
Cell Penetrating Peptide Conjugated Chitosan for Enhanced Delivery of Nucleic Acid


Abstract
:
1. Introduction
2. Cell Penetrating Peptides (CPPs) and Their Classification
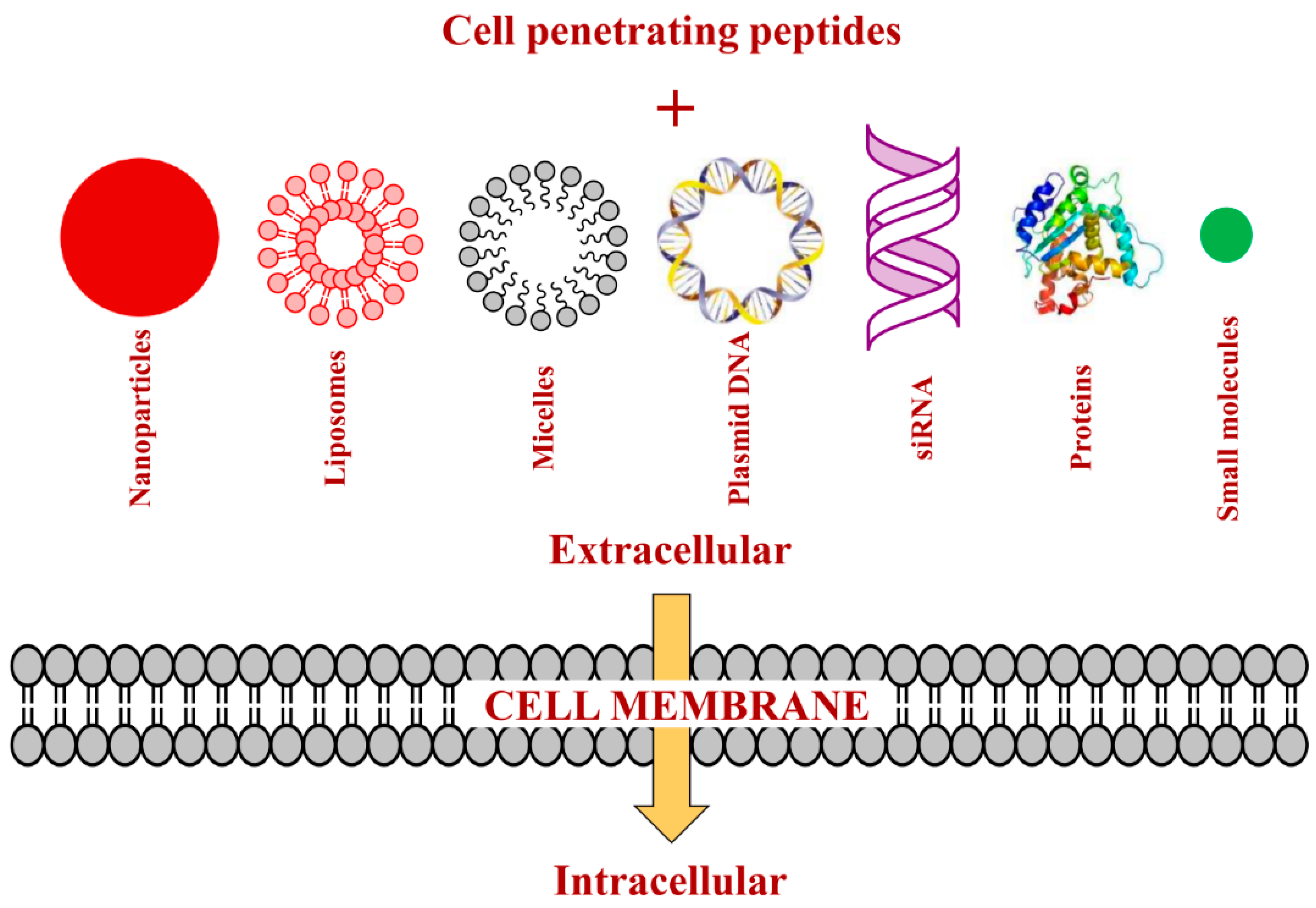
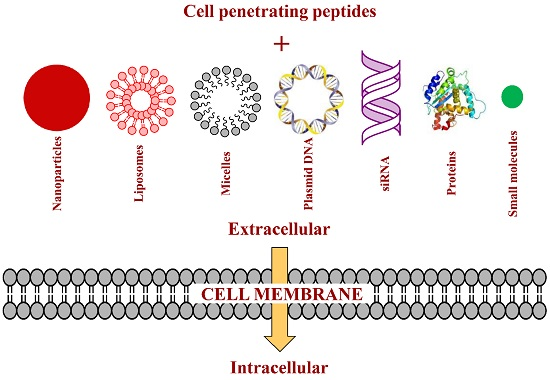
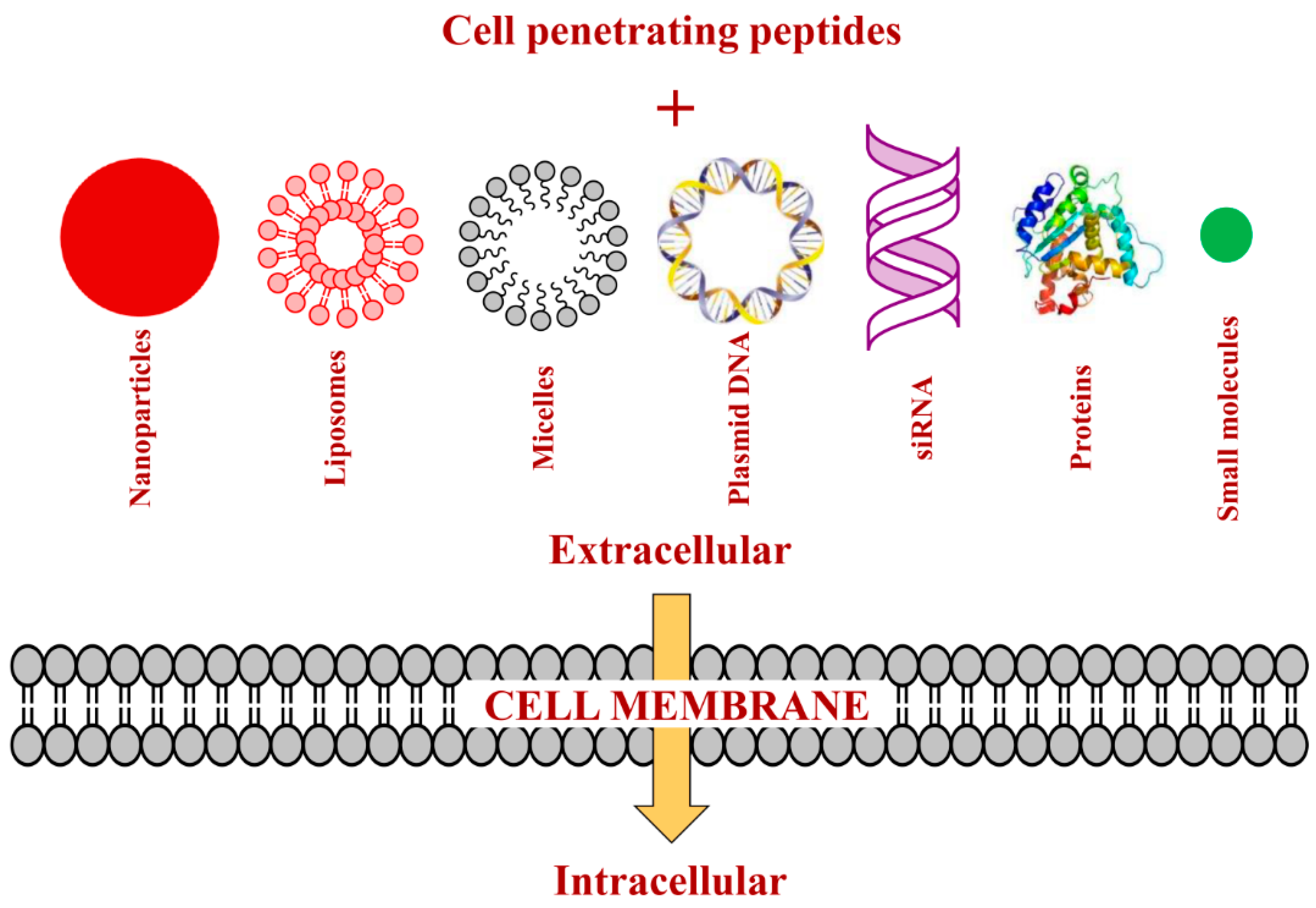
3. Strategies of Attaching CPPs to Cargos
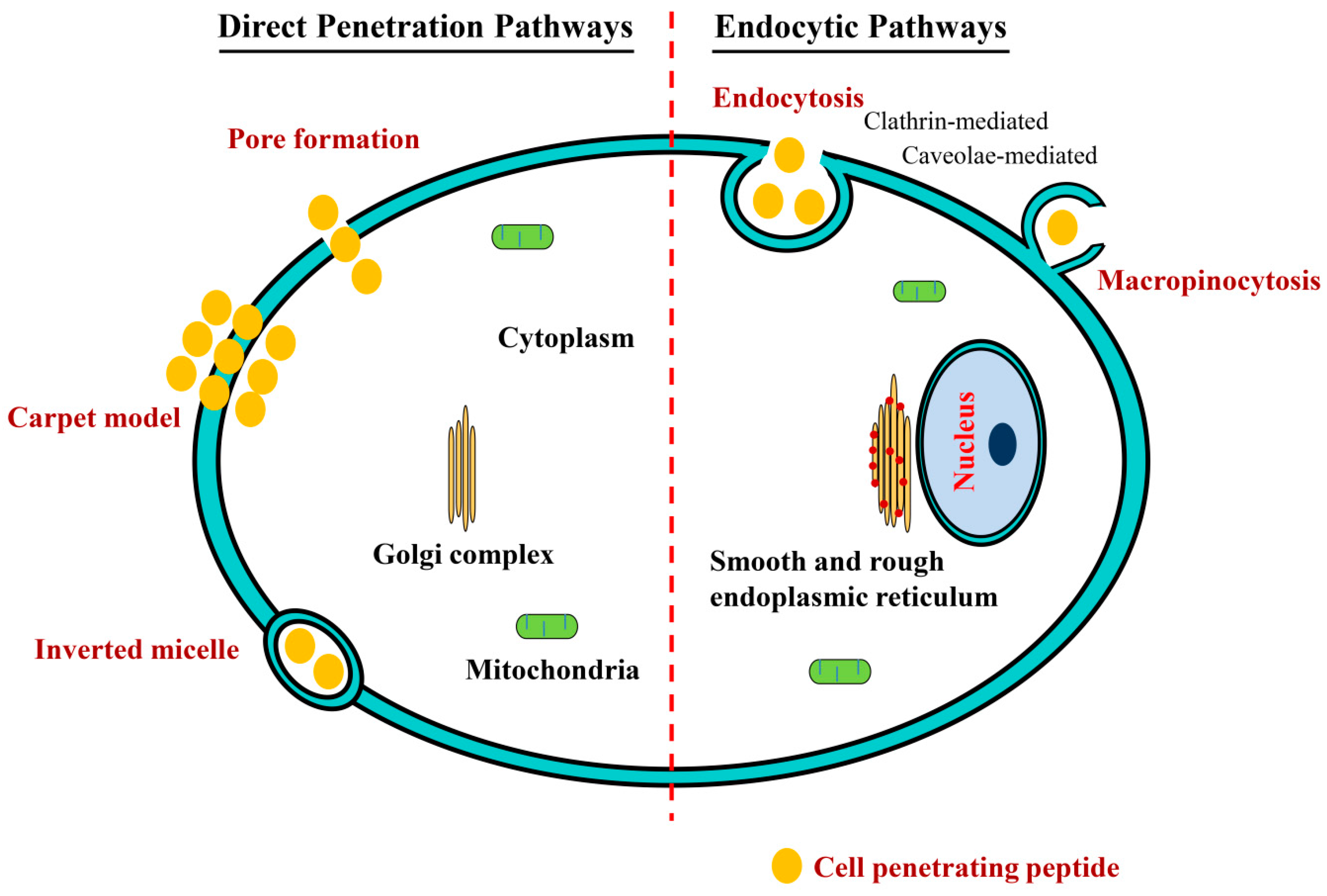
4. Mechanisms of Cellular Uptake of CPPs and Their Conjugates
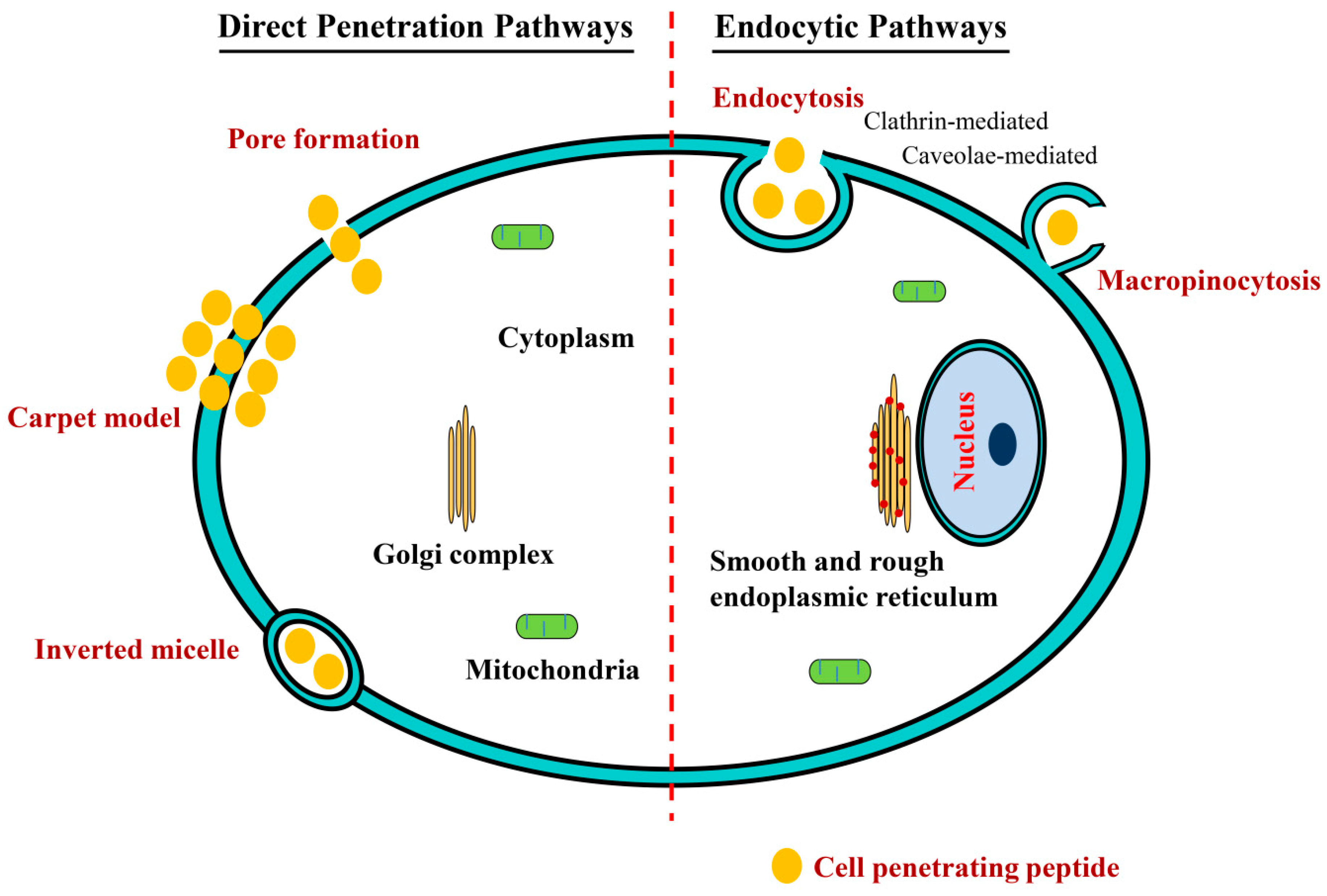
4.1. Direct Penetration Pathways
4.2. Endocytosis

{kind=link}
{kind=link}
{kind=link}
| Name | Origin | Sequence | Cellular Uptake Mechanism | Reference |
|---|---|---|---|---|
| TAT | Protein-derived from HIV-1 TAT protein | GRKKRRQRRRPPQ | Macropinocytosis, endocytosis, direct penetration | [69,72] |
| Penetratin | Protein-derived form Drosophila antennapedia | RQIKIWFQNRRMKWKK | Macropinocytosis, endocytosis, direct penetration | [69,71,72,113] |
| Transportan | Chimeric peptide of galanin and mastoparan | GWTLNSAGYLLGKINLKALAALAKKIL | Endocytosis, direct penetration | [72] |
| MAP | Synthetic peptide | KLALKLALKALKAALKLA | Energy dependent and energy independent endocytosis | [114,115] |
| VP22 | Protein-derived from HSV-1 | DAATATRGRSAASRPTERPRAPARSASRPRRPVD | Endocytosis | [116] |
| KALA | Synthetic peptide | WEAKLAKALAKALAKHLAKALAKALKACEA | Endocytosis | [69] |
| GALA | Synthetic peptide | WEAALAEALAAEALAEHLAEALAEALEALAA | Endocytosis | [69] |
| Pep-1 | Chimeric: HIV-reverse transcriptase/SV40 T-antigen | KETWWETWWTEWSQPKKKRKV | Direct penetration | [72] |
| MPG | Chimeric: HIV-gp41/SV40 T-antigen | GALFLGFLGAAGSTMGAWSQPKKKRKV | Direct penetration | [108] |
| Polyarginines | Synthetic peptide | Rn (6 < n < 12) | Macropinocytosis, endocytosis, direct penetration | [72] |
| CADY | Synthetic peptide | GLWRALWRLLRSLWRLLWRA | Direct penetration | [117] |
5. Recent Progress in CPP Conjugated Chitosan
5.1. CPP Conjugated Chitosan for DNA Delivery
| CPP | Chitosan Complex | Nucleic Acid | Cells/Model | Effect | Reference |
|---|---|---|---|---|---|
| Poly-l-arginine (PLR) | PEGylated PLR-grafted chitosan (PEG-CS-PLR) | siSVN, siGFP, siRFP | Hepa 1–6, A549, VK2 cells, and 293 T-GFP cells |
| [127] |
| Mice bearing B16F10-RFP tumors | |||||
| Octaarginine | Octaarginine-modified chitosan (R8-CS) | pGL3 | COS-1 cells |
| [128] |
| TAT | Chitosan-thioglycolic acid (CS-TGA) + Chitosan-TAT (CS-TAT) | pEGFP | HEK293 cells |
| [46] |
| Nonaarginine | Nonaarginine-modified chitosan (R9-chitosan) | siCypB | HeLa cells |
| [129] |
| TAT | TAT peptide-tagged PEGylated chitosan (CS-PEG-TAT) | siGLO | Neuro2a cells |
| [130] |
| Penetratin | Linoleic acid and penetratin dual-functionalized chitosan (CS-Lin-Pen) | pGFP, pβ-gal | HEK293, CHO, and HeLa |
| [51] |
| TAT | TAT-LHRH-chitosan conjugate (TLC) | pGL3 | BEL-7402, L02 cells |
| [131] |
| TAT | TAT tagged and folate modified N-succinyl-chitosan (TAT-Suc-FA) | pDNA | K562 cells |
| [132] |
5.2. CPP Conjugated Chitosan for siRNA Delivery
6. Conclusions and Future Prospects
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Friedmann, T.; Roblin, R. Gene therapy for human genetic disease? Science 1972, 175, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.; Geddes, D.M.; Alton, E.W. Barriers to and new approaches for gene therapy and gene delivery in cystic fibrosis. Adv. Drug Deliv. Rev. 2002, 54, 1373–1393. [Google Scholar] [CrossRef]
- Nathwani, A.C.; Davidoff, A.M.; Tuddenham, E.G. Prospects for gene therapy of haemophilia. Haemophilia 2004, 10, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Van Deutekom, J.C.; van Ommen, G.J. Advances in duchenne muscular dystrophy gene therapy. Nat. Rev. Genet. 2003, 4, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Cavazzana-Calvo, M.; Hacein-Bey, S.; de Saint Basile, G.; Gross, F.; Yvon, E.; Nusbaum, P.; Selz, F.; Hue, C.; Certain, S.; Casanova, J.L.; et al. Gene therapy of human severe combined immunodeficiency (scid)-X1 disease. Science 2000, 288, 669–672. [Google Scholar] [CrossRef] [PubMed]
