
Evodiamine Induces Apoptosis and Inhibits Migration of HCT-116 Human Colorectal Cancer Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
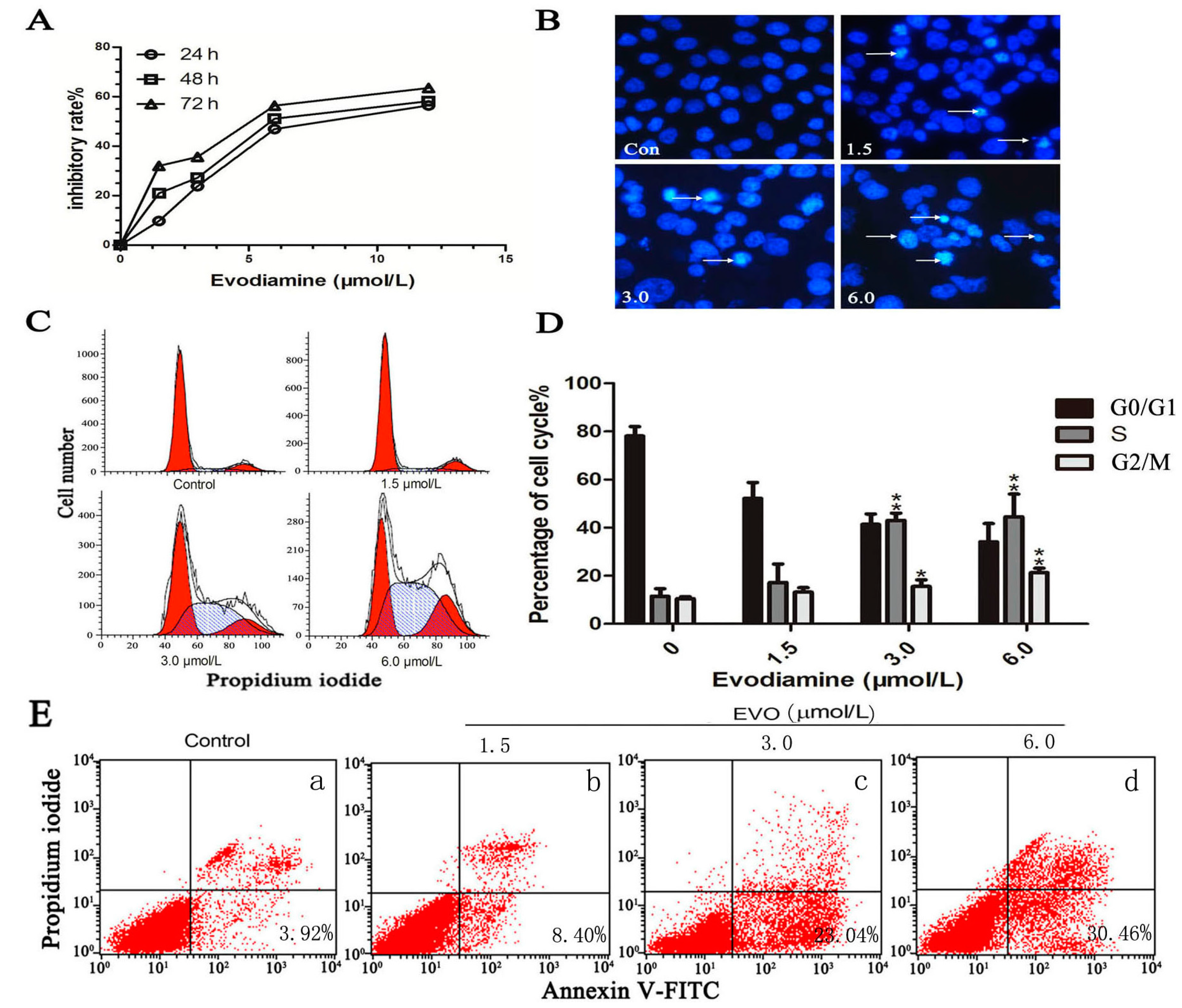
2.1. Evodiamine (EVO) Suppresses Cell Proliferation and Causes Cell Cycle Arrest in HCT-116 Cells
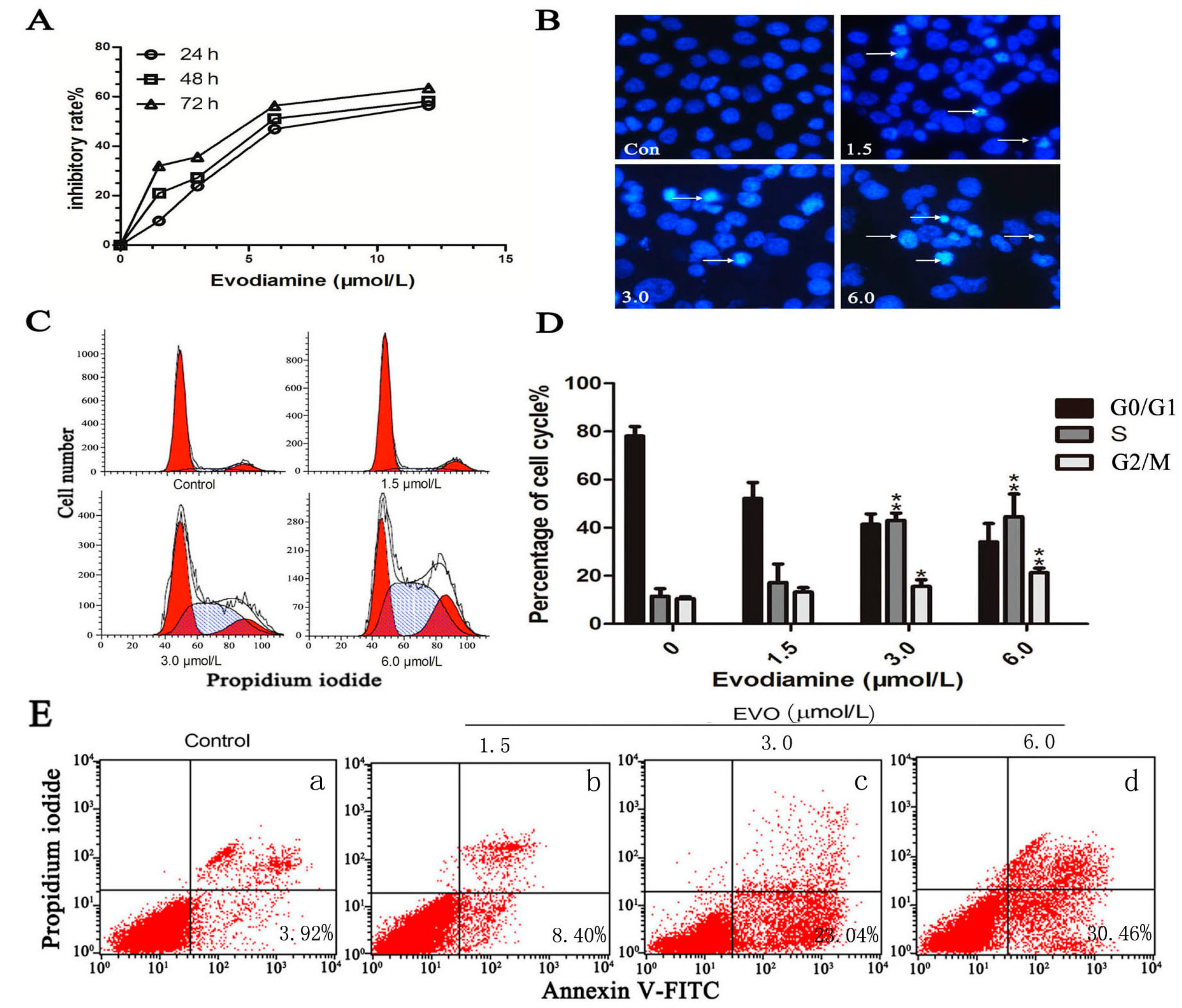
