
- Berezikov, E. Evolution of microRNA diversity and regulation in animals. Nature Rev. Genet. 2011, 12, 846–860. [Google Scholar] [CrossRef] [PubMed]
- Siomi, H.; Siomi, M.C. Posttranscriptional regulation of microRNA biogenesis in animals. Mol. Cell 2010, 38, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Nishikura, K. Editor meets silencer: Crosstalk between RNA editing and RNA interference. Nat. Rev. Mol. Cell Biol. 2006, 7, 919–931. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Sansam, C.L.; Singh, M.; Emeson, R.B. Altered RNA editing in mice lacking ADAR2 autoregulation. Mol. Cell. Biol. 2006, 26, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, Y.; Ito, K.; Sun, H.; Aizawa, H.; Kanazawa, I.; Kwak, S. Glutamate receptors: RNA editing and death of motor neurons. Nature 2004, 427, 801. [Google Scholar] [CrossRef] [PubMed]
- Paz, N.; Levanon, E.Y.; Amariglio, N.; Heimberger, A.B.; Ram, Z.; Constantini, S.; Barbash, Z.S.; Adamsky, K.; Safran, M.; Hirschberg, A.; et al. Altered adenosine-to-inosine RNA editing in human cancer. Genome Res. 2007, 17, 1586–1595. [Google Scholar] [CrossRef] [PubMed]
- Tomaselli, S.; Bonamassa, B.; Alisi, A.; Nobili, V.; Locatelli, F.; Gallo, A. ADAR enzyme and miRNA story: A nucleotide that can make the difference. Int. J. Mol. Sci. 2013, 14, 22796–22816. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, Y.; Tay, F.C.; Lam, D.H.; Sandanaraj, E.; Tang, C.; Ang, B.T.; Wang, S. Attenuated adenosine-to-inosine editing of microRNA-376a* promotes invasiveness of glioblastoma cells. J. Clin. Investig. 2012, 122, 4059–4076. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, Y.; Zinshteyn, B.; Sethupathy, P.; Iizasa, H.; Hatzigeorgiou, A.G.; Nishikura, K. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science 2007, 315, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Ota, H.; Sakurai, M.; Gupta, R.; Valente, L.; Wulff, B.E.; Ariyoshi, K.; Iizasa, H.; Davuluri, R.V.; Nishikura, K. ADAR1 forms a complex with Dicer to promote microRNA processing and RNA-induced gene silencing. Cell 2013, 153, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Eulalio, A.; Behm-Ansmant, I.; Izaurralde, E. P-bodies: At the crossroads of post-transcriptional pathways. Nat. Rev. Mol. Cell Biol. 2007, 8, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Jakymiw, A.; Pauley, K.M.; Li, S.; Ikeda, K.; Lian, S.; Eystathioy, T.; Satoh, M.; Fritzler, M.J.; Chan, E.K. The role of GW/P-bodies in RNA processing and silencing. J. Cell Sci. 2007, 120, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Valencia-Sanchez, M.A.; Hannon, G.J.; Parker, R. MicroRNA-dependent localization of targeted mRNAs to mammalian P-bodies. Nat. Cell Biol. 2005, 7, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Pillai, R.S.; Bhattacharyya, S.N.; Artus, C.G.; Zoller, T.; Cougot, N.; Basyuk, E.; Bertrand, E.; Filipowicz, W. Inhibition of translational initiation by let-7 microRNA in human cells. Science 2005, 309, 1573–1576. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.N.; Habermacher, R.; Martine, U.; Closs, E.I.; Filipowicz, W. Relief of microRNA-mediated translational repression in human cells subjected to stress. Cell 2006, 125, 1111–1124. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.P.; Doidge, R.; Tarr, A.W.; Jopling, C.L. The P body protein LSm1 contributes to stimulation of hepatitis C virus translation, but not replication, by microRNA-122. Nucleic Acids Res. 2014, 42, 1257–1269. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.R.; Quinton, L.J.; Blahna, M.T.; Neilson, J.R.; Fu, S.; Ivanov, A.R.; Wolf, D.A.; Mizgerd, J.P. Zcchc11-dependent uridylation of microRNA directs cytokine expression. Nat. Cell Biol. 2009, 11, 1157–1163. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.R.; Blahna, M.T.; Kozlowski, E.; Matsuura, K.Y.; Ferrari, J.D.; Morris, S.A.; Powers, J.T.; Daley, G.Q.; Quinton, L.J.; Mizgerd, J.P. Zcchc11 uridylates mature miRNAs to enhance neonatal IGF-1 expression, growth, and survival. PLoS Genet. 2012, 8, e1003105. [Google Scholar] [CrossRef] [PubMed]