- Adair, J.E.; Beard, B.C.; Trobridge, G.D.; Neff, T.; Rockhill, J.K.; Silbergeld, D.L.; Mrugala, M.M.; Kiem, H.P. Extended survival of glioblastoma patients after chemoprotective HSC gene therapy. Sci. Transl. Med. 2012, 4, 133ra157. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalian, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006, 314, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.F.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; Dudley, M.E.; Wunderlich, J.R.; Nahvi, A.V.; Helman, L.J.; Mackall, C.L.; et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol. 2011, 29, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Stewart, D.J.; Hilton, J.D.; Arnold, J.M.; Gregoire, J.; Rivard, A.; Archer, S.L.; Charbonneau, F.; Cohen, E.; Curtis, M.; Buller, C.E.; et al. Angiogenic gene therapy in patients with nonrevascularizable ischemic heart disease: A phase 2 randomized, controlled trial of ADVEGF121 (ADVEGF121) versus maximum medical treatment. Gene Ther. 2006, 13, 1503–1511. [Google Scholar] [CrossRef] [PubMed]
- Sundararaman, S.; Miller, T.J.; Pastore, J.M.; Kiedrowski, M.; Aras, R.; Penn, M.S. Plasmid-based transient human stromal cell-derived factor-1 gene transfer improves cardiac function in chronic heart failure. Gene Ther. 2011, 18, 867–873. [Google Scholar] [CrossRef] [PubMed]
- Alisky, J.M.; Davidson, B.L. Gene therapy for amyotrophic lateral sclerosis and other motor neuron diseases. Hum. Gene Ther. 2000, 11, 2315–2329. [Google Scholar] [CrossRef] [PubMed]
- Burton, E.A.; Glorioso, J.C.; Fink, D.J. Gene therapy progress and prospects: Parkinson’s disease. Gene Ther. 2003, 10, 1721–1727. [Google Scholar] [CrossRef] [PubMed]
- Tuszynski, M.H. Growth-factor gene therapy for neurodegenerative disorders. Lancet Neurol. 2002, 1, 51–57. [Google Scholar] [CrossRef]
- Bunnell, B.A.; Morgan, R.A. Gene therapy for infectious diseases. Clin. Microbiol. Rev. 1998, 11, 42–56. [Google Scholar] [PubMed]
- Hashiba, T.; Suzuki, M.; Nagashima, Y.; Suzuki, S.; Inoue, S.; Tsuburai, T.; Matsuse, T.; Ishigatubo, Y. Adenovirus-mediated transfer of heme oxygenase-1 cDNA attenuates severe lung injury induced by the influenza virus in mice. Gene Ther. 2001, 8, 1499–1507. [Google Scholar] [CrossRef] [PubMed]
- Ivacik, D.; Ely, A.; Ferry, N.; Arbuthnot, P. Sustained inhibition of hepatitis b virus replication in vivo using RNAi-activating lentiviruses. Gene Ther. 2015, 22, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Jiang, H.L.; Jere, D.; Park, I.K.; Cho, M.H.; Nah, J.W.; Choi, Y.J.; Akaike, T.; Cho, C.S. Chemical modification of chitosan as a gene carrier in vitro and in vivo. Prog. Polym. Sci. 2007, 32, 726–753. [Google Scholar] [CrossRef]
- Tamboli, V.; Mishra, G.P.; Mitrat, A.K. Polymeric vectors for ocular gene delivery. Ther. Deliv. 2011, 2, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J. Immune response following intraocular delivery of recombinant viral vectors. Gene Ther. 2003, 10, 977–982. [Google Scholar] [CrossRef] [PubMed]
- Dewey, R.A.; Morrissey, G.; Cowsill, C.M.; Stone, D.; Bolognani, F.; Dodd, N.J.; Southgate, T.D.; Klatzmann, D.; Lassmann, H.; Castro, M.G.; et al. Chronic brain inflammation and persistent herpes simplex virus 1 thymidine kinase expression in survivors of syngeneic glioma treated by adenovirus-mediated gene therapy: Implications for clinical trials. Nat. Med. 1999, 5, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Hacein-Bey-Abina, S.; von Kalle, C.; Schmidt, M.; le Deist, F.; Wulffraat, N.; McIntyre, E.; Radford, I.; Villeval, J.L.; Fraser, C.C.; Cavazzana-Calvo, M.; et al. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2003, 348, 255–256. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.E.; Ehrhardt, A.; Kay, M.A. Progress and problems with the use of viral vectors for gene therapy. Nat. Rev. Genet. 2003, 4, 346–358. [Google Scholar] [CrossRef] [PubMed]
- Niidome, T.; Huang, L. Gene therapy progress and prospects: Nonviral vectors. Gene Ther. 2002, 9, 1647–1652. [Google Scholar] [CrossRef] [PubMed]
- Leventis, R.; Silvius, J.R. Interactions of mammalian cells with lipid dispersions containing novel metabolizable cationic amphiphiles. Biochim. Biophys. Acta 1990, 1023, 124–132. [Google Scholar] [CrossRef]
- Felgner, P.L.; Gadek, T.R.; Holm, M.; Roman, R.; Chan, H.W.; Wenz, M.; Northrop, J.P.; Ringold, G.M.; Danielsen, M. Lipofection: A highly efficient, lipid-mediated DNA-transfection procedure. Proc. Natl. Acad. Sci. USA 1987, 84, 7413–7417. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Huang, L. A novel cationic liposome reagent for efficient transfection of mammalian cells. Biochem. Biophys. Res. Commun. 1991, 179, 280–285. [Google Scholar] [CrossRef]
- Lungwitz, U.; Breunig, M.; Blunk, T.; Gopferich, A. Polyethylenimine-based non-viral gene delivery systems. Eur. J. Pharm. Biopharm. 2005, 60, 247–266. [Google Scholar] [CrossRef] [PubMed]
