
Discovery of Akt Kinase Inhibitors through Structure-Based Virtual Screening and Their Evaluation as Potential Anticancer Agents

Abstract
:1. Introduction
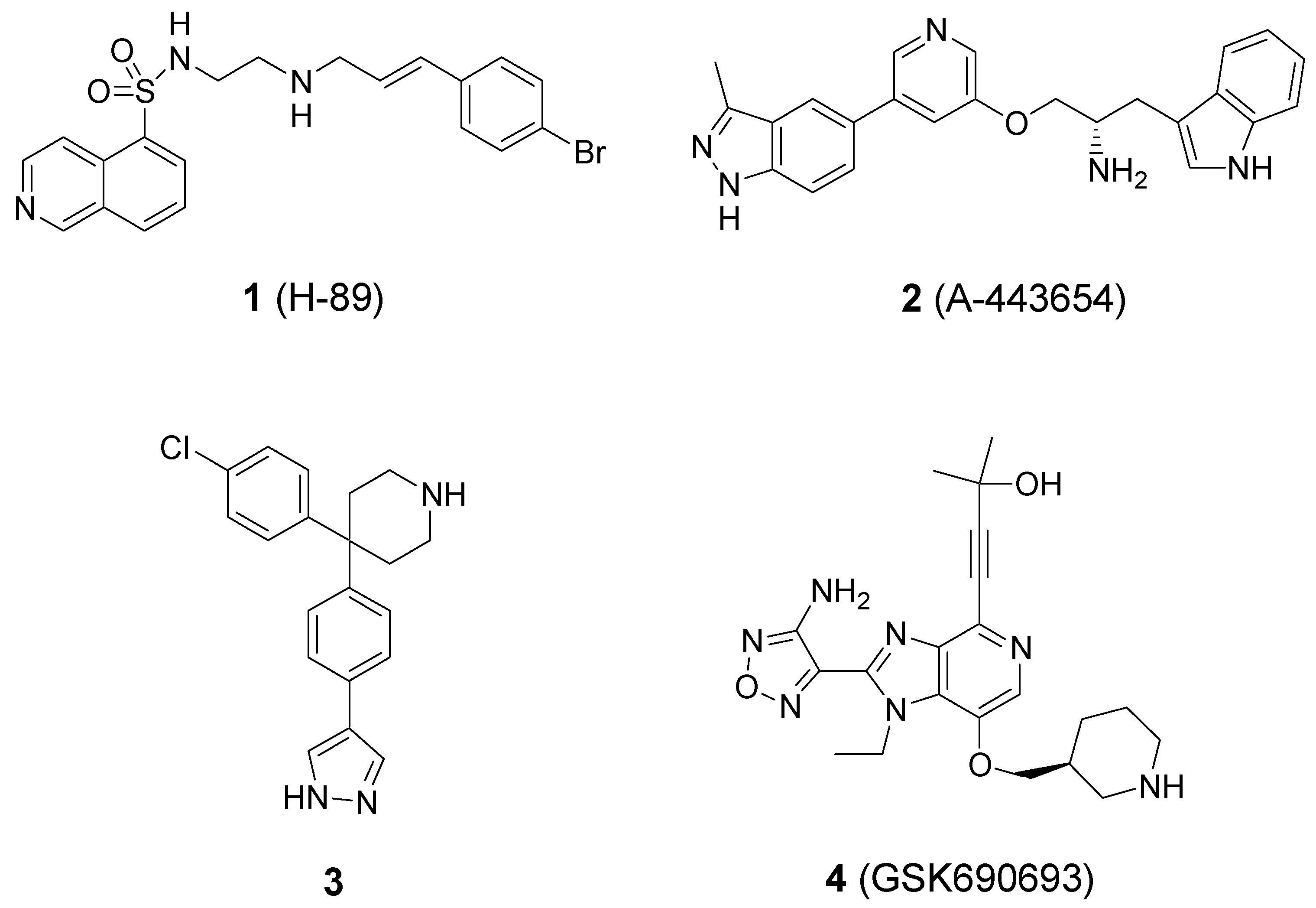
2. Results and Discussion
2.1. Computational Virtual Screening

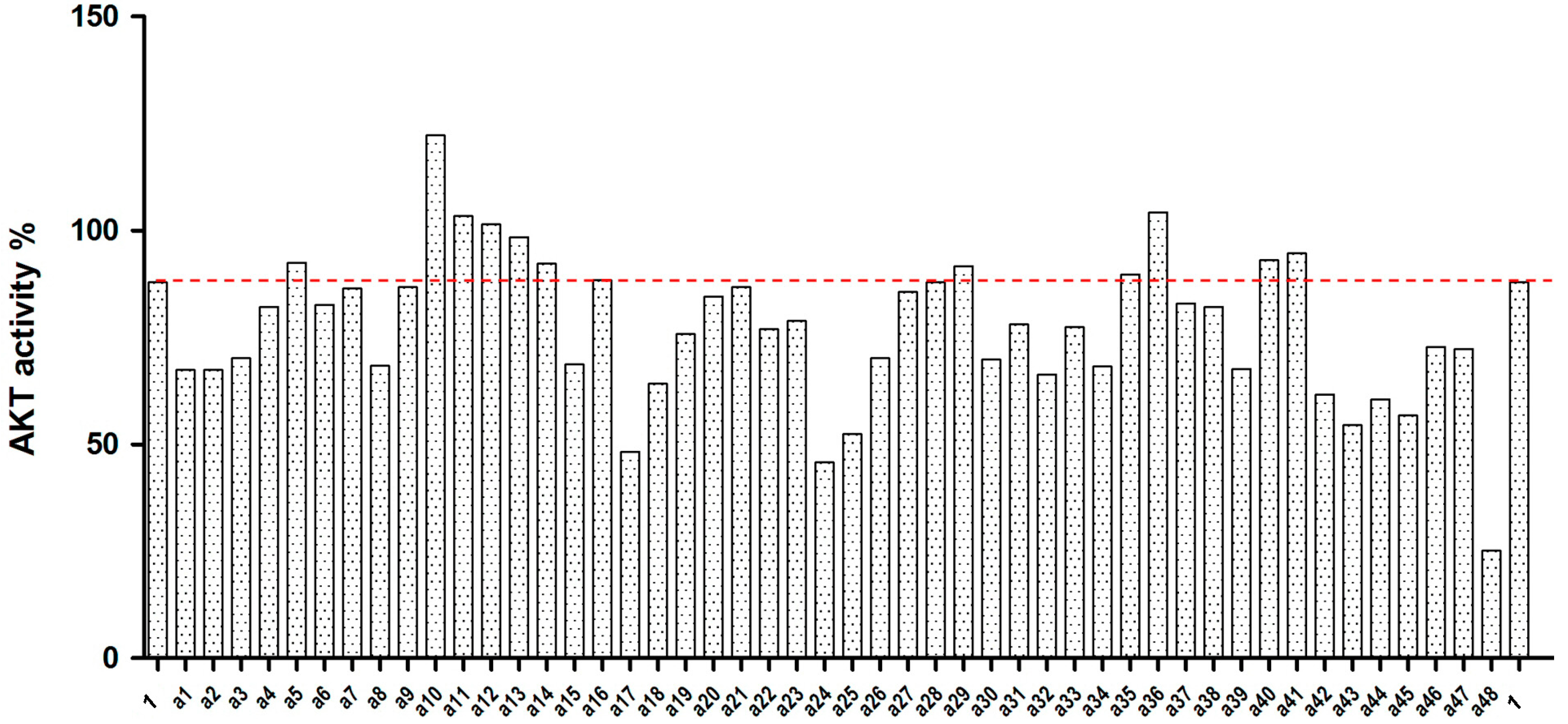
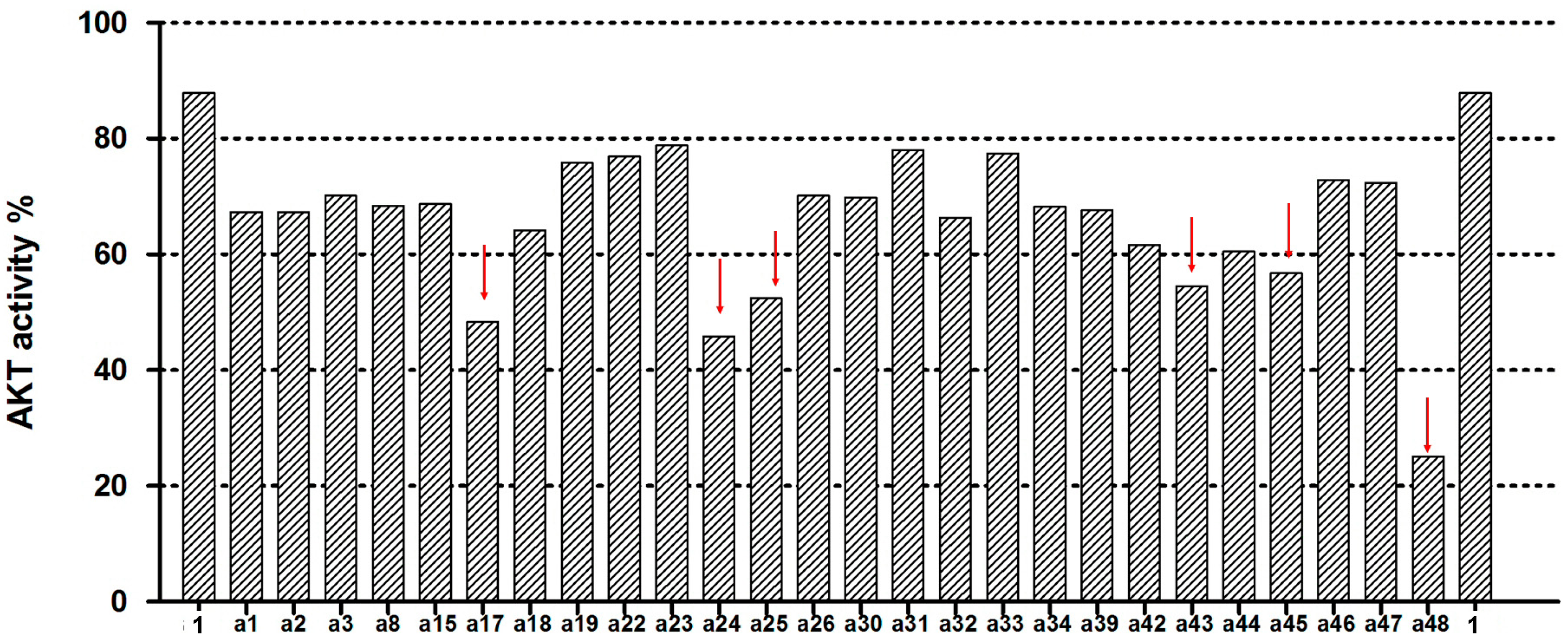
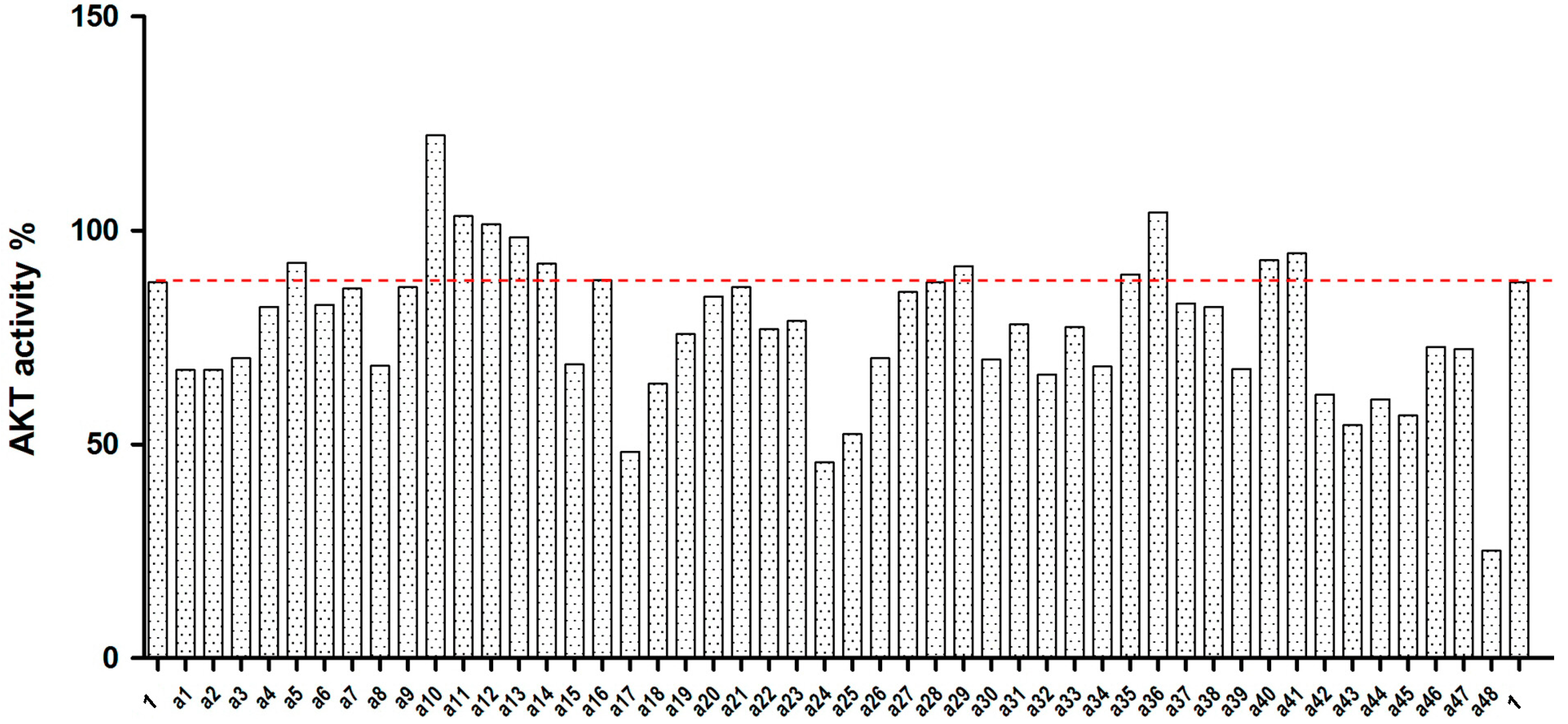
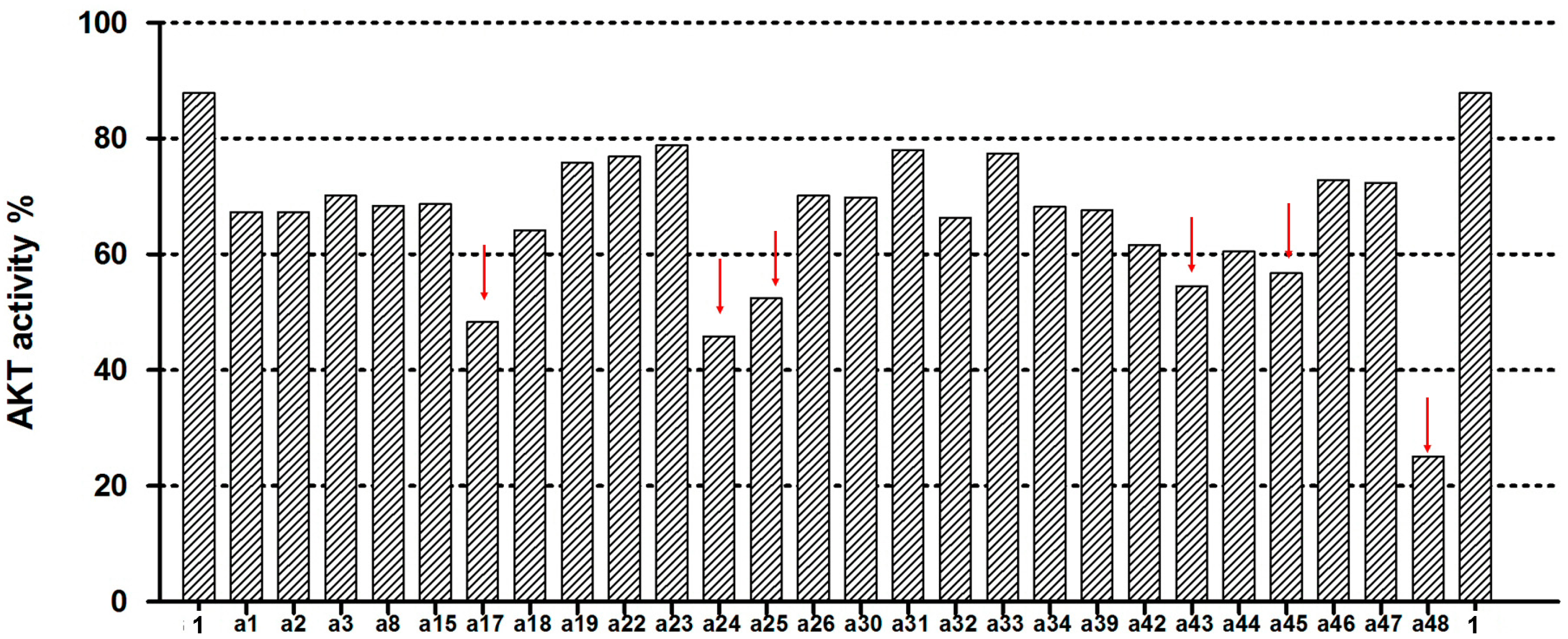
2.2. Akt Kinase Inhibition
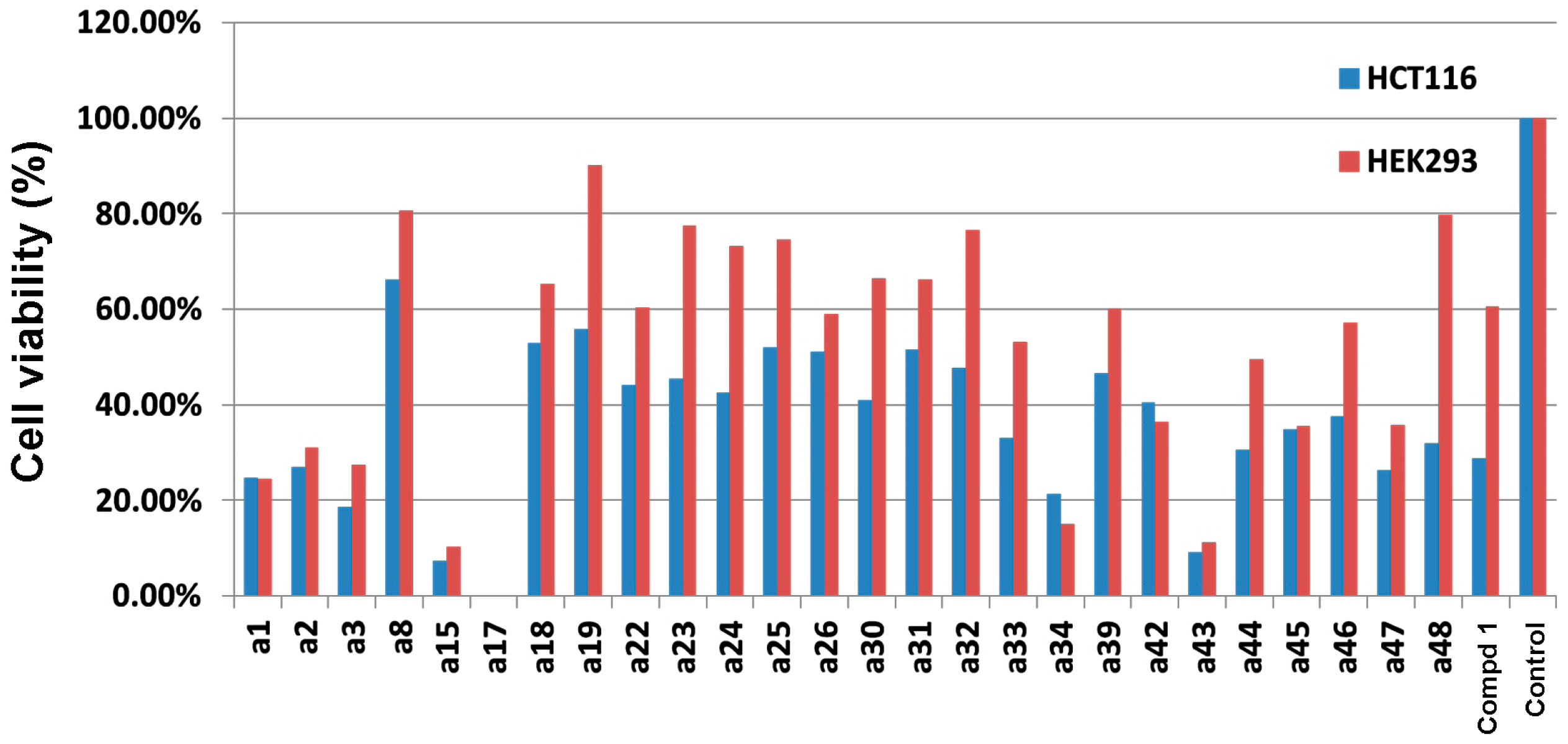
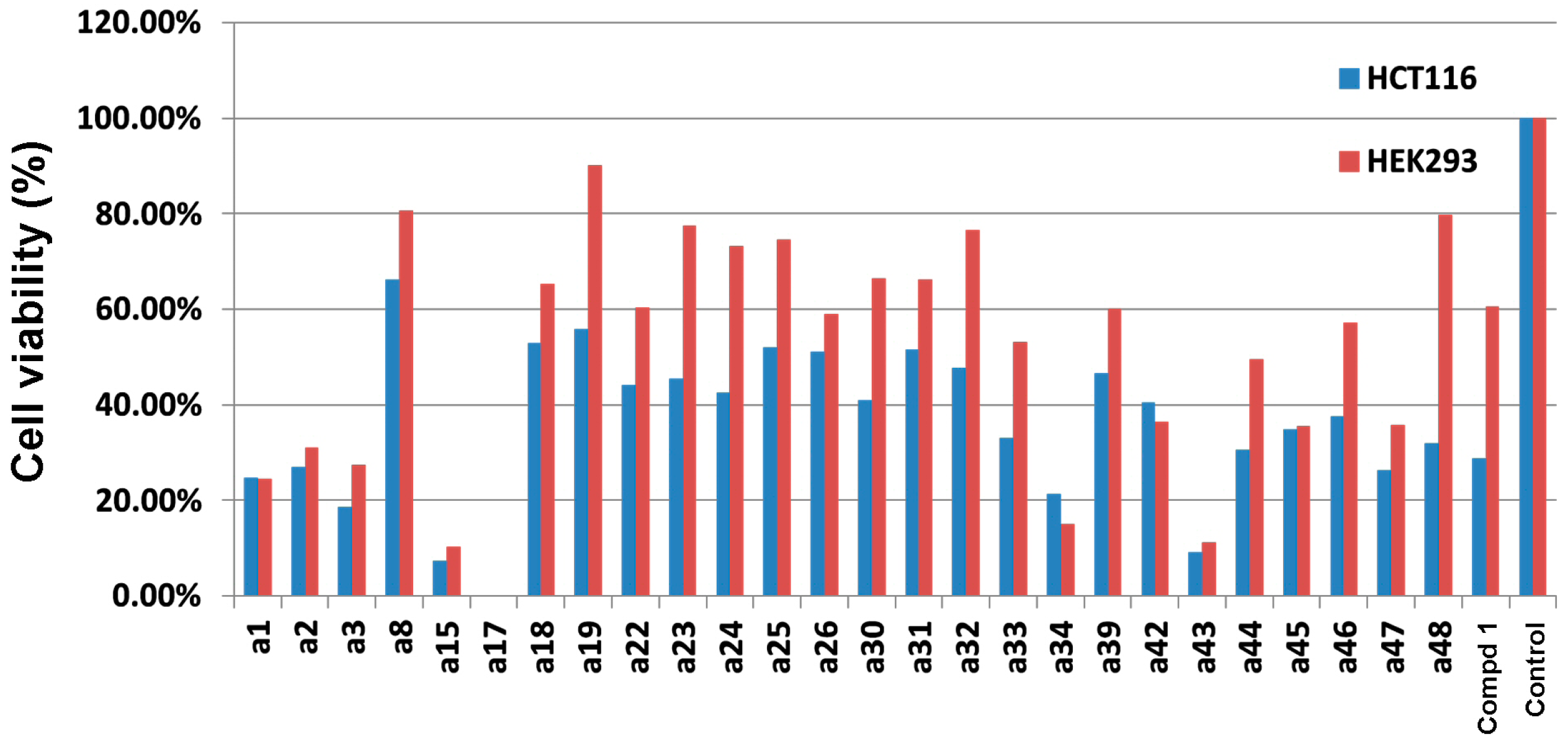
2.3. Cytotoxicity Evaluation on HCT-116 Cancer Cells and HEK-293 Normal Cells
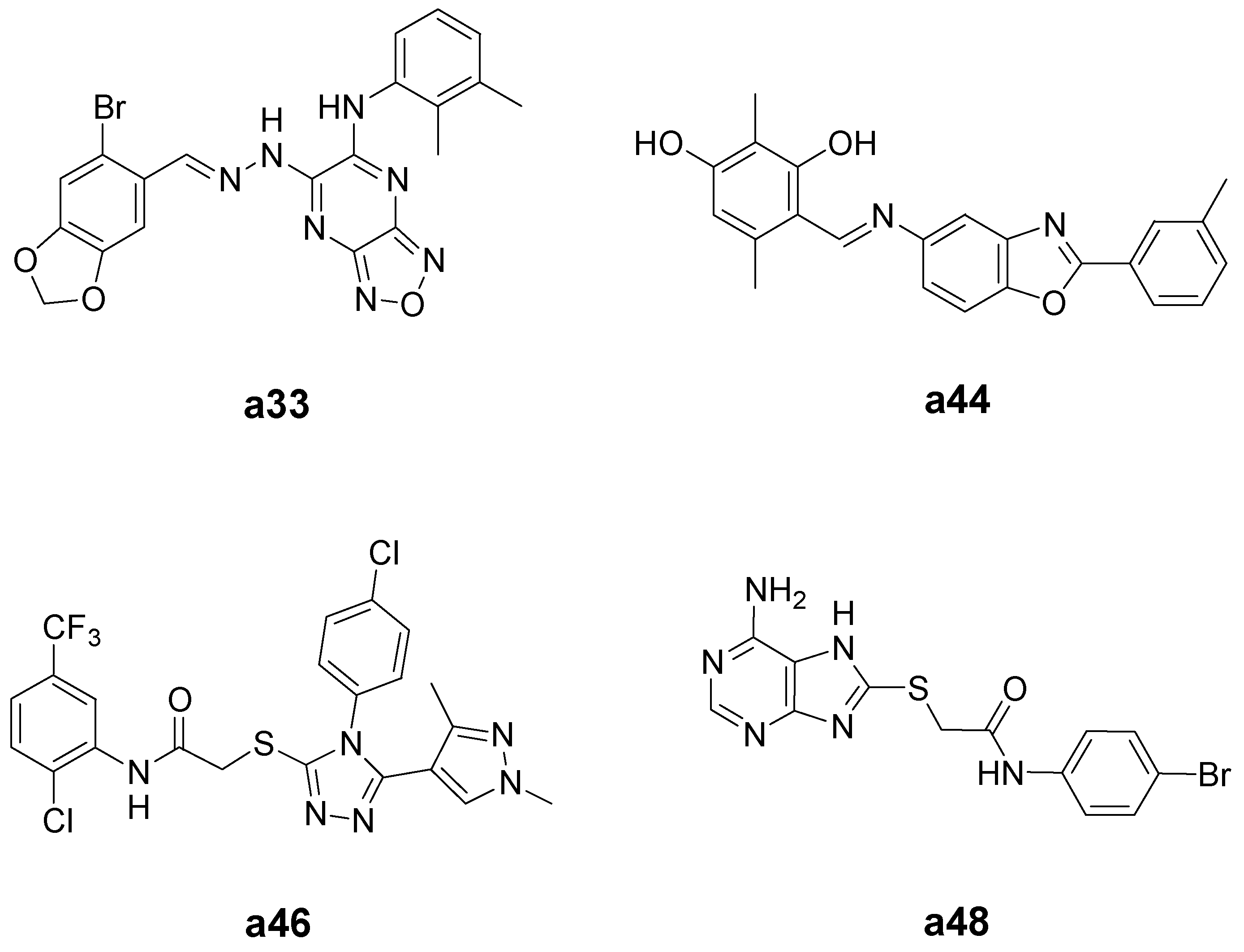

{kind=link}
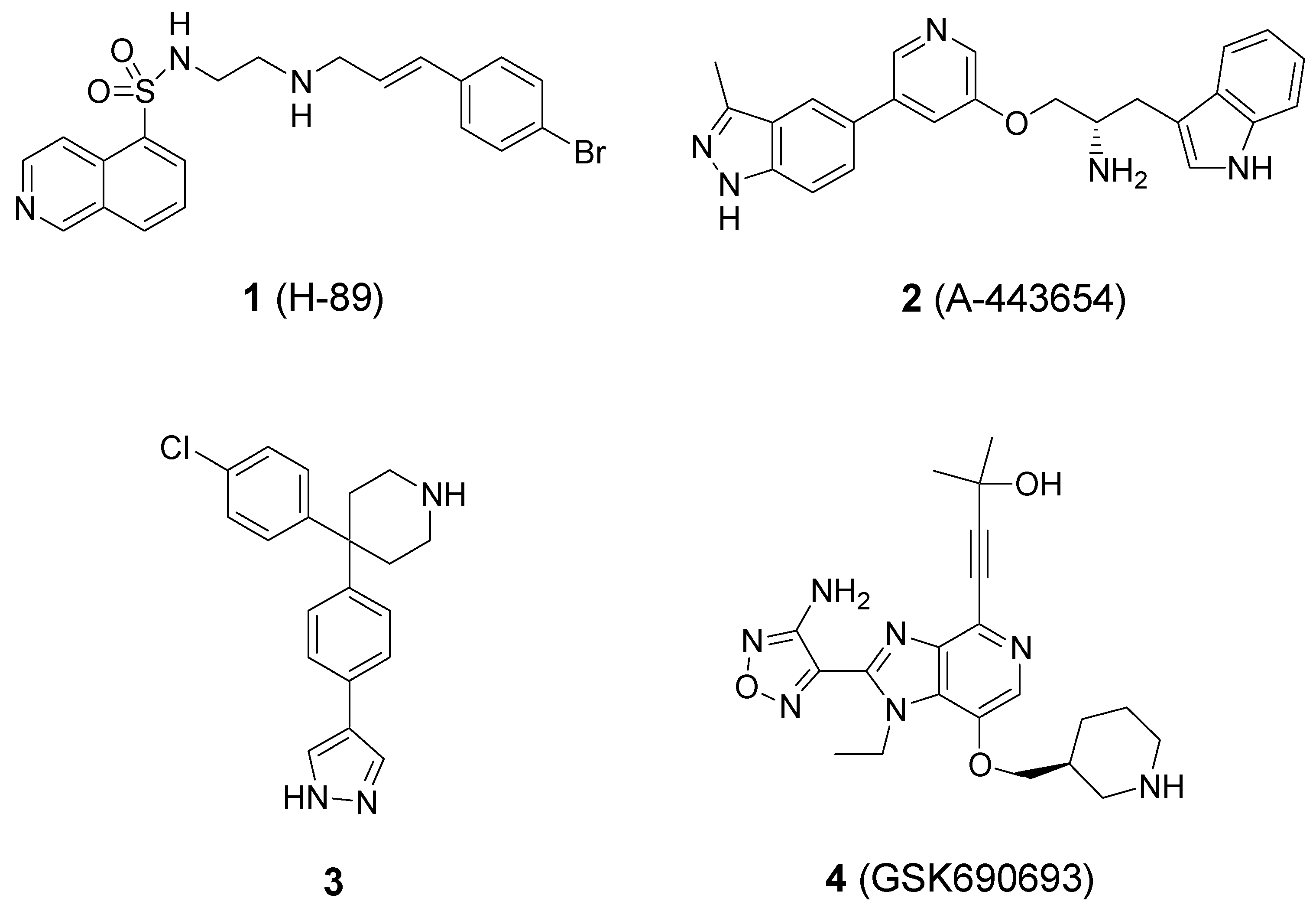
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (µM) a | Selectivity index b | |
|---|---|---|---|
| HCT-116 | HEK-293 | ||
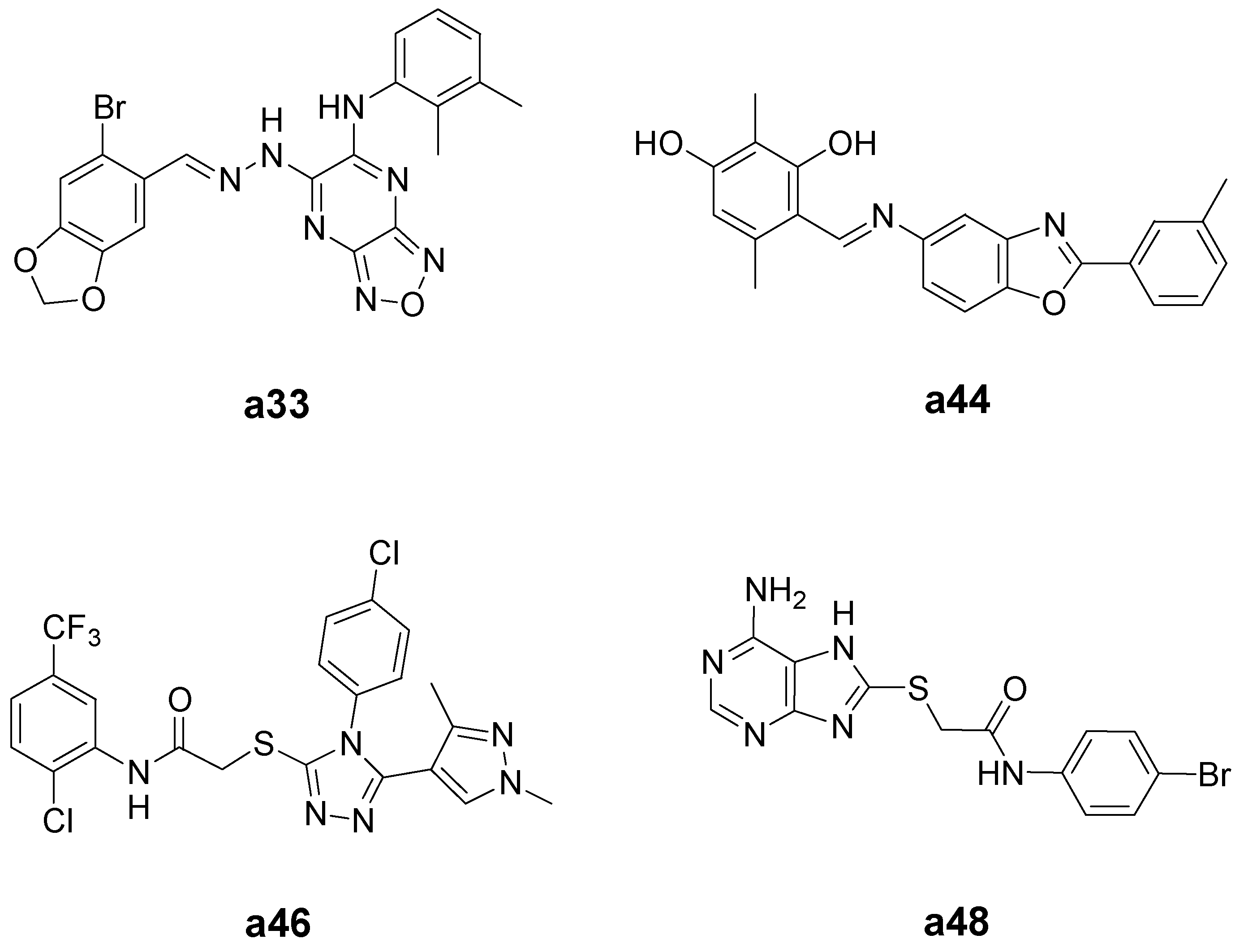