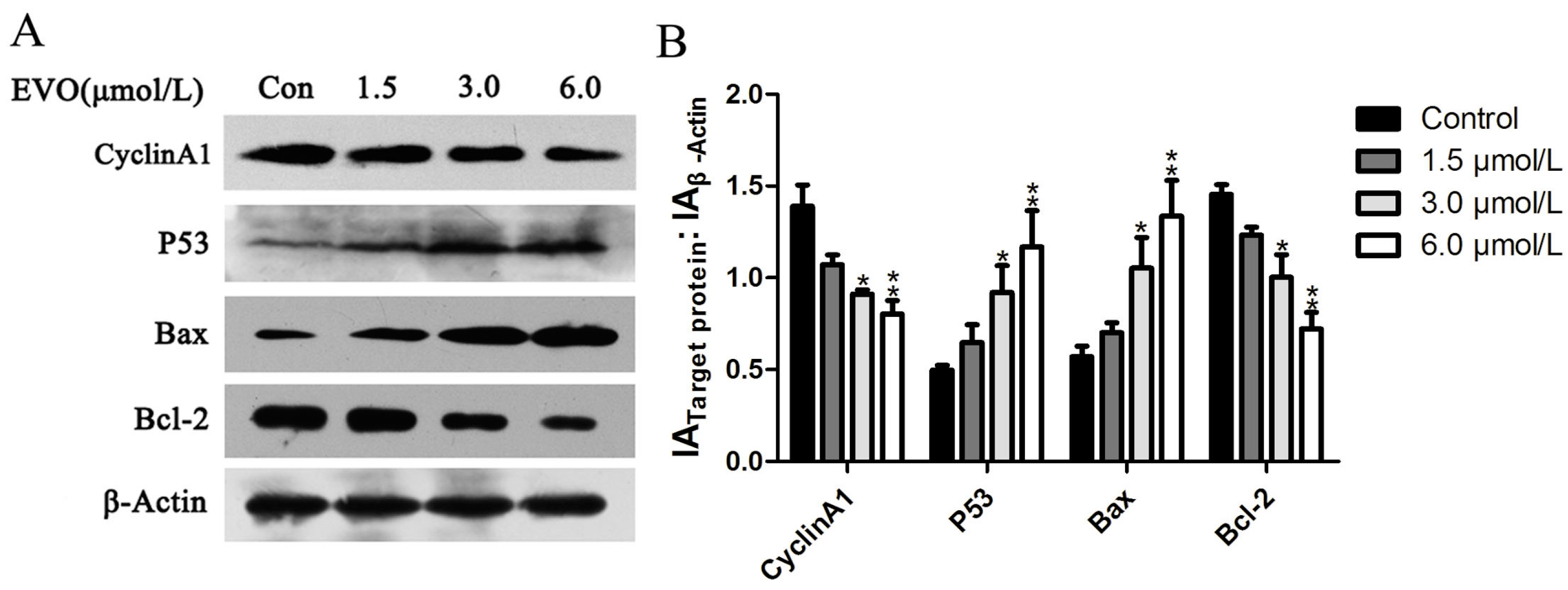
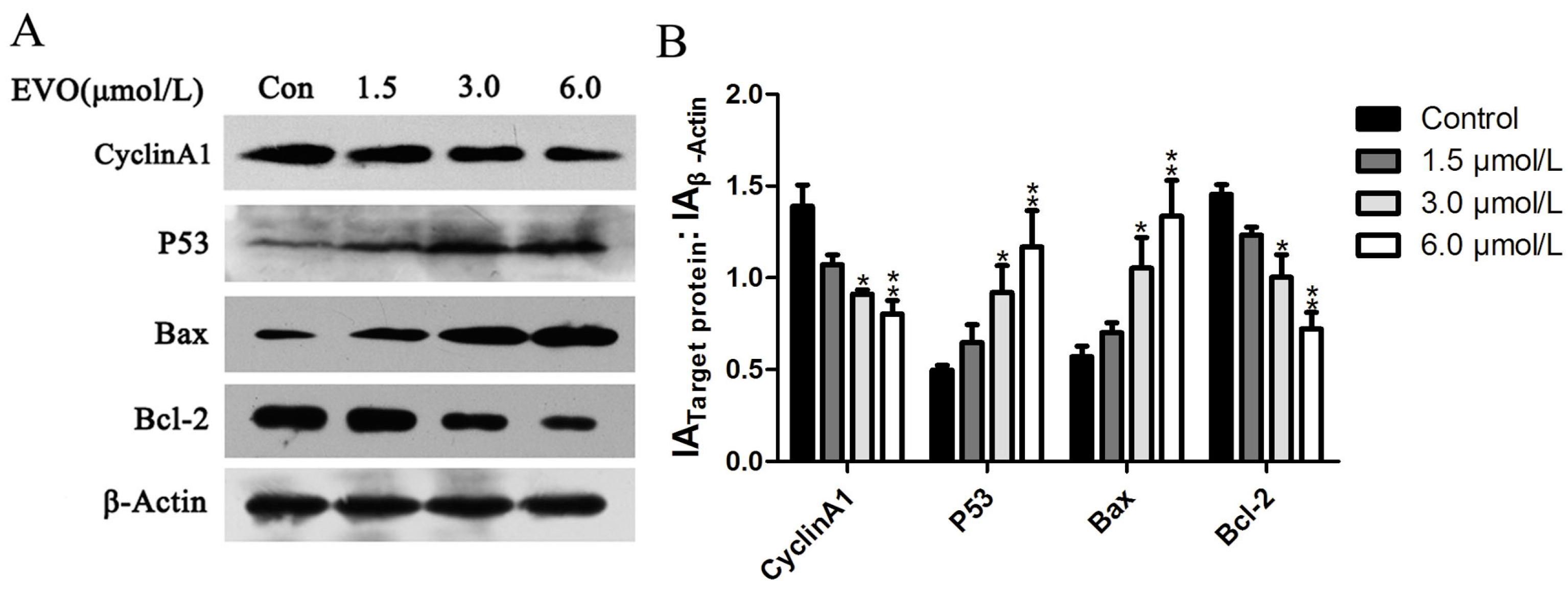
2.2. Effect of EVO on Cell Cycle Regulatory Protein (Cyclin