- Kodama, Y.; Nakamura, T.; Kurosaki, T.; Egashira, K.; Mine, T.; Nakagawa, H.; Muro, T.; Kitahara, T.; Higuchi, N.; Sasaki, H. Biodegradable nanoparticles composed of dendrigraft poly-l-lysine for gene delivery. Eur. J. Pharm. Biopharm. 2014, 87, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.M.; Pan, S.R.; Chen, M.W.; Wu, Y.; Wang, C.; Wen, Y.T.; Zeng, X.; Wu, C.B. A serum-resistant polyamidoamine-based polypeptide dendrimer for gene transfection. Biomaterials 2011, 32, 1619–1634. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.; Sun, W.; Kissel, T. Chitosan-based formulations for delivery of DNA and siRNA. Adv. Drug Deliv. Rev. 2010, 62, 12–27. [Google Scholar] [CrossRef] [PubMed]
- Boisguerin, P.; Deshayes, S.; Gait, M.J.; O’Donovan, L.; Godfrey, C.; Betts, C.A.; Wood, M.J.; Lebleu, B. Delivery of therapeutic oligonucleotides with cell penetrating peptides. Adv. Drug Deliv. Rev. 2015, 87, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Varum, K.M.; Anthonsen, M.W.; Grasdalen, H.; Smidsrod, O. 13C-N.m.r. Studies of the acetylation sequences in partially N-deacetylated chitins (chitosans). Carbohydr. Res. 1991, 217, 19–27. [Google Scholar] [CrossRef]
- Rane, K.D.; Hoover, D.G. Production of chitosan by fungi. Food Biotechnol. 1993, 7, 11–33. [Google Scholar] [CrossRef]
- Riva, R.; Ragelle, H.; des Rieux, A.; Duhem, N.; Jérôme, C.; Préat, V. Chitosan and chitosan derivatives in drug delivery and tissue engineering. In Chitosan for Biomaterials II; Jayakumar, R., Prabaharan, M., Muzzarelli, R.A.A., Eds.; Springer: Berlin, Germany; Heidelberg, Germany, 2011; Volume 244, pp. 19–44. [Google Scholar]
- Anthonsen, M.W.; Smidsrød, O. Hydrogen ion titration of chitosans with varying degrees of N-acetylation by monitoring induced 1H-NMR chemical shifts. Carbohydr. Polym. 1995, 26, 303–305. [Google Scholar] [CrossRef]
- Domard, A. PH and c.d. measurements on a fully deacetylated chitosan: Application to CuII—Polymer interactions. Int. J. Biol. Macromol. 1987, 9, 98–104. [Google Scholar] [CrossRef]
- Lee, M.K.; Chun, S.K.; Choi, W.J.; Kim, J.K.; Choi, S.H.; Kim, A.; Oungbho, K.; Park, J.S.; Ahn, W.S.; Kim, C.K. The use of chitosan as a condensing agent to enhance emulsion-mediated gene transfer. Biomaterials 2005, 26, 2147–2156. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.Z.; Zhu, K.J. The influence of multivalent phosphate structure on the properties of ionically cross-linked chitosan films for controlled drug release. Eur. J. Pharm. Biopharm. 2002, 54, 235–243. [Google Scholar] [CrossRef]
- Hu, F.Q.; Zhao, M.D.; Yuan, H.; You, J.; Du, Y.Z.; Zeng, S. A novel chitosan oligosaccharide-stearic acid micelles for gene delivery: Properties and in vitro transfection studies. Int. J. Pharm. 2006, 315, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Gihm, S.H.; Park, C.R.; Lee, K.Y.; Kim, T.W.; Kwon, I.C.; Chung, H.; Jeong, S.Y. Structural characteristics of size-controlled self-aggregates of deoxycholic acid-modified chitosan and their application as a DNA delivery carrier. Bioconjug. Chem. 2001, 12, 932–938. [Google Scholar] [CrossRef] [PubMed]
- Layek, B.; Singh, J. N-hexanoyl, N-octanoyl and N-decanoyl chitosans: Binding affinity, cell uptake, and transfection. Carbohydr. Polym. 2012, 89, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Layek, B.; Singh, J. Amino acid grafted chitosan for high performance gene delivery: Comparison of amino acid hydrophobicity on vector and polyplex characteristics. Biomacromolecules 2013, 14, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.G.; Zhang, X.; Sun, S.J.; Sun, G.J.; Yao, K.D.; Liang, D.C.; Guo, G.; Zhang, J.Y. N-alkylated chitosan as a potential nonviral vector for gene transfection. Bioconjug. Chem. 2003, 14, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Mandke, R.; Singh, J. Cationic nanomicelles for delivery of plasmids encoding interleukin-4 and interleukin-10 for prevention of autoimmune diabetes in mice. Pharm. Res. 2012, 29, 883–897. [Google Scholar] [CrossRef] [PubMed]
- Mandke, R.; Singh, J. Effect of acyl chain length and unsaturation on physicochemical properties and transfection efficiency of N-acyl-substituted low-molecular-weight chitosan. J. Pharm. Sci. 2012, 101, 268–282. [Google Scholar] [CrossRef] [PubMed]
- Rahmat, D.; Khan, M.I.; Shahnaz, G.; Sakloetsakun, D.; Perera, G.; Bernkop-Schnurch, A. Synergistic effects of conjugating cell penetrating peptides and thiomers on non-viral transfection efficiency. Biomaterials 2012, 33, 2321–2326. [Google Scholar] [CrossRef] [PubMed]