| a33 | 109.4 | 164.6 | 1.5 |
| a44 | 50.6 | 101.5 | 2.0 |
| a46 | 11.1 | 138.7 | 12.5 |
| a48 | 9.5 | 152.6 | 16.1 |
| H-89 c | 78.9 | 111.6 | 1.4 |
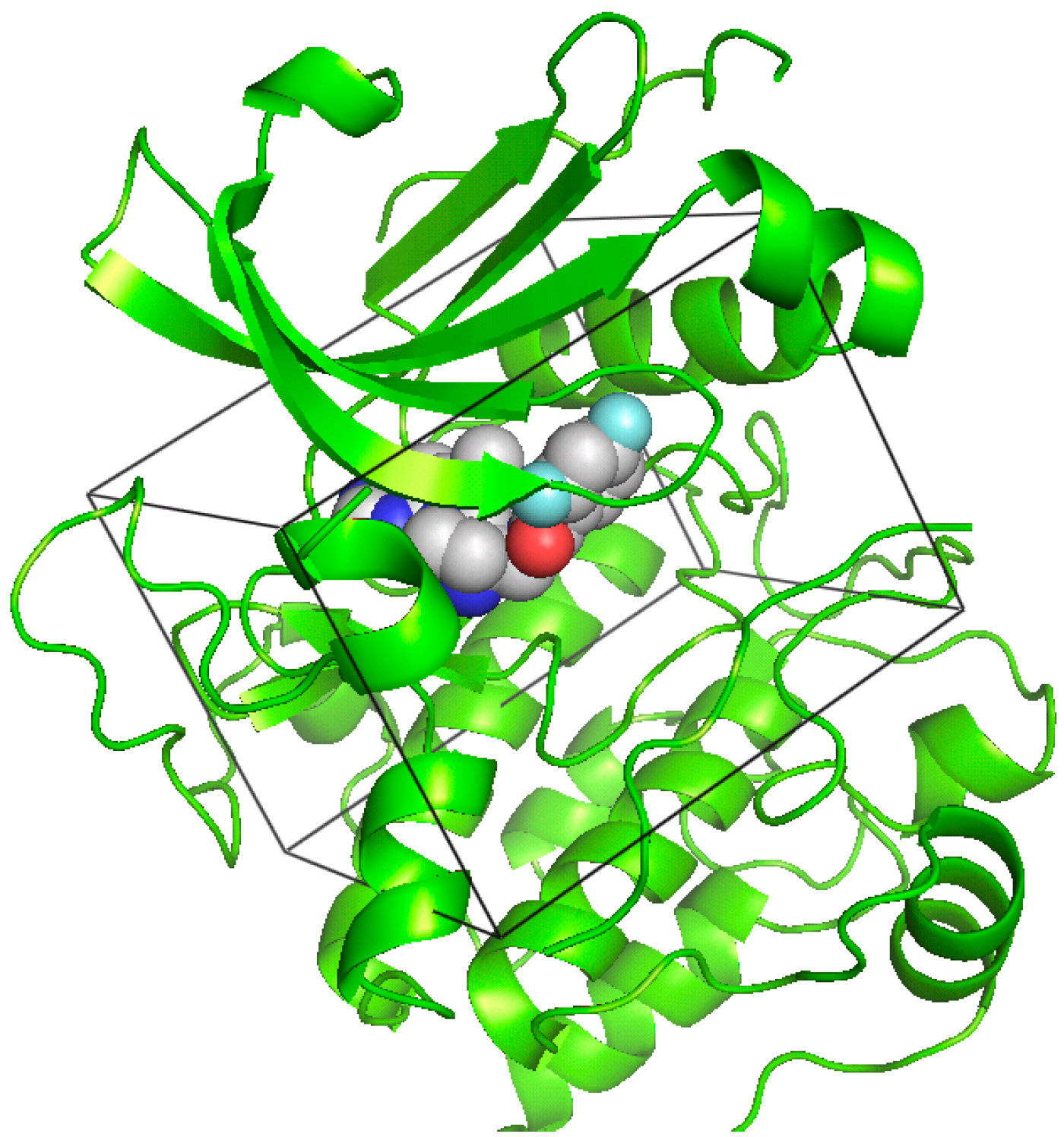
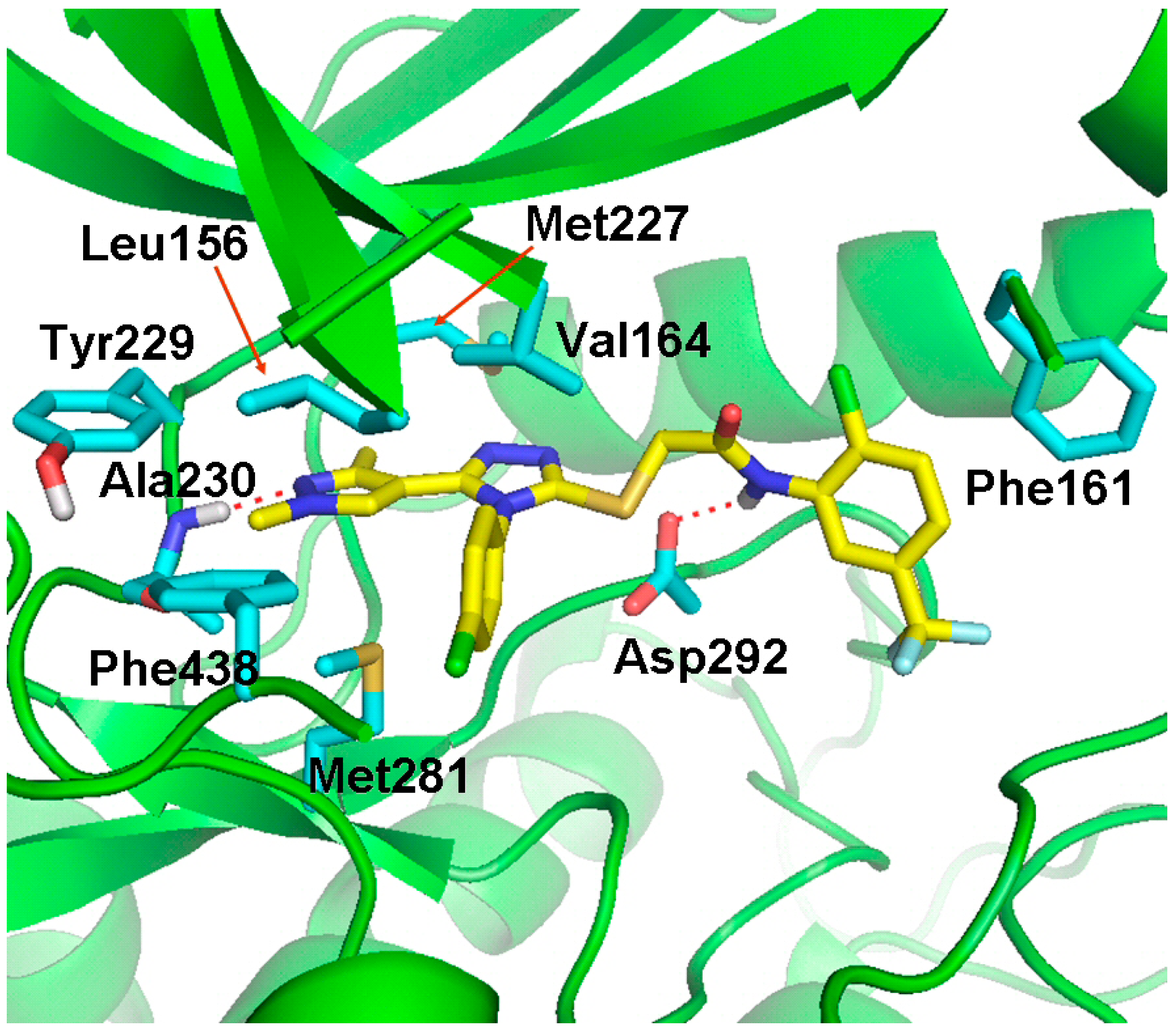
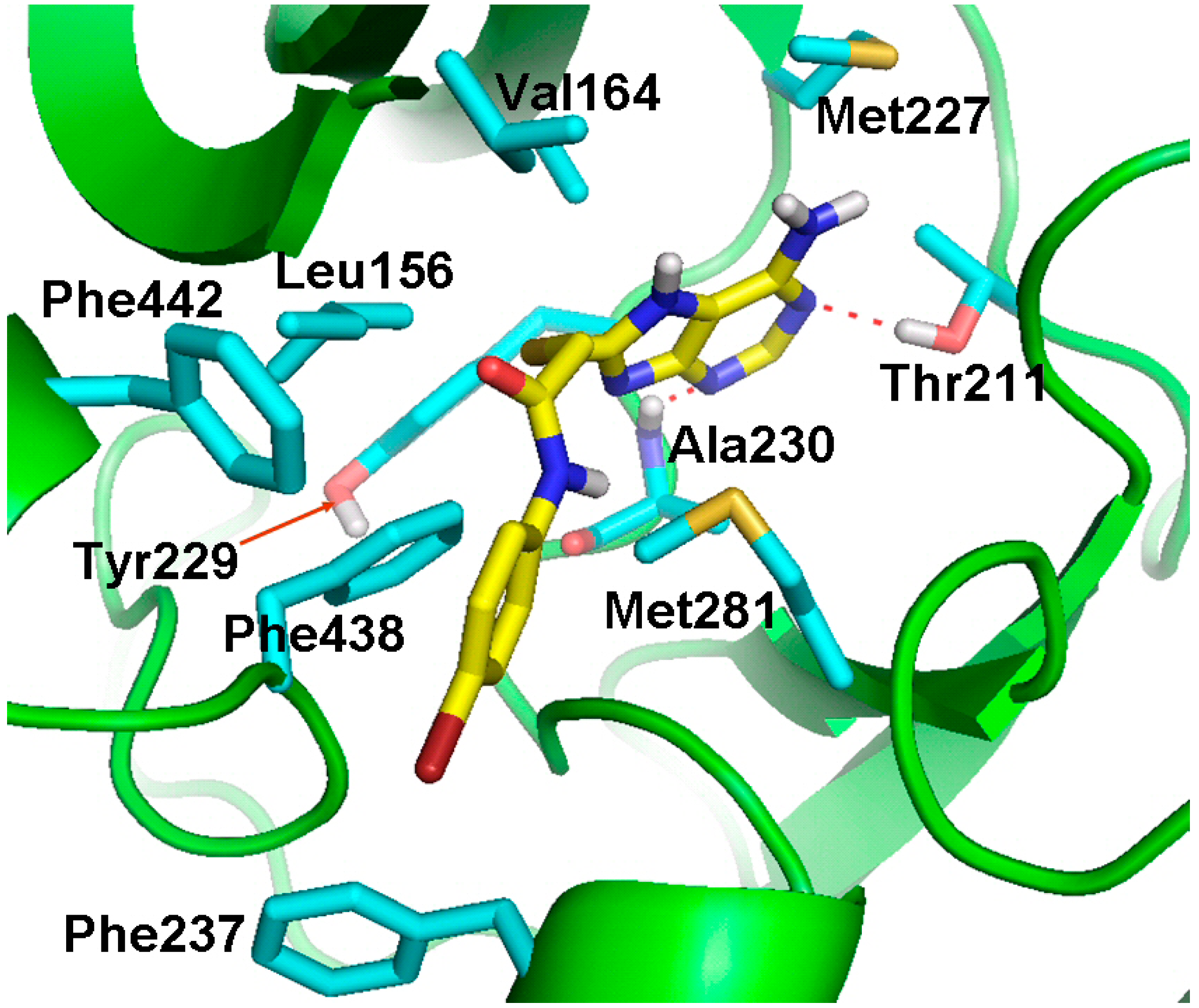
2.4. Molecular Docking Studies


3. Experimental Section
3.1. Virtual Screening
3.2. Molecular Docking Studies
3.3. Akt Kinase Inhibition Assay
3.4. Cytotoxic Evaluation of Compounds
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef]
- Hennessy, B.T.; Smith, D.L.; Ram, P.T.; Lu, Y.; Mills, G.B. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug Discov. 2005, 4, 988–1004. [Google Scholar] [CrossRef]
- Staal, S.P. Molecular cloning of the akt oncogene and its human homologues AKT1 and AKT2: Amplification of AKT1 in a primary human gastric adenocarcinoma. Proc. Natl. Acad. Sci. USA 1987, 84, 5034–5037. [Google Scholar] [CrossRef] [PubMed]