- Ustianenko, D.; Hrossova, D.; Potesil, D.; Chalupnikova, K.; Hrazdilova, K.; Pachernik, J.; Cetkovska, K.; Uldrijan, S.; Zdrahal, Z.; Vanacova, S. Mammalian DIS3L2 exoribonuclease targets the uridylated precursors of let-7 miRNAs. RNA 2013, 19, 1632–1638. [Google Scholar] [CrossRef] [PubMed]
- Heo, I.; Ha, M.; Lim, J.; Yoon, M.J.; Park, J.E.; Kwon, S.C.; Chang, H.; Kim, V.N. Mono-uridylation of pre-microRNA as a key step in the biogenesis of group II let-7 microRNAs. Cell 2012, 151, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Hagedorn, C.H.; Cullen, B.R. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA 2004, 10, 1957–1966. [Google Scholar] [CrossRef] [PubMed]
- Burns, D.M.; D’Ambrogio, A.; Nottrott, S.; Richter, J.D. CPEB and two poly(A) polymerases control miR-122 stability and p53 mRNA translation. Nature 2011, 473, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Katoh, T.; Sakaguchi, Y.; Miyauchi, K.; Suzuki, T.; Kashiwabara, S.; Baba, T.; Suzuki, T. Selective stabilization of mammalian microRNAs by 3' adenylation mediated by the cytoplasmic poly(A) polymerase GLD-2. Genes Dev. 2009, 23, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Melo, S.A.; Moutinho, C.; Ropero, S.; Calin, G.A.; Rossi, S.; Spizzo, R.; Fernandez, A.F.; Davalos, V.; Villanueva, A.; Montoya, G.; et al. A genetic defect in exportin-5 traps precursor microRNAs in the nucleus of cancer cells. Cancer Cell 2010, 18, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Cabello, D.; Callejas, S.; Benguria, A.; Moreno, A.; Alonso, J.; Palmero, I. Regulation of the microRNA processor DGCR8 by the tumor suppressor ING1. Cancer Res. 2010, 70, 1866–1874. [Google Scholar] [CrossRef] [PubMed]
- Sand, M.; Skrygan, M.; Georgas, D.; Arenz, C.; Gambichler, T.; Sand, D.; Altmeyer, P.; Bechara, F.G. Expression levels of the microRNA maturing microprocessor complex component DGCR8 and the RNA-induced silencing complex (RISC) components argonaute-1, argonaute-2, PACT, TARBP1, and TARBP2 in epithelial skin cancer. Mol. Carcinog. 2012, 51, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Chen, H.; Zhang, L.; Dakhova, O.; Zhang, Y.; Lewis, M.T.; Creighton, C.J.; Ittmann, M.M.; Xin, L. A dosage-dependent pleiotropic role of Dicer in prostate cancer growth and metastasis. Oncogene 2014, 33, 3099–3108. [Google Scholar] [CrossRef] [PubMed]
- Mullokandov, G.; Baccarini, A.; Ruzo, A.; Jayaprakash, A.D.; Tung, N.; Israelow, B.; Evans, M.J.; Sachidanandam, R.; Brown, B.D. High-throughput assessment of microRNA activity and function using microRNA sensor and decoy libraries. Nat. Methods 2012, 9, 840–846. [Google Scholar] [CrossRef] [PubMed]
- Perron, M.P.; Landry, P.; Plante, I.; Provost, P. Detection of human Dicer and Argonaute 2 catalytic activity. Methods Mol. Biol. 2011, 725, 121–141. [Google Scholar] [PubMed]
- Hock, J.; Weinmann, L.; Ender, C.; Rudel, S.; Kremmer, E.; Raabe, M.; Urlaub, H.; Meister, G. Proteomic and functional analysis of Argonaute-containing mRNA-protein complexes in human cells. EMBO Rep. 2007, 8, 1052–1060. [Google Scholar] [CrossRef] [PubMed]