- Park, I.K.; Ihm, J.E.; Park, Y.H.; Choi, Y.J.; Kim, S.I.; Kim, W.J.; Akaike, T.; Cho, C.S. Galactosylated chitosan (GC)-graft-poly(vinyl pyrrolidone) (PVP) as hepatocyte-targeting DNA carrier. Preparation and physicochemical characterization of GC-graft-PVP/DNA complex (1). J. Control. Release 2003, 86, 349–359. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, J.; Pan, Y.; Zhao, J.; Ren, L.; Liao, M.; Hu, Z.; Kong, L.; Wang, J. A novel pegylation of chitosan nanoparticles for gene delivery. Biotechnol. Appl. Biochem. 2007, 46, 197–204. [Google Scholar] [PubMed]
- Layek, B.; Haldar, M.K.; Sharma, G.; Lipp, L.; Mallik, S.; Singh, J. Hexanoic acid and polyethylene glycol double grafted amphiphilic chitosan for enhanced gene delivery: Influence of hydrophobic and hydrophilic substitution degree. Mol. Pharm. 2014, 11, 982–994. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; He, C.; Tang, C.; Yin, C. Effects of hydrophobic and hydrophilic modifications on gene delivery of amphiphilic chitosan based nanocarriers. Biomaterials 2011, 32, 4630–4638. [Google Scholar] [CrossRef] [PubMed]
- Layek, B.; Singh, J. Cell penetrating peptide conjugated polymeric micelles as a high performance versatile nonviral gene carrier. Biomacromolecules 2013, 14, 4071–4081. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Chen, J.; Xu, X.; Ding, Z.; Yang, Y.H.; Hua, Z.; Zhang, J. Galactosylated low molecular weight chitosan as DNA carrier for hepatocyte-targeting. Int. J. Pharm. 2003, 255, 57–68. [Google Scholar] [CrossRef]
- Layek, B.; Lipp, L.; Singh, J. Apc targeted micelle for enhanced intradermal delivery of hepatitis b DNA vaccine. J. Control. Release 2015, 207, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Lockey, R.; Mohapatra, S. Folate receptor-mediated cancer cell specific gene delivery using folic acid-conjugated oligochitosans. J. Nanosci. Nanotechnol. 2006, 6, 2860–2866. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the TAT protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef]
- Green, M.; Loewenstein, P.M. Autonomous functional domains of chemically synthesized human immunodeficiency virus TAT trans-activator protein. Cell 1988, 55, 1179–1188. [Google Scholar] [CrossRef]
- Derossi, D.; Joliot, A.H.; Chassaing, G.; Prochiantz, A. The third helix of the antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 1994, 269, 10444–10450. [Google Scholar] [PubMed]
- Joliot, A.; Pernelle, C.; Deagostini-Bazin, H.; Prochiantz, A. Antennapedia homeobox peptide regulates neural morphogenesis. Proc. Natl. Acad. Sci. USA 1991, 88, 1864–1868. [Google Scholar] [CrossRef] [PubMed]
- Vives, E.; Brodin, P.; Lebleu, B. A truncated HIV-1 TAT protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem. 1997, 272, 16010–16017. [Google Scholar] [CrossRef] [PubMed]
- Gupta, B.; Levchenko, T.S.; Torchilin, V.P. Intracellular delivery of large molecules and small particles by cell-penetrating proteins and peptides. Adv. Drug Deliv. Rev. 2005, 57, 637–651. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.M.; Harrison, S.D.; Maclean, D. Therapeutic applications of cell-penetrating peptides. Methods Mol. Biol. 2011, 683, 535–551. [Google Scholar] [PubMed]
- Sawant, R.; Torchilin, V. Intracellular transduction using cell-penetrating peptides. Mol. Biosyst. 2010, 6, 628–640. [Google Scholar] [CrossRef] [PubMed]
- Lewin, M.; Carlesso, N.; Tung, C.H.; Tang, X.W.; Cory, D.; Scadden, D.T.; Weissleder, R. TAT peptide-derivatized magnetic nanoparticles allow in vivo tracking and recovery of progenitor cells. Nat. Biotechnol. 2000, 18, 410–414. [Google Scholar] [PubMed]
- Torchilin, V.P. TAT peptide-mediated intracellular delivery of pharmaceutical nanocarriers. Adv. Drug Deliv. Rev. 2008, 60, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V.P.; Levchenko, T.S.; Rammohan, R.; Volodina, N.; Papahadjopoulos-Sternberg, B.; D’Souza, G.G. Cell transfection in vitro and in vivo with nontoxic TAT peptide-liposome-DNA complexes. Proc. Natl. Acad. Sci. USA 2003, 100, 1972–1977. [Google Scholar] [CrossRef] [PubMed]
- Copolovici, D.M.; Langel, K.; Eriste, E.; Langel, U. Cell-penetrating peptides: Design, synthesis, and applications. ACS Nano 2014, 8, 1972–1994. [Google Scholar] [CrossRef] [PubMed]
- Milletti, F. Cell-penetrating peptides: Classes, origin, and current landscape. Drug Discov. Today 2012, 17, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Bechara, C.; Sagan, S. Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett. 2013, 587, 1693–1702. [Google Scholar] [CrossRef] [PubMed]