A1), and p53/Bax/Bcl-2 in HCT-116 Cells
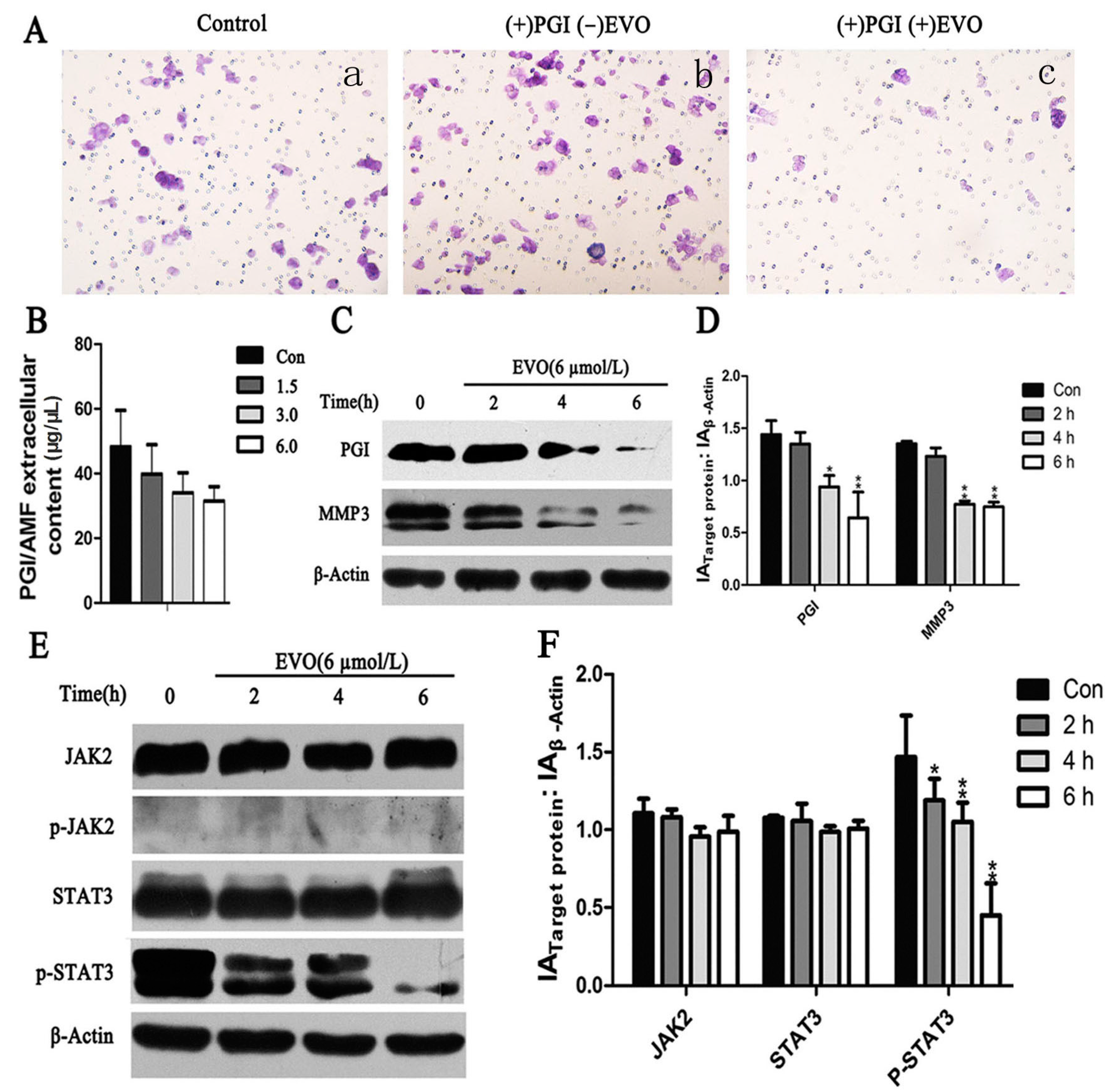
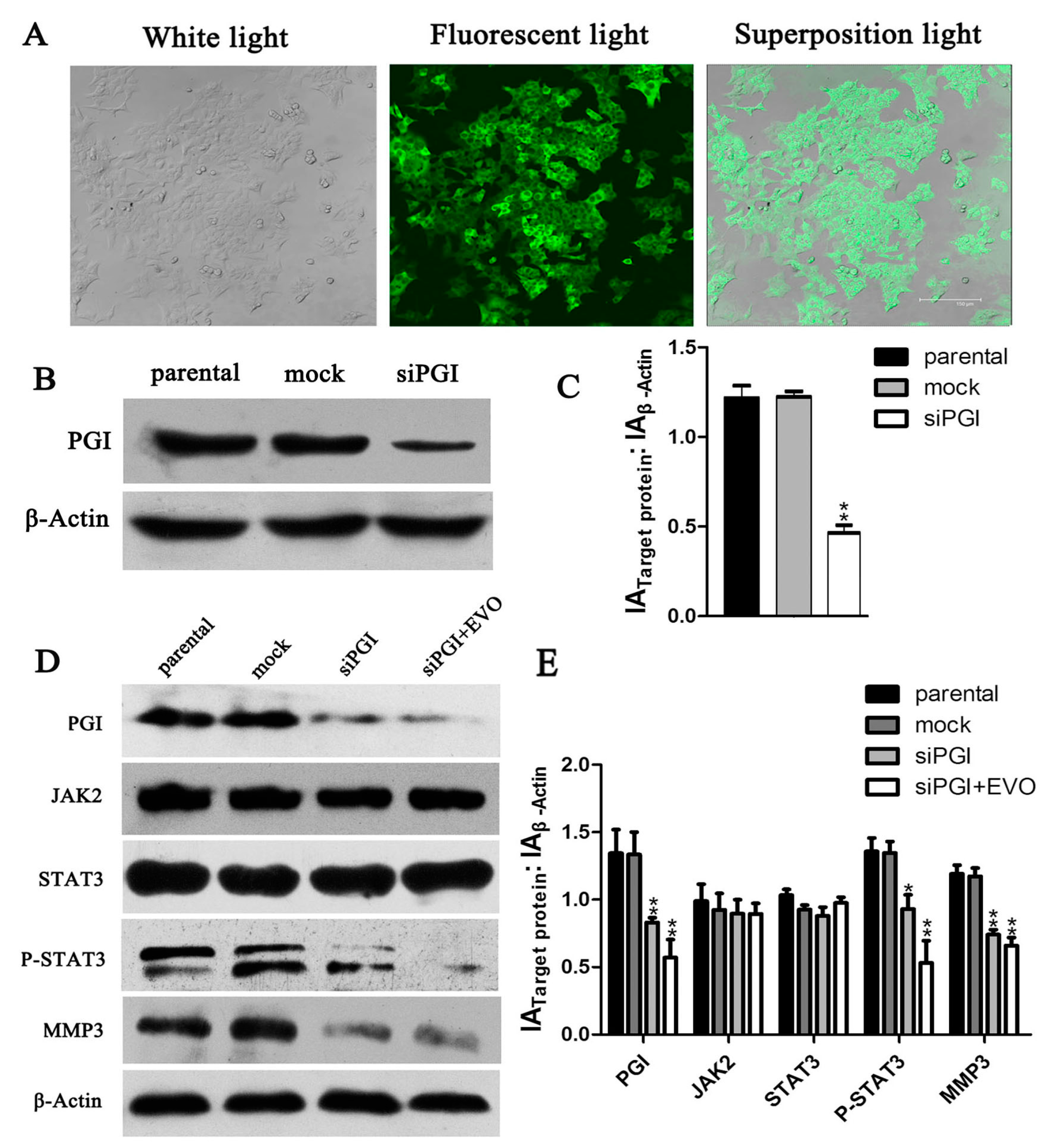
2.3. EVO Suppresses Expression