
The Role of the Neuroprotective Factor Npas4 in Cerebral Ischemia


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Npas4 Is a Member of the Basic Helix-Loop-Helix (bHLH)-PAS Transcription Factor Family
2.1. The bHLH-PAS Family of Transcriptional Regulators
2.2. Structure of the Mouse Npas4 Gene and Its Transcript

2.3. Domain Structure and Sequence Conservation within the Npas4 Protein
2.4. Npas4 and Its Interaction with Other bHLH-PAS Factors
3. Expression of Npas4
3.1. Npas4 Expression in Adult Tissues
3.2. Enriched Npas4 Expression in the Limbic System of the Brain
3.3. Subcellular Localization of Npas4
3.4. Cellular Distribution of Npas4
4. Regulation of Npas4 Expression by Neuronal Activity
4.1. Activity-Dependent Regulation of Npas4 Expression in Neurons
4.2. Nuclear Ca2+ Signaling and Regulation of Npas4 Expression
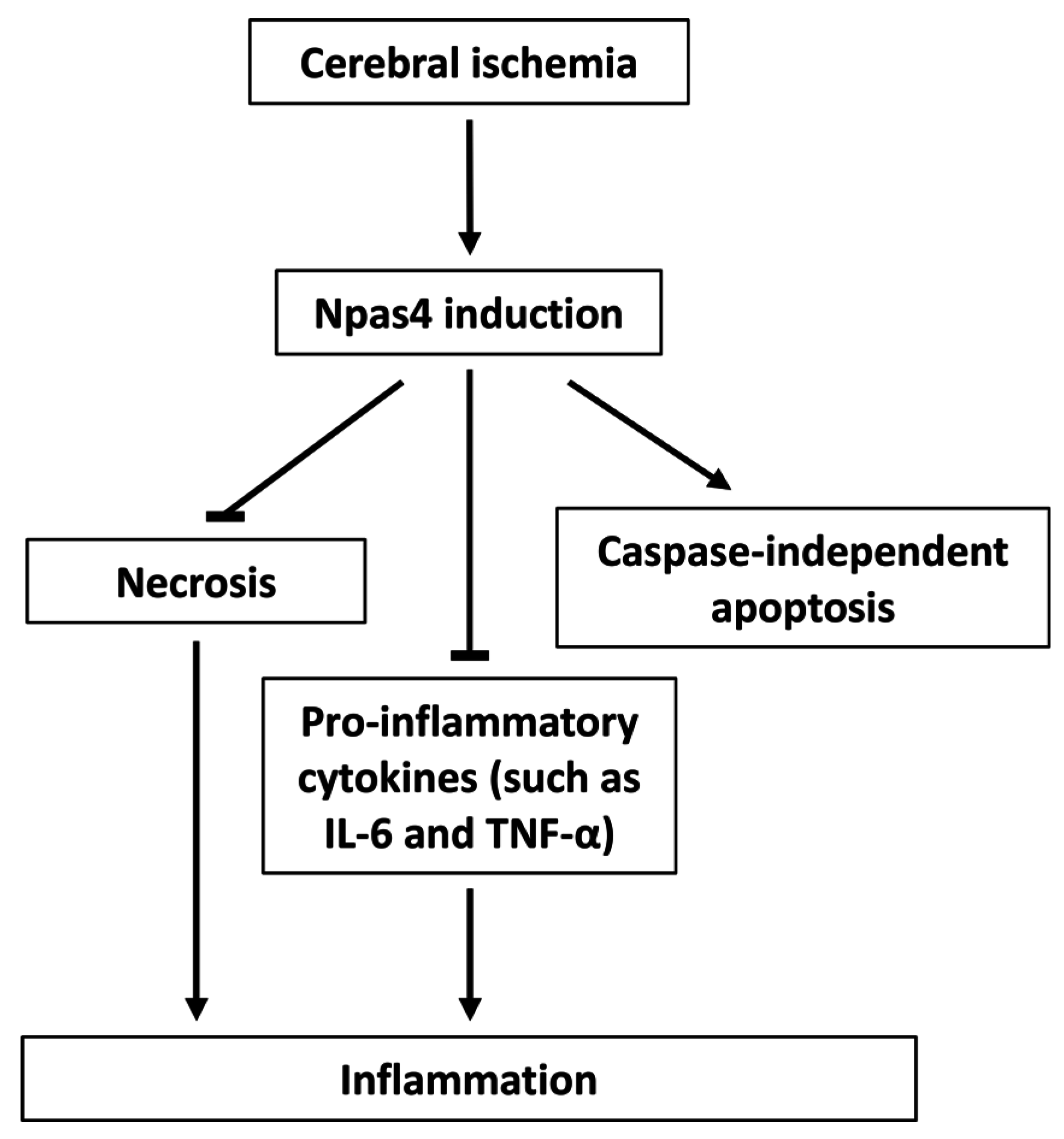
5. Npas4 Expression in Response to Cerebral Ischemia
6. Functions of Npas4 Following Ischemic Brain Injury
6.1. Neuroprotection
6.2. Apoptosis
6.3. Neuroinflammation
7. Conclusions
Author Contributions
Conflicts of Interest
References
- Donnan, G.A.; Fisher, M.; Macleod, M.; Davis, S.M. Stroke. Lancet 2008, 371, 1612–1623. [Google Scholar] [CrossRef]
- Go, A.S.; Mozaffarian, D.; Roger, V.L.; Benjamin, E.J.; Berry, J.D.; Blaha, M.J.; Dai, S.; Ford, E.S.; Fox, C.S.; Franco, S.; et al. Heart disease and stroke statistics-2014 update: A report from the american heart association. Circulation 2014, 129, E28–E292. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, R. Imaging-guided acute ischemic stroke therapy: From “time is brain” to “physiology is brain”. Am. J. Neuroradiol. 2006, 27, 728–735. [Google Scholar] [PubMed]
- Flood, W.D.; Moyer, R.W.; Tsykin, A.; Sutherland, G.R.; Koblar, S.A. Nxf and fbxo33: Novel seizure-responsive genes in mice. Eur. J. Neurosci. 2004, 20, 1819–1826. [Google Scholar] [CrossRef] [PubMed]
- Ooe, N.; Saito, K.; Mikami, N.; Nakatuka, I.; Kaneko, H. Identification of a novel basic helix-loop-helix-pas factor, NXF, reveals a Sim2 competitive, positive regulatory role in dendritic-cytoskeleton modulator drebrin gene expression. Mol. Cell. Biol. 2004, 24, 608–616. [Google Scholar] [CrossRef] [PubMed]
- Moser, M.; Knoth, R.; Bode, C.; Patterson, C. LE-PAS, a novel Arnt-dependent HLH-PAS protein, is expressed in limbic tissues and transactivates the CNS midline enhancer element. Mol. Brain Res. 2004, 128, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Bloodgood, B.L.; Hauser, J.L.; Lapan, A.D.; Koon, A.C.; Kim, T.-K.; Hu, L.S.; Malik, A.N.; Greenberg, M.E. Activity-dependent regulation of inhibitory synapse development by Npas4. Nature 2008, 455, 1198–1204. [Google Scholar] [CrossRef] [PubMed]
- Bloodgood, B.L.; Sharma, N.; Browne, H.A.; Trepman, A.Z.; Greenberg, M.E. The activity-dependent transcription factor Npas4 regulates domain-specific inhibition. Nature 2013, 503, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, I.; Mardinly, A.R.; Gabel, H.W.; Bazinet, J.E.; Couch, C.H.; Tzeng, C.P.; Harmin, D.A.; Greenberg, M.E. Npas4 regulates excitatory-inhibitory balance within neural circuits through cell-type-specific gene programs. Cell 2014, 157, 1216–1229. [Google Scholar] [CrossRef] [PubMed]
- Pruunsild, P.; Sepp, M.; Orav, E.; Koppel, I.; Timmusk, T. Identification of cis-elements and transcription factors regulating neuronal activity-dependent transcription of human bdnf gene. J. Neurosci. 2011, 31, 3295–3308. [Google Scholar] [CrossRef] [PubMed]