- Deshayes, S.; Morris, M.C.; Divita, G.; Heitz, F. Cell-penetrating peptides: Tools for intracellular delivery of therapeutics. Cell. Mol. Life Sci. 2005, 62, 1839–1849. [Google Scholar] [CrossRef] [PubMed]
- Shi, N.Q.; Qi, X.R.; Xiang, B.; Zhang, Y. A survey on “trojan horse” peptides: Opportunities, issues and controlled entry to “troy”. J. Control. Release 2014, 194, 53–70. [Google Scholar] [CrossRef] [PubMed]
- Farkhani, S.M.; Valizadeh, A.; Karami, H.; Mohammadi, S.; Sohrabi, N.; Badrzadeh, F. Cell penetrating peptides: Efficient vectors for delivery of nanoparticles, nanocarriers, therapeutic and diagnostic molecules. Peptides 2014, 57, 78–94. [Google Scholar] [CrossRef] [PubMed]
- Koren, E.; Torchilin, V.P. Cell-penetrating peptides: Breaking through to the other side. Trends Mol. Med. 2012, 18, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Bolhassani, A.; Rafati, S. Non-Viral Delivery Systems in Gene Therapy and Vaccine Development; Intech Open Access Publisher: Rijeka, Croatia, 2011. [Google Scholar]
- Pooga, M.; Lindgren, M.; Hallbrink, M.; Brakenhielm, E.; Langel, U. Galanin-based peptides, galparan and transportan, with receptor-dependent and independent activities. Ann. N. Y. Acad. Sci. 1998, 863, 450–453. [Google Scholar] [CrossRef] [PubMed]
- Mager, I.; Eiriksdottir, E.; Langel, K.; el Andaloussi, S.; Langel, U. Assessing the uptake kinetics and internalization mechanisms of cell-penetrating peptides using a quenched fluorescence assay. Biochim. Biophys. Acta 2010, 1798, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Davidson, T.J.; Harel, S.; Arboleda, V.A.; Prunell, G.F.; Shelanski, M.L.; Greene, L.A.; Troy, C.M. Highly efficient small interfering RNA delivery to primary mammalian neurons induces microRNA-like effects before mRNA degradation. J. Neurosci. 2004, 24, 10040–10046. [Google Scholar] [CrossRef] [PubMed]
- Muratovska, A.; Eccles, M.R. Conjugate for efficient delivery of short interfering RNA (siRNA) into mammalian cells. FEBS Lett. 2004, 558, 63–68. [Google Scholar] [CrossRef]
- Chiu, Y.L.; Ali, A.; Chu, C.Y.; Cao, H.; Rana, T.M. Visualizing a correlation between siRNA localization, cellular uptake, and RNAi in living cells. Chem. Biol. 2004, 11, 1165–1175. [Google Scholar] [CrossRef] [PubMed]
- Meade, B.R.; Dowdy, S.F. Exogenous siRNA delivery using peptide transduction domains/cell penetrating peptides. Adv. Drug Deliv. Rev. 2007, 59, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Deshayes, S.; Gerbal-Chaloin, S.; Morris, M.C.; Aldrian-Herrada, G.; Charnet, P.; Divita, G.; Heitz, F. On the mechanism of non-endosomial peptide-mediated cellular delivery of nucleic acids. Biochim. Biophys. Acta 2004, 1667, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Gros, E.; Deshayes, S.; Morris, M.C.; Aldrian-Herrada, G.; Depollier, J.; Heitz, F.; Divita, G. A non-covalent peptide-based strategy for protein and peptide nucleic acid transduction. Biochim. Biophys. Acta 2006, 1758, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Crombez, L.; Aldrian-Herrada, G.; Konate, K.; Nguyen, Q.N.; McMaster, G.K.; Brasseur, R.; Heitz, F.; Divita, G. A new potent secondary amphipathic cell-penetrating peptide for siRNA delivery into mammalian cells. Mol. Ther. 2009, 17, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Kurzawa, L.; Pellerano, M.; Morris, M.C. PEP and CADY-mediated delivery of fluorescent peptides and proteins into living cells. Biochim. Biophys. Acta 2010, 1798, 2274–2285. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Vidal, P.; Chaloin, L.; Heitz, F.; Divita, G. A new peptide vector for efficient delivery of oligonucleotides into mammalian cells. Nucleic Acids Res. 1997, 25, 2730–2736. [Google Scholar] [CrossRef] [PubMed]
- Crombez, L.; Morris, M.C.; Deshayes, S.; Heitz, F.; Divita, G. Peptide-based nanoparticle for ex vivo and in vivo drug delivery. Curr. Pharm. Des. 2008, 14, 3656–3665. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Depollier, J.; Mery, J.; Heitz, F.; Divita, G. A peptide carrier for the delivery of biologically active proteins into mammalian cells. Nat. Biotechnol. 2001, 19, 1173–1176. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, P.; El-Andaloussi, S.; Sutlu, T.; Johansson, H.; Langel, U. Delivery of short interfering RNA using endosomolytic cell-penetrating peptides. FASEB J. 2007, 21, 2664–2671. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.J.; Christensen, L.V.; Jo, S.; Yockman, J.W.; Jeong, J.H.; Kim, Y.H.; Kim, S.W. Cholesteryl oligoarginine delivering vascular endothelial growth factor siRNA effectively inhibits tumor growth in colon adenocarcinoma. Mol. Ther. 2006, 14, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Pooga, M.; Kut, C.; Kihlmark, M.; Hallbrink, M.; Fernaeus, S.; Raid, R.; Land, T.; Hallberg, E.; Bartfai, T.; Langel, U. Cellular translocation of proteins by transportan. FASEB J. 2001, 15, 1451–1453. [Google Scholar] [CrossRef] [PubMed]