- Zinda, M.J.; Johnson, M.A.; Paul, J.D.; Horn, C.; Konicek, B.W.; Lu, Z.H.; Sandusky, G.; Thomas, J.E.; Neubauer, B.L.; Lai, M.T.; et al. AKT-1, -2, and -3 are expressed in both normal and tumor tissues of the lung, breast, prostate and colon. Clin. Cancer Res. 2001, 7, 2475–2479. [Google Scholar]
- Yuan, Z.Q.; Sun, M.; Feldman, R.I.; Wang, G.; Ma, X.; Jiang, C.; Coppola, D.; Nicosia, S.V.; Cheng, J.Q. Frequent activation of AKT2 and induction of apoptosis by inhibition of phosphoinositide-3-OH kinase/Akt pathway in human ovarian cancer. Oncogene 2000, 19, 2324–2330. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.Q.; Lindsley, C.W.; Cheng, G.Z.; Yang, H.; Nicosia, S.V. The Akt/PKB pathway: Molecular target for cancer drug discovery. Oncogene 2005, 24, 7482–7492. [Google Scholar] [CrossRef] [PubMed]
- Testa, J.R.; Tsichlis, P.N. Akt signaling in normal and malignant cells. Oncogene 2005, 24, 7391–7393. [Google Scholar] [CrossRef] [PubMed]
- Reuveni, H.; Livnah, N.; Geiger, T.; Klein, S.; Ohne, O.; Cohen, I.; Benhar, M.; Gellerman, G.; Levitzki, A. Toward a PKB inhibitor: Modification of a selective PKA inhibitor by rational design. Biochemistry 2002, 41, 10304–10314. [Google Scholar] [CrossRef] [PubMed]
- Woods, K.W.; Fischer, J.P.; Claiborne, A.; Li, T.; Thomas, S.A.; Zhu, G.; Diebold, R.B.; Liu, X.; Shi, Y.; Klinghofer, V.; et al. Synthesis and SAR of indazole-pyridine based protein kinase B/Akt inhibitors. Bioorg. Med. Chem. 2006, 14, 6832–6846. [Google Scholar]
- Saxty, G.; Woodhead, S.J.; Berdini, V.; Davies, T.G.; Verdonk, M.L.; Wyatt, P.G.; Boyle, R.G.; Barford, D.; Downham, R.; Garrett, M.D.; et al. Identification