of Phosphoglucose Isomerase (PGI), p-STAT3, MMP3, and PGI-Induced Migration in HCT-116 Cells
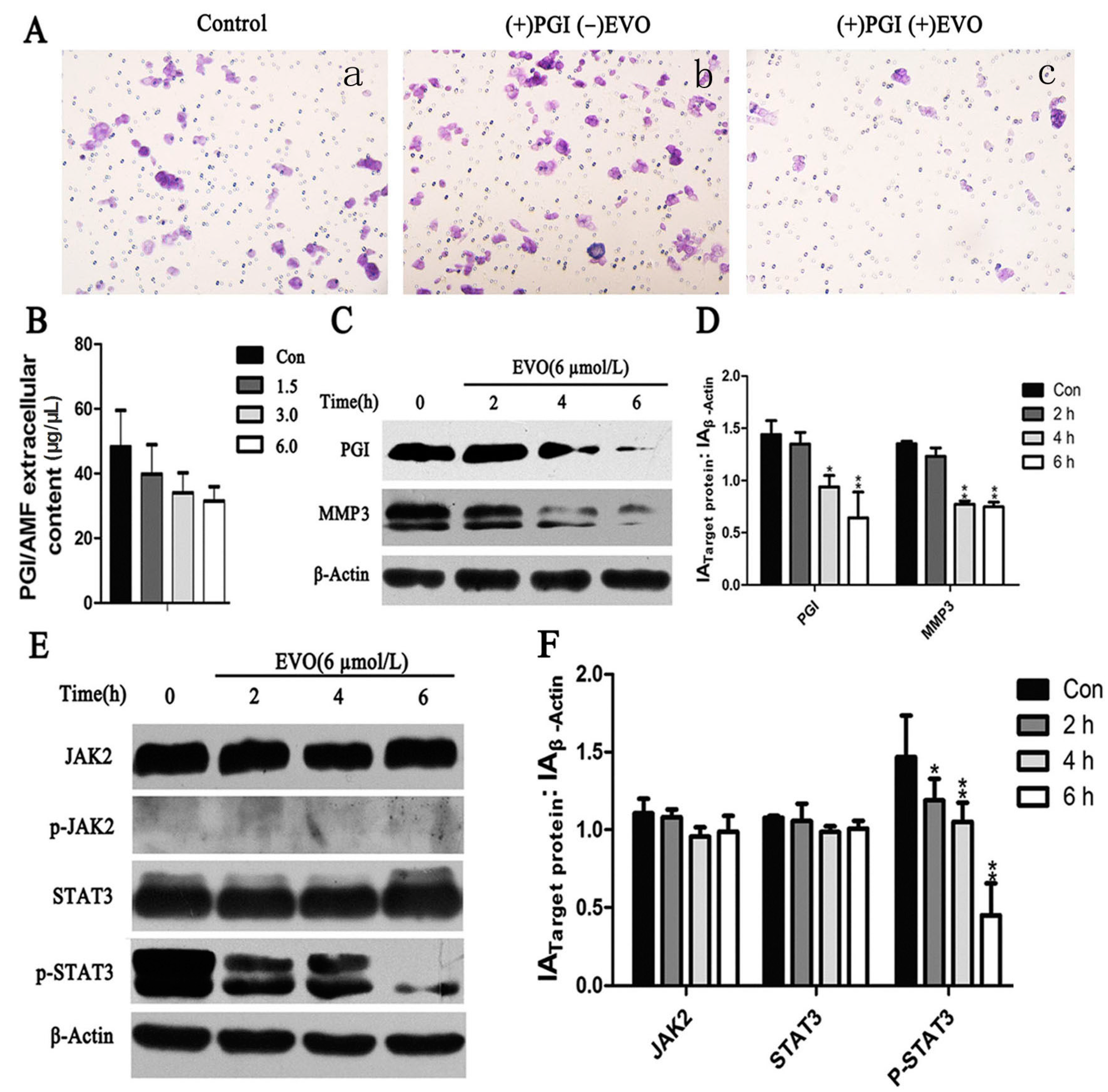