- Deshayes, S.; Konate, K.; Aldrian, G.; Crombez, L.; Heitz, F.; Divita, G. Structural polymorphism of non-covalent peptide-based delivery systems: Highway to cellular uptake. Biochim. Biophys. Acta 2010, 1798, 2304–2314. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, S.B.; Pereira, M.P.; Kelley, S.O. Recent advances in the use of cell-penetrating peptides for medical and biological applications. Adv. Drug Deliv. Rev. 2009, 61, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Mae, M.; Langel, U. Cell-penetrating peptides as vectors for peptide, protein and oligonucleotide delivery. Curr. Opin. Pharmacol. 2006, 6, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Derossi, D.; Calvet, S.; Trembleau, A.; Brunissen, A.; Chassaing, G.; Prochiantz, A. Cell internalization of the third helix of the antennapedia homeodomain is receptor-independent. J. Biol. Chem. 1996, 271, 18188–18193. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K.; Yoneyama, S.; Murase, O.; Miyajima, K. Transbilayer transport of ions and lipids coupled with mastoparan X translocation. Biochemistry 1996, 35, 8450–8456. [Google Scholar] [CrossRef] [PubMed]
- Pouny, Y.; Rapaport, D.; Mor, A.; Nicolas, P.; Shai, Y. Interaction of antimicrobial dermaseptin and its fluorescently labeled analogues with phospholipid membranes. Biochemistry 1992, 31, 12416–12423. [Google Scholar] [CrossRef] [PubMed]
- Alves, I.D.; Goasdoue, N.; Correia, I.; Aubry, S.; Galanth, C.; Sagan, S.; Lavielle, S.; Chassaing, G. Membrane interaction and perturbation mechanisms induced by two cationic cell penetrating peptides with distinct charge distribution. Biochim. Biophys. Acta 2008, 1780, 948–959. [Google Scholar] [CrossRef] [PubMed]
- Joanne, P.; Galanth, C.; Goasdoue, N.; Nicolas, P.; Sagan, S.; Lavielle, S.; Chassaing, G.; El Amri, C.; Alves, I.D. Lipid reorganization induced by membrane-active peptides probed using differential scanning calorimetry. Biochim. Biophys. Acta 2009, 1788, 1772–1781. [Google Scholar] [CrossRef] [PubMed]
- Derossi, D.; Chassaing, G.; Prochiantz, A. Trojan peptides: The penetratin system for intracellular delivery. Trends Cell Biol. 1998, 8, 84–87. [Google Scholar] [CrossRef]
- Lundberg, P.; Langel, U. A brief introduction to cell-penetrating peptides. J. Mol. Recognit. 2003, 16, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Shai, Y. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by α-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim. Biophys. Acta 1999, 1462, 55–70. [Google Scholar] [CrossRef]
- Yang, L.; Harroun, T.A.; Weiss, T.M.; Ding, L.; Huang, H.W. Barrel-stave model or toroidal model? A case study on melittin pores. Biophys. J. 2001, 81, 1475–1485. [Google Scholar] [CrossRef]
- Deshayes, S.; Morris, M.C.; Divita, G.; Heitz, F. Interactions of primary amphipathic cell penetrating peptides with model membranes: Consequences on the mechanisms of intracellular delivery of therapeutics. Curr. Pharm. Des. 2005, 11, 3629–3638. [Google Scholar] [CrossRef] [PubMed]
- Herce, H.D.; Garcia, A.E.; Litt, J.; Kane, R.S.; Martin, P.; Enrique, N.; Rebolledo, A.; Milesi, V. Arginine-rich peptides destabilize the plasma membrane, consistent with a pore formation translocation mechanism of cell-penetrating peptides. Biophys. J. 2009, 97, 1917–1925. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.T. Macropinocytosis: Searching for an endocytic identity and role in the uptake of cell penetrating peptides. J. Cell. Mol. Med. 2007, 11, 670–684. [Google Scholar] [CrossRef] [PubMed]