- Turchinovich, A.; Burwinkel, B. Distinct AGO1 and AGO2 associated miRNA profiles in human cells and blood plasma. RNA Biol. 2012, 9, 1066–1075. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, C.C.; Cheng, H.H.; Tewari, M. MicroRNA profiling: Approaches and considerations. Nat. Rev. Genet. 2012, 13, 358–369. [Google Scholar] [CrossRef] [PubMed]
- Lagos-Quintana, M.; Rauhut, R.; Lendeckel, W.; Tuschl, T. Identification of novel genes coding for small expressed RNAs. Science 2001, 294, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Varallyay, E.; Burgyan, J.; Havelda, Z. MicroRNA detection by northern blotting using locked nucleic acid probes. Nat. Protoc. 2008, 3, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Leshkowitz, D.; Horn-Saban, S.; Parmet, Y.; Feldmesser, E. Differences in microRNA detection levels are technology and sequence dependent. RNA 2013, 19, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Git, A.; Dvinge, H.; Salmon-Divon, M.; Osborne, M.; Kutter, C.; Hadfield, J.; Bertone, P.; Caldas, C. Systematic comparison of microarray profiling, real-time PCR, and next-generation sequencing technologies for measuring differential microRNA expression. RNA 2010, 16, 991–1006. [Google Scholar] [CrossRef] [PubMed]
- Mestdagh, P.; Hartmann, N.; Baeriswyl, L.; Andreasen, D.; Bernard, N.; Chen, C.; Cheo, D.; D’Andrade, P.; DeMayo, M.; Dennis, L.; et al. Evaluation of quantitative miRNA expression platforms in the microRNA quality control (miRQC) study. Nat. Methods 2014, 11, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Lee, E.J.; Jiang, J.; Sarkar, A.; Yang, L.; Elton, T.S.; Chen, C. Real-time PCR quantification of precursor and mature microRNA. Methods 2008, 44, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Wang, X. A PCR-based platform for microRNA expression profiling studies. RNA 2009, 15, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Mestdagh, P.; Derveaux, S.; Vandesompele, J. Whole-genome RT-qPCR microRNA expression profiling. Methods Mol. Biol. 2012, 815, 121–130. [Google Scholar] [PubMed]
- Friedlander, M.R.; Chen, W.; Adamidi, C.; Maaskola, J.; Einspanier, R.; Knespel, S.; Rajewsky, N. Discovering microRNAs from deep sequencing data using miRDeep. Nat. Biotechnol. 2008, 26, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.F.; Ansel, K.M. Construction of small RNA cDNA libraries for deep sequencing. Methods Mol. Biol. 2010, 667, 93–111. [Google Scholar] [PubMed]
- Kloosterman, W.P.; Wienholds, E.; de Bruijn, E.; Kauppinen, S.; Plasterk, R.H. In situ detection of miRNAs in animal embryos using LNA-modified oligonucleotide probes. Nat. Methods 2006, 3, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Baldwin, D.A.; Kloosterman, W.P.; Kauppinen, S.; Plasterk, R.H.; Mourelatos, Z. RAKE and LNA-ISH reveal microRNA expression and localization in archival human brain. RNA 2006, 12, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Soe, M.J.; Moller, T.; Dufva, M.; Holmstrom, K. A sensitive alternative for microRNA in situ hybridizations using probes of 2'-O-methyl RNA + LNA. J. Histochem. Cytochem. 2011, 59, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Monroy-Contreras, R.; Vaca, L. Molecular beacons: Powerful tools for imaging RNA in living cells. J. Nucleic Acids 2011, 2011, 741723. [Google Scholar] [CrossRef] [PubMed]
- Bratu, D.P.; Catrina, I.E.; Marras, S.A. Tiny molecular beacons for in vivo mRNA detection. Methods Mol. Biol. 2011, 714, 141–157. [Google Scholar] [PubMed]