- Bibel, M.; Barde, Y.A. Neurotrophins: Key regulators of cell fate and cell shape in the vertebrate nervous system. Genes Dev. 2000, 14, 2919–2937. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.J.; Reichardt, L.F. Neurotrophins: Roles in neuronal development and function. Annu. Rev. Neurosci. 2001, 24, 677–736. [Google Scholar] [CrossRef] [PubMed]
- Ramamoorthi, K.; Fropf, R.; Belfort, G.M.; Fitzmaurice, H.L.; McKinney, R.M.; Neve, R.L.; Otto, T.; Lin, Y. Npas4 regulates a transcriptional program in CA3 required for contextual memory formation. Science 2011, 334, 1669–1675. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Koike, H.; Ibi, D.; Toth, E.; Mizoguchi, H.; Nitta, A.; Yoneyama, M.; Ogita, K.; Yoneda, Y.; Nabeshima, T.; et al. Chronic restraint stress impairs neurogenesis and hippocampus-dependent fear memory in mice: Possible involvement of a brain-specific transcription factor Npas4. J. Neurochem. 2010, 114, 1840–1851. [Google Scholar] [CrossRef] [PubMed]
- Ploski, J.E.; Monsey, M.S.; Tam, N.; DiLeone, R.J.; Schafe, G.E. The neuronal pas domain protein 4 (Npas4) is required for new and reactivated fear memories. PLoS ONE 2011, 6, e23760. [Google Scholar] [CrossRef] [PubMed]
- Coutellier, L.; Beraki, S.; Ardestani, P.M.; Saw, N.L.; Shamloo, M. Npas4: A neuronal transcription factor with a key role in social and cognitive functions relevant to developmental disorders. PLoS ONE 2012, 7, e46604. [Google Scholar] [CrossRef] [PubMed]
- Sklar, P.; Ripke, S.; Scott, L.J.; Andreassen, O.A.; Cichon, S.; Craddock, N.; Edenberg, H.J.; Nurnberger, J.I., Jr.; Rietschel, M.; Blackwood, D.; et al. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat. Genet. 2011, 43, 977–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebert, D.H.; Greenberg, M.E. Activity-dependent neuronal signalling and autism spectrum disorder. Nature 2013, 493, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Floor, K.; Baroy, T.; Misceo, D.; Kanavin, O.J.; Fannemel, M.; Frengen, E. A 1 mb de novo deletion within 11q13.1q13.2 in a boy with mild intellectual disability and minor dysmorphic features. Eur. J. Med. Genet. 2012, 55, 695–699. [Google Scholar] [CrossRef] [PubMed]
- Jaehne, E.J.; Klaric, T.S.; Koblar, S.A.; Baune, B.T.; Lewis, M.D. Effects of Npas4 deficiency on anxiety, depression-like, cognition and sociability behaviour. Behav. Brain Res. 2015, 281, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Leong, W.K.; Klaric, T.S.; Lin, Y.; Lewis, M.D.; Koblar, S.A. Upregulation of the neuronal Per-Arnt-Sim domain protein 4 (Npas4) in the rat corticolimbic system following focal cerebral ischemia. Eur. J. Neurosci. 2013, 37, 1875–1884. [Google Scholar] [CrossRef] [PubMed]
- Shamloo, M.; Soriano, L.; von Schack, D.; Rickhag, M.; Chin, D.J.; Gonzalez-Zulueta, M.; Gido, G.; Urfer, R.; Wieloch, T.; Nikolich, K. Npas4, a novel helix-loop-helix pas domain protein, is regulated in response to cerebral ischemia. Eur. J. Neurosci. 2006, 24, 2705–2720. [Google Scholar] [CrossRef] [PubMed]
- Hester, I.; McKee, S.; Pelletier, P.; Thompson, C.; Storbeck, C.; Mears, A.; Schulz, J.B.; Hakim, A.A.; Sabourin, L.A. Transient expression of Nxf, a bHLH-PAS transactivator induced by neuronal preconditioning, confers neuroprotection in cultured cells. Brain Res. 2007, 1135, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ooe, N.; Motonaga, K.; Kobayashi, K.; Saito, K.; Kaneko, H. Functional characterization of basic helix-loop-helix-PAS type transcription factor NXF in vivo putative involvement in an “on demand” neuroprotection system. J. Biol. Chem. 2009, 284, 1057–1063. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-J.; Zou, M.; Lu, L.; Lau, D.; Ditzel, D.A.W.; Delucinge-Vivier, C.; Aso, Y.; Descombes, P.; Bading, H. Nuclear calcium signaling controls expression of a large gene pool: Identification of a gene program for acquired neuroprotection induced by synaptic activity. PLoS Genet. 2009, 5, e1000604. [Google Scholar] [CrossRef] [PubMed]