- Mayor, S.; Pagano, R.E. Pathways of clathrin-independent endocytosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Falcone, S.; Cocucci, E.; Podini, P.; Kirchhausen, T.; Clementi, E.; Meldolesi, J. Macropinocytosis: Regulated coordination of endocytic and exocytic membrane traffic events. J. Cell Sci. 2006, 119, 4758–4769. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.T.; Sayers, E.J. Cell entry of cell penetrating peptides: Tales of tails wagging dogs. J. Control. Release 2012, 161, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Madani, F.; Lindberg, S.; Langel, U.; Futaki, S.; Graslund, A. Mechanisms of cellular uptake of cell-penetrating peptides. J. Biophys. 2011, 2011, 414729. [Google Scholar] [CrossRef] [PubMed]
- Mueller, J.; Kretzschmar, I.; Volkmer, R.; Boisguerin, P. Comparison of cellular uptake using 22 CPPs in 4 different cell lines. Bioconjug. Chem. 2008, 19, 2363–2374. [Google Scholar] [CrossRef] [PubMed]
- Walrant, A.; Bechara, C.; Alves, I.D.; Sagan, S. Molecular partners for interaction and cell internalization of cell-penetrating peptides: How identical are they? Nanomedicine 2012, 7, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Lundin, P.; Johansson, H.; Guterstam, P.; Holm, T.; Hansen, M.; Langel, U.; El Andaloussi, S. Distinct uptake routes of cell-penetrating peptide conjugates. Bioconjug. Chem. 2008, 19, 2535–2542. [Google Scholar] [CrossRef] [PubMed]
- Duchardt, F.; Fotin-Mleczek, M.; Schwarz, H.; Fischer, R.; Brock, R. A comprehensive model for the cellular uptake of cationic cell-penetrating peptides. Traffic 2007, 8, 848–866. [Google Scholar] [CrossRef] [PubMed]
- Dupont, E.; Prochiantz, A.; Joliot, A. Penetratin story: An overview. Methods Mol. Biol. 2015, 1324, 29–37. [Google Scholar] [PubMed]
- Lindgren, M.; Hallbrink, M.; Prochiantz, A.; Langel, U. Cell-penetrating peptides. Trends Pharmacol. Sci. 2000, 21, 99–103. [Google Scholar] [CrossRef]
- Oehlke, J.; Scheller, A.; Wiesner, B.; Krause, E.; Beyermann, M.; Klauschenz, E.; Melzig, M.; Bienert, M. Cellular uptake of an α-helical amphipathic model peptide with the potential to deliver polar compounds into the cell interior non-endocytically. Biochim. Biophys. Acta 1998, 1414, 127–139. [Google Scholar] [CrossRef]
- Nishi, K.; Saigo, K. Cellular internalization of green fluorescent protein fused with herpes simplex virus protein VP22 via a lipid raft-mediated endocytic pathway independent of caveolae and Rho family GTPases but dependent on dynamin and Arf6. J. Biol. Chem. 2007, 282, 27503–27517. [Google Scholar] [CrossRef] [PubMed]
- Konate, K.; Rydstrom, A.; Divita, G.; Deshayes, S. Everything you always wanted to know about CADY-mediated siRNA delivery* (* but afraid to ask). Curr. Pharm. Des. 2013, 19, 2869–2877. [Google Scholar] [CrossRef] [PubMed]
- Sparks, J.; Slobodkin, G.; Matar, M.; Congo, R.; Ulkoski, D.; Rea-Ramsey, A.; Pence, C.; Rice, J.; McClure, D.; Polach, K.J.; et al. Versatile cationic lipids for siRNA delivery. J. Control. Release 2012, 158, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Sparrow, J.T.; Edwards, V.V.; Tung, C.; Logan, M.J.; Wadhwa, M.S.; Duguid, J.; Smith, L.C. Synthetic peptide-based DNA complexes for nonviral gene delivery. Adv. Drug Deliv. Rev. 1998, 30, 115–131. [Google Scholar] [PubMed]
- Singha, K.; Namgung, R.; Kim, W.J. Polymers in small-interfering RNA delivery. Oligonucleotides 2011, 21, 133–147. [Google Scholar]
- Dassie, J.P.; Liu, X.Y.; Thomas, G.S.; Whitaker, R.M.; Thiel, K.W.; Stockdale, K.R.; Meyerholz, D.K.; McCaffrey, A.P.; McNamara, J.O., II; Giangrande, P.H. Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMA-expressing tumors. Nat. Biotechnol. 2009, 27, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Cuellar, T.L.; Barnes, D.; Nelson, C.; Tanguay, J.; Yu, S.F.; Wen, X.; Scales, S.J.; Gesch, J.; Davis, D.; van Brabant Smith, A.; et al. Systematic evaluation of antibody-mediated siRNA delivery using an industrial platform of thiomab-siRNA conjugates. Nucleic Acids Res. 2015, 43, 1189–1203. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; Halder, S.; Nizak, C.; Krishnan, Y. Recombinant antibody mediated delivery of organelle-specific DNA pH sensors along endocytic pathways. Nanoscale 2014, 6, 1144–1152. [Google Scholar] [CrossRef] [PubMed]