of novel inhibitors of protein kinase B using fragment-based lead discovery. J. Med. Chem. 2007, 50, 2293–2296. [Google Scholar]
- Rhodes, N.; Heerding, D.A.; Duckett, D.R.; Eberwein, D.J.; Knick, V.B.; Lansing, T.J.; McConnell, R.T.; Gilmer, T.M.; Zhang, S.Y.; Robell, K.; et al. Characterization of an AKT kinase inhibitor with potent pharmacodynamic and antitumor activity. Cancer Res. 2008, 68, 2366–2374. [Google Scholar]
- Shoichet, B.K. Virtual screening of chemical libraries. Nature 2004, 432, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C. The process of structure-based drug design. Chem. Biol. 2003, 10, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Lyne, P.D. Structure-based virtual screening: An overview. Drug Discov. Today 2002, 7, 1047–1055. [Google Scholar] [CrossRef] [PubMed]
- Kuntz, I.D. Structure-based strategies for drug design and discovery. Science 1992, 257, 1078–1082. [Google Scholar] [CrossRef] [PubMed]
- Meng, E.C.; Shoichet, B.K.; Kuntz, I.D. Automated docking with grid-based energy evaluation. J. Comput. Chem. 1992, 13, 505–524. [Google Scholar] [CrossRef]
- Markowitz, J.; Chen, I.; Gitti, R.; Baldisseri, D.M.; Pan, Y.; Udan, R.; Carrier, F.; MacKerell, A.D., Jr.; Weber, D.J. Identification and characterization of small molecule inhibitors of the calcium-dependent S100B-p53 tumor suppressor interaction. J. Med. Chem. 2004, 47, 5085–5093. [Google Scholar] [CrossRef] [PubMed]
- Vangrevelinghe, E.; Zimmermann, K.; Schoepfer, J.; Portmann, R.; Fabbro, D.; Furet, P. Discovery of a potent and selective protein kinase CK2 inhibitor by high-throughput docking. J. Med. Chem. 2003, 46, 2656–2662. [Google Scholar] [CrossRef] [PubMed]