- Choy, F.C.; Klaric, T.S.; Leong, W.K.; Koblar, S.A.; Lewis, M.D. Reduction of the neuroprotective transcription factor Npas4 results in increased neuronal necrosis, inflammation and brain lesion size following ischaemia. J. Cereb. Blood Flow Metab. 2015. [Google Scholar] [CrossRef]
- Crews, S.T.; Fan, C.M. Remembrance of things pas: Regulation of development by bHLH-PAS proteins. Curr. Opin. Genet. Dev. 1999, 9, 580–587. [Google Scholar] [CrossRef]
- Kewley, R.J.; Whitelaw, M.L.; Chapman-Smith, A. The mammalian basic helix-loop-helix/PAS family of transcriptional regulators. Int. J. Biochem. Cell Biol. 2004, 36, 189–204. [Google Scholar] [CrossRef]
- Guillemin, K.; Krasnow, M.A. The hypoxic response: Huffing and hifing. Cell 1997, 89, 9–12. [Google Scholar] [CrossRef]
- Ryan, H.E.; Lo, J.; Johnson, R.S. Hif-1α is required for solid tumor formation and embryonic vascularization. Embo J. 1998, 17, 3005–3015. [Google Scholar] [CrossRef] [PubMed]
- Lees, M.J.; Whitelaw, M.L. Multiple roles of ligand in transforming the dioxin receptor to an active basic helix-loop-helix/PAS transcription factor complex with the nuclear protein arnt. Mol. Cell. Biol. 1999, 19, 5811–5822. [Google Scholar] [CrossRef] [PubMed]
- Michaud, J.L.; Rosenquist, T.; May, N.R.; Fan, C.M. Development of neuroendocrine lineages requires the bHLH-PAS transcription factor sim1. Genes Dev. 1998, 12, 3264–3275. [Google Scholar] [CrossRef] [PubMed]
- Chapman-Smith, A.; Whitelaw, M.L. Novel DNA binding by a basic helix-loop-helix protein—The role of the dioxin receptor pas domain. J. Biol. Chem. 2006, 281, 12535–12545. [Google Scholar] [CrossRef] [PubMed]
- Fairman, R.; BeranSteed, R.K.; Handel, T.M. Heteronuclear (1H-, 13C, 15N) nmr assignments and secondary structure of the basic region-helix-loop-helix domain of E47. Protein Sci. 1997, 6, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Nambu, J.R.; Lewis, J.O.; Wharton, K.A.; Crews, S.T. The drosophila single-minded gene encodes a helix-loop-helix protein that acts as a master regulator of CNS midline development. Cell 1991, 67, 1157–1167. [Google Scholar] [CrossRef]
- Taylor, B.L.; Zhulin, I.B. Pas domains: Internal sensors of oxygen, redox potential, and light. Microbiol. Mol. Biol. Rev. 1999, 63, 479–506. [Google Scholar] [PubMed]
- Reiszporszasz, S.; Probst, M.R.; Fukunaga, B.N.; Hankinson, O. Identification of functional domains of the aryl-hydrocarbon receptor nuclear translocator protein (ARNT). Mol. Cell. Biol. 1994, 14, 6075–6086. [Google Scholar] [CrossRef]
- Hirose, K.; Morita, M.; Ema, M.; Mimura, J.; Hamada, H.; Fujii, H.; Saijo, Y.; Gotoh, O.; Sogawa, K.; FujiiKuriyama, Y. cDNA cloning and tissue-specific expression of a novel basic helix-loop-helix/PAS factor (Arnt2) with close sequence similarity to the aryl hydrocarbon receptor nuclear translocator (Arnt). Mol. Cell. Biol. 1996, 16, 1706–1713. [Google Scholar] [CrossRef] [PubMed]
- Ooe, N.; Saito, K.; Kaneko, H. Characterization of functional heterodirner partners in brain for a bHLH-PAS factor NXF. Biochim. Biophys. Acta 2009, 1789, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Schoenhard, J.A.; Eren, M.; Johnson, C.H.; Vaughan, D.E. Alternative splicing yields novel BMAL2 variants: Tissue distribution and functional characterization. Am. J. Physiol. Cell Physiol. 2002, 283, C103–C114. [Google Scholar] [CrossRef] [PubMed]