- Regberg, J.; Srimanee, A.; Langel, U. Applications of cell-penetrating peptides for tumor targeting and future cancer therapies. Pharmaceuticals 2012, 5, 991–1007. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Zhang, W.; Shirley, S.A.; Kong, X.; Hellermann, G.R.; Lockey, R.F.; Mohapatra, S.S. Thiolated chitosan/DNA nanocomplexes exhibit enhanced and sustained gene delivery. Pharm. Res. 2007, 24, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, T.; Bravo-Osuna, I.; Vauthier, C.; Ponchel, G.; Loretz, B.; Bernkop-Schnurch, A. Development and in vitro evaluation of a thiomer-based nanoparticulate gene delivery system. Biomaterials 2007, 28, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Noh, S.M.; Park, M.O.; Shim, G.; Han, S.E.; Lee, H.Y.; Huh, J.H.; Kim, M.S.; Choi, J.J.; Kim, K.; Kwon, I.C.; et al. Pegylated poly-l-arginine derivatives of chitosan for effective delivery of siRNA. J. Control. Release 2010, 145, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Li, Z.; Liu, W.; Lam, W.; Sun, P.; Kao, R.Y.; Luk, K.D.; Lu, W.W. Octaarginine-modified chitosan as a nonviral gene delivery vector: Properties and in vitro transfection efficiency. J. Nanopart. Res. 2011, 13, 693–702. [Google Scholar] [CrossRef]
- Park, S.; Jeong, E.J.; Lee, J.; Rhim, T.; Lee, S.K.; Lee, K.Y. Preparation and characterization of nonaarginine-modified chitosan nanoparticles for siRNA delivery. Carbohydr. Polym. 2013, 92, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, M.; Tomaro-Duchesneau, C.; Saha, S.; Kahouli, I.; Prakash, S. Development and characterization of chitosan-PEG-TAT nanoparticles for the intracellular delivery of siRNA. Int. J. Nanomed. 2013, 8, 2041–2052. [Google Scholar]
- Liu, L.; Dong, X.; Zhu, D.; Song, L.; Zhang, H.; Leng, X.G. TAT-LHRH conjugated low molecular weight chitosan as a gene carrier specific for hepatocellular carcinoma cells. Int. J. Nanomed. 2014, 9, 2879–2889. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.Y.; Gu, J.W.; Hou, D.P.; Jing, H.Y.; Wang, J.; Guo, Y.Z.; Katsumi, H.; Sakane, T.; Yamamoto, A. Synthesis of TAT tagged and folate modified N-succinyl-chitosan self-assembly nanoparticles as a novel gene vector. Int. J. Biol. Macromol. 2015, 72, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Nakase, I.; Takeuchi, T.; Tanaka, G.; Futaki, S. Methodological and cellular aspects that govern the internalization mechanisms of arginine-rich cell-penetrating peptides. Adv. Drug Deliv. Rev. 2008, 60, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.; Chou, J.C.; Lee, H.J. Cellular internalization of fluorescent proteins via arginine-rich intracellular delivery peptide in plant cells. Plant Cell Physiol. 2005, 46, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Futaki, S.; Suzuki, T.; Ohashi, W.; Yagami, T.; Tanaka, S.; Ueda, K.; Sugiura, Y. Arginine-rich peptides. An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J. Biol. Chem. 2001, 276, 5836–5840. [Google Scholar] [CrossRef] [PubMed]
- Castanotto, D.; Rossi, J.J. The promises and pitfalls of RNA-interference-based therapeutics. Nature 2009, 457, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Alexis, F.; Pridgen, E.; Molnar, L.K.; Farokhzad, O.C. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol. Pharm. 2008, 5, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Kanasty, R.L.; Whitehead, K.A.; Vegas, A.J.; Anderson, D.G. Action and reaction: The biological response to siRNA and its delivery vehicles. Mol. Ther. 2012, 20, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Petros, R.A.; DeSimone, J.M. Strategies in the design of nanoparticles for therapeutic applications. Nat. Rev. Drug Discov. 2010, 9, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Whitehead, K.A.; Langer, R.; Anderson, D.G. Knocking down barriers: Advances in siRNA delivery. Nat. Rev. Drug Discov. 2009, 8, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Katas, H.; Nik Dzulkefli, N.N.S.; Sahudin, S. Synthesis of a new potential conjugated TAT-peptide-chitosan nanoparticles carrier via disulphide linkage. J. Nanomater. 2012, 2012, 7. [Google Scholar] [CrossRef]
- Xie, W.; Liu, J.; Qiu, M.; Yuan, J.; Xu, A. Design, synthesis and biological activity of cell-penetrating peptide-modified octreotide analogs. J. Pept. Sci. 2010, 16, 105–109. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Layek, B.; Lipp, L.; Singh, J. Cell Penetrating Peptide Conjugated Chitosan for Enhanced Delivery of Nucleic Acid. Int. J. Mol. Sci. 2015, 16, 28912-28930. https://doi.org/10.3390/ijms161226142
Layek B, Lipp L, Singh J. Cell Penetrating Peptide Conjugated Chitosan for Enhanced Delivery of Nucleic Acid. International Journal of Molecular Sciences. 2015; 16(12):28912-28930. https://doi.org/10.3390/ijms161226142
Chicago/Turabian StyleLayek, Buddhadev, Lindsey Lipp, and Jagdish Singh. 2015. "Cell Penetrating Peptide Conjugated Chitosan for Enhanced Delivery of Nucleic Acid" International Journal of Molecular Sciences 16, no. 12: 28912-28930. https://doi.org/10.3390/ijms161226142