- Baker, M.B.; Bao, G.; Searles, C.D. In vitro quantification of specific microRNA using molecular beacons. Nucleic Acids Res. 2012, 40, e13. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.Y.; Lee, J.; Joo, J.Y.; Lee, Y.S.; Heo, H.; Ko, J.J.; Kim, S. A color-tunable molecular beacon to sense miRNA-9 expression during neurogenesis. Sci. Rep. 2014, 4, 4626. [Google Scholar] [PubMed]
- Kang, W.J.; Cho, Y.L.; Chae, J.R.; Lee, J.D.; Choi, K.J.; Kim, S. Molecular beacon-based bioimaging of multiple microRNAs during myogenesis. Biomaterials 2011, 32, 1915–1922. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Kim, H.S.; Woo, J.; Hwang, E.H.; Cho, K.W.; Kim, S.; Moon, W.K. Real-time imaging of the epithelial-mesenchymal transition using microRNA-200a sequence-based molecular beacon-conjugated magnetic nanoparticles. PLoS One 2014, 9, e102164. [Google Scholar] [CrossRef] [PubMed]
- Dean, Z.S.; Riahi, R.; Wong, P.K. Spatiotemporal dynamics of microRNA during epithelial collective cell migration. Biomaterials 2015, 37, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Zhang, A.M.; Ma, H.; Lin, S.; Wang, X.X.; Sun, J.G.; Chen, Z.T. Novel molecular beacons to monitor microRNAs in non-small-cell lung cancer. Mol. Cell. Probes 2012, 26, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.W.; Song, I.C.; Lee, D.S.; Kim, S. Smart magnetic fluorescent nanoparticle imaging probes to monitor microRNAs. Small 2010, 6, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Choi, K.J.; Lee, M.; Jo, M.H.; Kim, S. Molecular imaging of a cancer-targeting theragnostics probe using a nucleolin aptamer- and microRNA-221 molecular beacon-conjugated nanoparticle. Biomaterials 2012, 33, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Yang, J.; Park, J.; Kim, S.; Kim, N.H.; Yook, J.I.; Suh, J.S.; Haam, S.; Huh, Y.M. Consecutive targetable smart nanoprobe for molecular recognition of cytoplasmic microRNA in metastatic breast cancer. ACS Nano 2012, 6, 8525–8535. [Google Scholar] [CrossRef] [PubMed]
- Maroof, H.; Salajegheh, A.; Smith, R.A.; Lam, A.K. MicroRNA-34 family, mechanisms of action in cancer: A review. Curr. Cancer Drug Targets 2014, 14, 737–751. [Google Scholar] [CrossRef] [PubMed]
- Bader, A.G. miR-34-a microRNA replacement therapy is headed to the clinic. Front. Genet. 2012, 3, 120. [Google Scholar] [CrossRef] [PubMed]
- Massoud, T.F.; Gambhir, S.S. Molecular imaging in living subjects: Seeing fundamental biological processes in a new light. Genes Dev. 2003, 17, 545–580. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Gibbs, S.L.; Lee, J.H.; Kim, S.H.; Ashitate, Y.; Liu, F.; Hyun, H.; Park, G.; Xie, Y.; Bae, S.; et al. Targeted zwitterionic near-infrared fluorophores for improved optical imaging. Nat. Biotechnol. 2013, 31, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Prescher, J.A.; Contag, C.H. Guided by the light: Visualizing biomolecular processes in living animals with bioluminescence. Curr. Opin.Chem. Biol. 2010, 14, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Serganova, I.; Mayer-Kukuck, P.; Huang, R.; Blasberg, R. Molecular imaging: Reporter gene imaging. Handb. Exp. Pharmacol. 2008, 167–223. [Google Scholar]
- Ko, H.Y.; Hwang, D.W.; Lee, D.S.; Kim, S. A reporter gene imaging system for monitoring microRNA biogenesis. Nat. Protoc. 2009, 4, 1663–1669. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.Y.; Lee, Y.S.; Kim, S. Bioluminescence reporter gene-based detection of microRNAs. Methods Mol. Biol. 2014, 1098, 85–95. [Google Scholar] [PubMed]