- Wenger, R.H. Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. FASEB J. 2002, 16, 1151–1162. [Google Scholar] [CrossRef] [PubMed]
- Klaric, T.; Lardelli, M.; Key, B.; Koblar, S.; Lewis, M. Activity-dependent expression of neuronal PAS domain-containing protein 4 (Npas4a) in the develop zebrafish brain. Front. Neuroanat. 2014, 8, 148. [Google Scholar] [PubMed]
- Isaacson, R.L. A fuzzy limbic system. Behav. Brain Res. 1992, 52, 129–131. [Google Scholar] [CrossRef]
- Kotter, R.; Meyer, N. The limbic system—A review of its empirical foundation. Behav. Brain Res. 1992, 52, 105–127. [Google Scholar] [CrossRef]
- Bachevalier, J.; Alvarado, M.C.; Malkova, L. Memory and socioemotional behavior in monkeys after hippocampal damage incurred in infancy or in adulthood. Biol. Psychiatry 1999, 46, 329–339. [Google Scholar] [CrossRef]
- Bachevalier, J.; Malkova, L. The amygdala and development of social cognition: Theoretical comment on Bauman, Toscano, Mason, Lavenex, and Amaral (2006). Behav. Neurosci. 2006, 120, 989–991. [Google Scholar] [CrossRef] [PubMed]
- Prentice, L.M.; de Tassigny, X.D.A.; McKinney, S.; de Algara, T.R.; Yap, D.; Turashvili, G.; Poon, S.; Sutcliffe, M.; Allard, P.; Burleigh, A.; et al. The testosterone-dependent and independent transcriptional networks in the hypothalamus of Gpr54 and Kiss1 knockout male mice are not fully equivalent. BMC Genom. 2011, 12. [Google Scholar] [CrossRef] [PubMed]
- Unfried, C.; Burbach, G.; Korf, H.-W.; von Gall, C. Melatonin receptor 1-dependent gene expression in the mouse pars tuberalis as revealed by cdna microarray analysis and in situ hybridization. J. Pineal Res. 2010, 48, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, S.-I.; Takahashi, H.; Nishimura, N.; Kinoshita, M.; Asahina, R.; Kitsuki, M.; Tatsumi, K.; Furukawa-Hibi, Y.; Hirai, H.; Nagai, T.; et al. Npas4 regulates Mdm2 and thus Dcx in experience-dependent dendritic spine development of newborn olfactory bulb interneurons. Cell Rep. 2014, 8, 843–857. [Google Scholar] [CrossRef] [PubMed]
- Klaric, T.S.; Thomas, P.Q.; Dottori, M.; Leong, W.K.; Koblar, S.A.; Lewis, M.D. A reduction in Npas4 expression results in delayed neural differentiation of mouse embryonic stem cells. Stem Cell Res. Ther. 2014, 5, 64. [Google Scholar] [CrossRef] [PubMed]
- Bertolino, M.; Llinas, R.R. The central role of voltage-activated and receptor-operated calcium channels in neuronal cells. Annu. Rev. Pharmacol. Toxicol. 1992, 32, 399–421. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, G.E.; Bading, H. Nuclear calcium: A key regulator of gene expression. Biometals 1998, 11, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, G.E.; Bading, H. The Yin and Yang of NMDA receptor signalling. Trends Neurosci. 2003, 26, 81–89. [Google Scholar] [CrossRef]
- Lanahan, A.; Worley, P. Immediate-early genes and synaptic function. Neurobiol. Learn. Mem. 1998, 70, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Ooe, N.; Kobayashi, K.; Motonaga, K.; Saito, K.; Kaneko, H. Dynamic regulation of bHLH-PAS-type transcription factor NXF gene expression and neurotrophin dependent induction of the transcriptional control activity. Biochem. Biophys. Res. Commun. 2009, 378, 761–765. [Google Scholar] [CrossRef] [PubMed]