- Doman, T.N.; McGovern, S.L.; Witherbee, B.J.; Kasten, T.P.; Kurumbail, R.; Stallings, W.C.; Connolly, D.T.; Shoichet, B.K. Molecular docking and high-throughput screening for novel inhibitors of protein tyrosine phosphatase-1B. J. Med. Chem. 2002, 45, 2213–2221. [Google Scholar] [CrossRef] [PubMed]
- Shoichet, B.K.; Stroud, R.M.; Santi, D.V.; Kuntz, I.D.; Perry, K.M. Structure-based discovery of inhibitors of thymidylate synthase. Science 1993, 259, 1445–1450. [Google Scholar] [CrossRef] [PubMed]
- Freeman-Cook, K.D.; Autry, C.; Borzillo, G.; Gordon, D.; Barbacci-Tobin, E.; Bernardo, V.; Briere, D.; Clark, T.; Corbett, M.; Jakubczak, J.; et al. Design of selective, ATP-competitive inhibitors of Akt. J. Med. Chem. 2010, 53, 4615–4622. [Google Scholar]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chuang, C.-H.; Cheng, T.-C.; Leu, Y.-L.; Chuang, K.-H.; Tzou, S.-C.; Chen, C.-S. Discovery of Akt Kinase Inhibitors through Structure-Based Virtual Screening and Their Evaluation as Potential Anticancer Agents. Int. J. Mol. Sci. 2015, 16, 3202-3212. https://doi.org/10.3390/ijms16023202
Chuang C-H, Cheng T-C, Leu Y-L, Chuang K-H, Tzou S-C, Chen C-S. Discovery of Akt Kinase Inhibitors through Structure-Based Virtual Screening and Their Evaluation as Potential Anticancer Agents. International Journal of Molecular Sciences. 2015; 16(2):3202-3212. https://doi.org/10.3390/ijms16023202
Chicago/Turabian StyleChuang, Chih-Hung, Ta-Chun Cheng, Yu-Ling Leu, Kuo-Hsiang Chuang, Shey-Cherng Tzou, and Chien-Shu Chen. 2015. "Discovery of Akt Kinase Inhibitors through Structure-Based Virtual Screening and Their Evaluation as Potential Anticancer Agents" International Journal of Molecular Sciences 16, no. 2: 3202-3212. https://doi.org/10.3390/ijms16023202