- Brown, B.D.; Venneri, M.A.; Zingale, A.; Sergi, L.S.; Naldini, L. Endogenous microRNA regulation suppresses transgene expression in hematopoietic lineages and enables stable gene transfer. Nat. Med. 2006, 12, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Miyaki, S.; Yokoyama, S.; Omori, S.; Inoue, A.; Horiuchi, M.; Asahara, H. Real-time functional imaging for monitoring miR-133 during myogenic differentiation. Int. J. Biochem. Cell Biol. 2009, 41, 2225–2231. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.D.; Naldini, L. Exploiting and antagonizing microRNA regulation for therapeutic and experimental applications. Nat. Rev. Genet. 2009, 10, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Kang, W.J.; Cho, Y.L.; Chae, J.R.; Lee, J.D.; Ali, B.A.; Al-Khedhairy, A.A.; Lee, C.H.; Kim, S. Dual optical biosensors for imaging microRNA-1 during myogenesis. Biomaterials 2012, 33, 6430–6437. [Google Scholar] [CrossRef] [PubMed]
- Lee, JY.; Kim, S.; Hwang, D.W.; Jeong, J.M.; Chung, J.K.; Lee, M.C.; Lee, D.S. Development of a dual-luciferase reporter system for in vivo visualization of MicroRNA biogenesis and posttranscriptional regulation. J. Nucl. Med. 2008, 49, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Ko, M.H.; Kim, S.; Hwang, D.W.; Ko, H.Y.; Kim, Y.H.; Lee, D.S. Bioimaging of the unbalanced expression of microRNA9 and microRNA9* during the neuronal differentiation of P19 cells. FEBS J. 2008, 275, 2605–2616. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, F.; Patrawala, L.; Osaki, M.; Takahashi, R.U.; Yamamoto, Y.; Kosaka, N.; Kawamata, M.; Kelnar, K.; Bader, A.G.; Brown, D.; et al. Systemic delivery of synthetic microRNA-16 inhibits the growth of metastatic prostate tumors via downregulation of multiple cell-cycle genes. Mol. Ther. 2010, 18, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, Y.H.; Lee, D.S.; Chung, J.K.; Kim, S. In vivo imaging of functional targeting of miR-221 in papillary thyroid carcinoma. J. Nucl. Med. 2008, 49, 1686–1693. [Google Scholar] [CrossRef] [PubMed]
- Ko, HY.; Lee, D.S.; Kim, S. Noninvasive imaging of microRNA124a-mediated repression of the chromosome 14 ORF 24 gene during neurogenesis. FEBS J. 2009, 276, 4854–4865. [Google Scholar] [CrossRef] [PubMed]
- Laloo, B.; Simon, D.; Veillat, V.; Lauzel, D.; Guyonnet-Duperat, V.; Moreau-Gaudry, F.; Sagliocco, F.; Grosset, C. Analysis of post-transcriptional regulations by a functional, integrated, and quantitative method. Mol. Cell. Proteomics 2009, 8, 1777–1788. [Google Scholar] [CrossRef] [PubMed]
- Maurel, M.; Jalvy, S.; Ladeiro, Y.; Combe, C.; Vachet, L.; Sagliocco, F.; Bioulac-Sage, P.; Pitard, V.; Jacquemin-Sablon, H.; Zucman-Rossi, J.; et al. A functional screening identifies five microRNAs controlling glypican-3: Role of miR-1271 down-regulation in hepatocellular carcinoma. Hepatology 2013, 57, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Ezzine, S.; Vassaux, G.; Pitard, B.; Barteau, B.; Malinge, J.M.; Midoux, P.; Pichon, C.; Baril, P. RILES, a novel method for temporal analysis of the in vivo regulation of miRNA expression. Nucleic Acids Res. 2013, 41, e192. [Google Scholar] [CrossRef] [PubMed]
- Pichard, V.; Aubert, D.; Boni, S.; Battaglia, S.; Ivacik, D.; Nguyen, T.H.; Arbuthnot, P.; Ferry, N. Specific micro RNA-regulated TetR-KRAB transcriptional control of transgene expression in viral vector-transduced cells. PLoS One 2012, 7, e51952. [Google Scholar] [CrossRef] [PubMed]