- Coba, M.P.; Valor, L.M.; Kopanitsa, M.V.; Afinowi, N.O.; Grant, S.G.N. Kinase networks integrate profiles of N-methyl-d-aspartate receptor-mediated gene expression in hippocampus. J. Biol. Chem. 2008, 283, 34101–34107. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-J.; Steijaert, M.N.; Lau, D.; Schuetz, G.; Delucinge-Vivier, C.; Descombes, P.; Bading, H. Decoding NMDA receptor signaling: Identification of genomic programs specifying neuronal survival and death. Neuron 2007, 53, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Madabhushi, R.; Gao, F.; Pfenning, A.R.; Pan, L.; Yamakawa, S.; Seo, J.; Rueda, R.; Phan, T.X.; Yamakawa, H.; Pao, P.-C. Activity-induced DNA breaks govern the expression of neuronal early-response genes. Cell 2015, 161, 1592–1605. [Google Scholar] [CrossRef] [PubMed]
- Hande, K. Etoposide: Four decades of development of a topoisomerase II inhibitor. Eur. J. Cancer 1998, 34, 1514–1521. [Google Scholar] [CrossRef]
- Carrion, A.M.; Link, W.A.; Ledo, F.; Mellstrom, B.; Naranjo, J.R. Dream is a Ca2+-regulated transcriptional repressor. Nature 1999, 398, 80–84. [Google Scholar] [PubMed]
- Mellstroem, B.; Sahun, I.; Ruiz-Nuno, A.; Murtra, P.; Gomez-Villafuertes, R.; Savignac, M.; Oliveros, J.C.; Gonzalez, P.; Kastanauskaite, A.; Knafo, S.; et al. Dream controls the on/off switch of specific activity-dependent transcription pathways. Mol. Cell. Biol. 2014, 34, 877–887. [Google Scholar] [CrossRef] [PubMed]
- Lindblad-Toh, K.; Garber, M.; Zuk, O.; Lin, M.F.; Parker, B.J.; Washietl, S.; Kheradpour, P.; Ernst, J.; Jordan, G.; Mauceli, E.; et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature 2011, 478, 476–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunyak, V.V.; Rosenfeld, M.G. No rest for REST: REST/NRSF regulation of neurogenesis. Cell 2005, 121, 499–501. [Google Scholar] [CrossRef] [PubMed]
- Bersten, D.C.; Wright, J.A.; McCarthy, P.J.; Whitelaw, M.L. Regulation of the neuronal transcription factor Npas4 by rest and micrornas. Biochim. Biophys. Acta 2014, 1839, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci. 1999, 22, 391–397. [Google Scholar] [CrossRef]
- Mitsios, N.; Gaffney, J.; Kumar, P.; Krupinski, J.; Kumar, S.; Slevin, M. Pathophysiology of acute ischaemic stroke: An analysis of common signalling mechanisms and identification of new molecular targets. Pathobiology 2006, 73, 159–175. [Google Scholar] [CrossRef] [PubMed]
- Catsicas, M.; Pequignot, Y.; Clarke, P.G.H. Rapid onset of neuronal death induced by blockade of either axoplasmic-transport or action-potentials in afferent-fibers during brain-development. J. Neurosci. 1992, 12, 4642–4650. [Google Scholar] [PubMed]
- Sherrard, R.M.; Bower, A.J. Role of afferents in the development and cell survival of the vertebrate nervous system. Clin. Exp. Pharmacol. Physiol. 1998, 25, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Bok, J.; Wang, Q.; Huang, H.; Green, S.H. CaMKII and CaMKIV mediate distinct prosurvival signaling pathways in response to depolarization in neurons. Mol. Cell. Neurosci. 2007, 36, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Papadia, S.; Stevenson, P.; Hardingham, N.R.; Bading, H.; Hardingham, G.E. Nuclear Ca2+ and the camp response element-binding protein family mediate a late phase of activity-dependent neuroprotection. J. Neurosci. 2005, 25, 4279–4287. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, M.; Ehrenreich, H. Brain derived proteins as markers of acute stroke: Their relation to pathophysiology, outcome prediction and neuroprotective drug monitoring. Restor. Neurol. Neurosci. 2003, 21, 177–190. [Google Scholar] [PubMed]
- Matsushima, K.; Hogan, M.J.; Hakim, A.M. Cortical spreading depression protects against subsequent focal cerebral ischemia in rats. J. Cereb. Blood Flow Metab. 1996, 16, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, K.; Schmidt-Kastner, R.; Hogan, M.J.; Hakim, A.M. Cortical spreading depression activates trophic factor expression, in neurons and astrocytes and protects against subsequent focal brain ischemia. Brain Res. 1998, 807, 47–60. [Google Scholar] [CrossRef]
- Lipsky, R.H.; Marini, A.M. Brain-derived neurotrophic factor in neuronal survival and behavior-related plasticity. Ann. N. Y. Acad. Sci. 2007, 1122, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Carnahan, J.; Greenberg, M.E. Requirement for bdnf in activity-dependent survival of cortical-neurons. Science 1994, 263, 1618–1623. [Google Scholar] [CrossRef] [PubMed]
- Alderson, R.F.; Alterman, A.L.; Barde, Y.A.; Lindsay, R.M. Brain-derived neurotrophic factor increases survival and differentiated functions of rat septal cholinergic neurons in culture. Neuron 1990, 5, 297–306. [Google Scholar] [CrossRef]
- Golstein, P.; Kroemer, G. Redundant cell death mechanisms as relics and backups. Cell Death Differ. 2005, 12, 1490–1496. [Google Scholar] [CrossRef] [PubMed]
- Proskuryakov, S.Y.; Konoplyannikov, A.G.; Gabai, V.L. Necrosis: A specific form of programmed cell death? Exp. Cell Res. 2003, 283, 1–16. [Google Scholar] [CrossRef]
- Brouns, R.; de Deyn, P.P. The complexity of neurobiological processes in acute ischemic stroke. Clin. Neurol. Neurosurg. 2009, 111, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Kreutzberg, G.W. Microglia: A sensor for pathological events in the CNS. Trends Neurosci. 1996, 19, 312–318. [Google Scholar] [CrossRef]
- Thomas, W.E. Brain macrophages: Evaluation of microglia and their functions. Brain Res. Rev. 1992, 17, 61–74. [Google Scholar] [CrossRef]
- Wood, P.L. Microglia as a unique cellular target in the treatment of stroke: Potential neurotoxic mediators produced by activated microglia. Neurol. Res. 1995, 17, 242–248. [Google Scholar] [PubMed]
- Zhang, N.; Komine-Kobayashi, M.; Tanaka, R.; Liu, M.Z.; Mizuno, Y.; Urabe, T. Edaravone reduces early accumulation of oxidative products and sequential inflammatory responses after transient focal ischemia in mice brain. Stroke 2005, 36, 2220–2225. [Google Scholar] [CrossRef] [PubMed]
- Gunther, A.; Kuppers-Tiedt, L.; Schneider, P.M.; Kunert, I.; Berrouschot, J.; Schneider, D.; Rossner, S. Reduced infarct volume and differential effects on glial cell activation after hyperbaric oxygen treatment in rat permanent focal cerebral ischaemia. Eur. J. Neurosci. 2005, 21, 3189–3194. [Google Scholar] [CrossRef] [PubMed]
- Giulian, D.; Corpuz, M.; Chapman, S.; Mansouri, M.; Robertson, C. Reactive mononuclear phagocytes release neurotoxins after ischemic and traumatic injury to the central-nervous-system. J. Neurosci. Res. 1993, 36, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Lehnardt, S.; Massillon, L.; Follett, P.; Jensen, F.E.; Ratan, R.; Rosenberg, P.A.; Volpe, J.J.; Vartanian, T. Activation of innate immunity in the CNS triggers neurodegeneration through a toll-like receptor 4-dependent pathway. Proc. Natl. Acad. Sci. USA 2003, 100, 8514–8519. [Google Scholar] [CrossRef] [PubMed]