- Amendola, M.; Giustacchini, A.; Gentner, B.; Naldini, L. A double-switch vector system positively regulates transgene expression by endogenous microRNA expression (miR-ON vector). Mol. Ther. 2013, 21, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Stieger, K.; Belbellaa, B.; Le Guiner, C.; Moullier, P.; Rolling, F. In vivo gene regulation using tetracycline-regulatable systems. Adv. Drug Deliv. Rev. 2009, 61, 527–541. [Google Scholar] [CrossRef] [PubMed]
- Mullick, A.; Xu, Y.; Warren, R.; Koutroumanis, M.; Guilbault, C.; Broussau, S.; Malenfant, F.; Bourget, L.; Lamoureux, L.; Lo, R.; et al. The cumate gene-switch: A system for regulated expression in mammalian cells. BMC Biotechnol. 2006, 6, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, N.; Williams, A.H.; Maxeiner, J.M.; Bezprozvannaya, S.; Shelton, J.M.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. microRNA-206 promotes skeletal muscle regeneration and delays progression of Duchenne muscular dystrophy in mice. J. Clin. Investig. 2012, 122, 2054–2065. [Google Scholar] [CrossRef] [PubMed]
- Novak, J.; Kruzliak, P.; Bienertova-Vasku, J.; Slaby, O.; Novak, M. MicroRNA-206: A promising theranostic marker. Theranostics 2014, 4, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.D.; Gentner, B.; Cantore, A.; Colleoni, S.; Amendola, M.; Zingale, A.; Baccarini, A.; Lazzari, G.; Galli, C.; Naldini, L. Endogenous microRNA can be broadly exploited to regulate transgene expression according to tissue, lineage and differentiation state. Nat. Biotechnol. 2007, 25, 1457–1467. [Google Scholar] [CrossRef] [PubMed]
- Kelly, E.J.; Hadac, E.M.; Greiner, S.; Russell, S.J. Engineering microRNA responsiveness to decrease virus pathogenicity. Nat. Med. 2008, 14, 1278–1283. [Google Scholar] [CrossRef] [PubMed]
- Le Guiner, C.; Stieger, K.; Toromanoff, A.; Guilbaud, M.; Mendes-Madeira, A.; Devaux, M.; Guigand, L.; Cherel, Y.; Moullier, P.; Rolling, F.; et al. Transgene regulation using the tetracycline-inducible TetR-KRAB system after AAV-mediated gene transfer in rodents and nonhuman primates. PLoS One 2014, 9, e102538. [Google Scholar] [CrossRef] [PubMed]
- Grohmann, M.; Paulmann, N.; Fleischhauer, S.; Vowinckel, J.; Priller, J.; Walther, D.J. A mammalianized synthetic nitroreductase gene for high-level expression. BMC Cancer 2009, 9, 301. [Google Scholar] [CrossRef] [PubMed]
- Chien, K.R. Regenerative medicine and human models of human disease. Nature 2008, 453, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Baril, P.; Martin-Duque, P.; Vassaux, G. Visualization of gene expression in the live subject using the Na/I symporter as a reporter gene: Applications in biotherapy. Br. J. Pharmacol. 2010, 159, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Dohan, O.; de la Vieja, A.; Paroder, V.; Riedel, C.; Artani, M.; Reed, M.; Ginter, C.S.; Carrasco, N. The sodium/iodide Symporter (NIS): Characterization, regulation, and medical significance. Endocr. Rev. 2003, 24, 48–77. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baril, P.; Ezzine, S.; Pichon, C. Monitoring the Spatiotemporal Activities of miRNAs in Small Animal Models Using Molecular Imaging Modalities. Int. J. Mol. Sci. 2015, 16, 4947-4972. https://doi.org/10.3390/ijms16034947
Baril P, Ezzine S, Pichon C. Monitoring the Spatiotemporal Activities of miRNAs in Small Animal Models Using Molecular Imaging Modalities. International Journal of Molecular Sciences. 2015; 16(3):4947-4972. https://doi.org/10.3390/ijms16034947
Chicago/Turabian StyleBaril, Patrick, Safia Ezzine, and Chantal Pichon. 2015. "Monitoring the Spatiotemporal Activities of miRNAs in Small Animal Models Using Molecular Imaging Modalities" International Journal of Molecular Sciences 16, no. 3: 4947-4972. https://doi.org/10.3390/ijms16034947