- Denes, A.; Vidyasagar, R.; Feng, J.; Narvainen, J.; McColl, B.W.; Kauppinen, R.A.; Allan, S.M. Proliferating resident microglia after focal cerebral ischaemia in mice. J. Cereb. Blood Flow Metab. 2007, 27, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, K.; Kohsaka, S. Microglia: Neuroprotective and neurotrophic cells in the central nervous system. Curr. Drug Targets Cardiovasc. Haematol. Disord. 2004, 4, 65–84. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J. Microglia as neuroprotective, immunocompetent cells of the CNS. Glia 2002, 40, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Rempe, D.A. Targeting astrocytes for stroke therapy. Neurotherapeutics 2010, 7, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Pekny, M.; Nilsson, M. Astrocyte activation and reactive gliosis. Glia 2005, 50, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Hewett, S.J.; Muir, J.K.; Lobner, D.; Symons, A.; Choi, D.W. Potentiation of oxygen-glucose deprivation-induced neuronal death after induction of inos. Stroke 1996, 27, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Donohue, P.J.; Richards, C.M.; Brown, S.A.N.; Hanscom, H.N.; Buschman, J.; Thangada, S.; Hla, T.; Williams, M.S.; Winkles, J.A. TWEAK is an endothelial cell growth and chemotactic factor that also potentiates FGF-2 and VEGF-a mitogenic activity. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Yepes, M.; Brown, S.A.N.; Moore, E.G.; Smith, E.P.; Lawrence, D.A.; Winkles, J.A. A soluble Fn14-Fc decoy receptor reduces infarct volume in a murine model of cerebral ischemia. Am. J. Pathol. 2005, 166, 511–520. [Google Scholar] [CrossRef]
- Lakhan, S.E.; Kirchgessner, A.; Hofer, M. Inflammatory mechanisms in ischemic stroke: Therapeutic approaches. J. Transl. Med. 2009, 7, 97. [Google Scholar] [CrossRef] [PubMed]
- Lucas, S.M.; Rothwell, N.J.; Gibson, R.M. The role of inflammation in cns injury and disease. Br. J. Pharmacol. 2006, 147, S232–S240. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Sawamoto, K.; Suzuki, S.; Suzuki, N.; Adachi, K.; Kawase, T.; Mihara, M.; Ohsugi, Y.; Abe, K.; Okano, H. Blockade of interleukin-6 signaling aggravates ischemic cerebral damage in mice: Possible involvement of Stat3 activation in the protection of neurons. J. Neurochem. 2005, 94, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Berghe, T.V.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Chinenov, Y.; Kerppola, T.K. Close encounters of many kinds: Fos-jun interactions that mediate transcription regulatory specificity. Oncogene 2001, 20, 2438–2452. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Prywes, R. Activation of the c-fos enhancer by the erk MAP kinase pathway through two sequence elements: The c-fos AP-1 and p62TCF sites. Oncogene 2000, 19, 1379–1385. [Google Scholar] [CrossRef] [PubMed]
- Shafarenko, M.; Amanullah, A.; Gregory, B.; Liebermann, D.A.; Hoffman, B. Fos modulates myeloid cell survival and differentiation and partially abrogates the c-Myc block in terminal myeloid differentiation. Blood 2004, 103, 4259–4267. [Google Scholar] [CrossRef] [PubMed]
- Appierto, V.; Villani, M.G.; Cavadini, E.; Lotan, R.; Vinson, C.; Formelli, F. Involvement of c-Fos in fenretinide-induced apoptosis in human ovarian carcinoma cells. Cell Death Differ. 2004, 11, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Ray, N.; Kuwahara, M.; Takada, Y.; Maruyama, K.; Kawaguchi, T.; Tsubone, H.; Ishikawa, H.; Matsuo, K. c-Fos suppresses systemic inflammatory response to endotoxin. Int. Immunol. 2006, 18, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.S., Jr. Series introduction: The transcription factor NF-ΚB and human disease. J. Clin. Investig. 2001, 107, 3. [Google Scholar] [CrossRef] [PubMed]
- Thomasova, D.; Mulay, S.R.; Bruns, H.; Anders, H.-J. p53-independent roles of MDM2 in NF-κB signaling: Implications for cancer therapy, wound healing, and autoimmune diseases. Neoplasia 2012, 14, 1097–1101. [Google Scholar] [CrossRef] [PubMed]
- Colton, C.A. Heterogeneity of microglial activation in the innate immune response in the brain. J. Neuroimmune Pharmacol. 2009, 4, 399–418. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, Y.; Chiba, K. Role of microglial M1/M2 polarization in relapse and remission of psychiatric disorders and diseases. Pharmaceuticals 2014, 7, 1028–1048. [Google Scholar] [CrossRef] [PubMed]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflamm. 2014, 11, 98. [Google Scholar] [CrossRef] [PubMed]
- O’Collins, V.E.; Macleod, M.R.; Donnan, G.A.; Horky, L.L.; van der Worp, B.H.; Howells, D.W. 1026 experimental treatments in acute stroke. Ann. Neurol. 2006, 59, 467–477. [Google Scholar] [PubMed]
- Iadecola, C.; Anrather, J. The immunology of stroke: From mechanisms to translation. Nat. Med. 2011, 17, 796–808. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.K. Investigational anti-inflammatory agents for the treatment of ischaemic brain injury. Expert Opin. Investig. Drugs 2005, 14, 393–409. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, G.; Granger, D.N. Cell adhesion molecules and ischemic stroke. Neurol. Res. 2008, 30, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Prestigiacomo, C.J.; Kim, S.C.; Connolly, E.S.; Liao, H.; Yan, S.F.; Pinsky, D.J. Cd18-mediated neutrophil recruitment contributes to the pathogenesis of reperfused but not nonreperfused stroke. Stroke 1999, 30, 1110–1116. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.L.; Chopp, M.; Jiang, N.; Tang, W.X.; Prostak, J.; Manning, A.M.; Anderson, D.C. Anti-intercellular adhesion molecule-1 antibody reduces ischemic cell damage after transient but not permanent middle cerebral artery occlusion in the wistar rat. Stroke 1995, 26, 1438–1442. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, Z.G.; Zhang, R.L.; Lu, M.; Krams, M.; Chopp, M. Effects of a selective CD11b/CD18 antagonist and recombinant human tissue plasminogen activator treatment alone and in combination in a rat embolic model of stroke. Stroke 2003, 34, 1790–1795. [Google Scholar] [CrossRef] [PubMed]
- Sherman, D.G.; Bes, A.; Easton, J.D.; Hacke, W.; Kaste, M.; Polmar, S.H.; Zivin, J.A.; Fieschi, C.; Miller, P.; Schoenfeld, D.; et al. Use of anti-icam-1 therapy in ischemic stroke—Results of the enlimomab acute stroke trial. Neurology 2001, 57, 1428–1434. [Google Scholar]
- Becker, K.J. Anti-leukocyte antibodies: Leukarrest (hu23f2g) and enlimomab (r6.5) in acute stroke. Curr. Med. Res. Opin. 2002, 18, s18–s22. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Liu, L.; Zhang, S.; Nanda, A.; Li, G. Role of inflammation and its mediators in acute ischemic stroke. J. Cardiovasc. Transl. Res. 2013, 6, 834–851. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.R.; Ritzel, R.; McCullough, L.D.; Liu, F. Microglia and ischemic stroke: A double-edged sword. Int. J. Physiol. Pathophysiol. Pharmacol. 2013, 5, 73–90. [Google Scholar] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choy, F.C.; Klarić, T.S.; Koblar, S.A.; Lewis, M.D. The Role of the Neuroprotective Factor Npas4 in Cerebral Ischemia. Int. J. Mol. Sci. 2015, 16, 29011-29028. https://doi.org/10.3390/ijms161226144
Choy FC, Klarić TS, Koblar SA, Lewis MD. The Role of the Neuroprotective Factor Npas4 in Cerebral Ischemia. International Journal of Molecular Sciences. 2015; 16(12):29011-29028. https://doi.org/10.3390/ijms161226144
Chicago/Turabian StyleChoy, Fong Chan, Thomas S. Klarić, Simon A. Koblar, and Martin D. Lewis. 2015. "The Role of the Neuroprotective Factor Npas4 in Cerebral Ischemia" International Journal of Molecular Sciences 16, no. 12: 29011-29028. https://doi.org/10.3390/ijms161226144