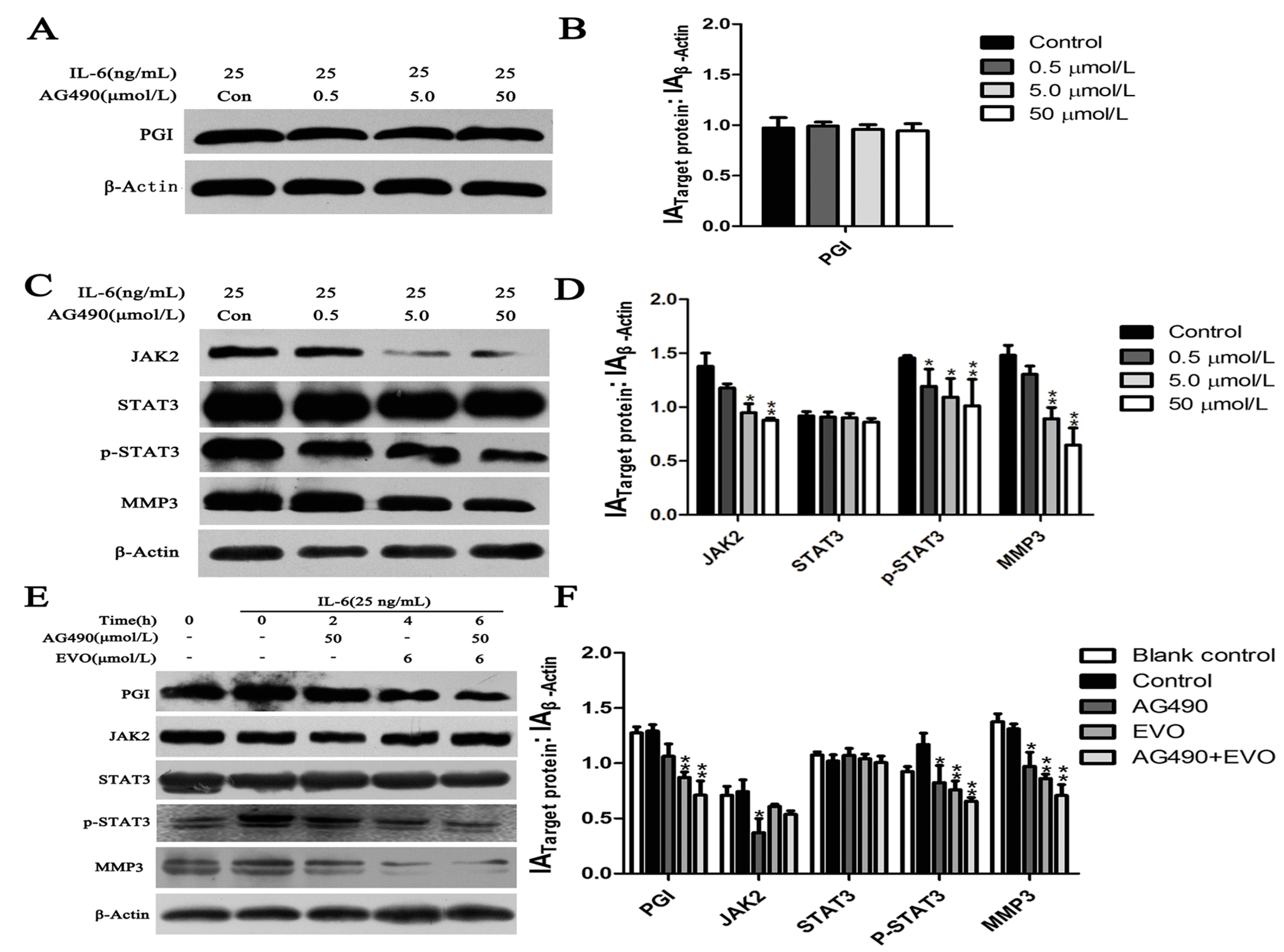
2.4. Effect of the JAK2-Specific Inhibitor AG490 on JAK2/STAT3 Signaling, PGI and MMP3

2.5. The Inhibitory Effects of EVO on PGI, JAK2/STAT3 Signaling, and MMP3 Were Stronger than that of AG490
2.6. EVO Abolishes PGI-Mediated STAT3/MMP3 Signal Transduction

3. Discussion
4. Experimental Section
4.1. Cell Culture
4.2. PGI Silencing
4.3. Reagents and Antibodies
4.4. Cell Viability Analysis
4.5. Cell Cycle and Apoptosis Rate Analysis
4.6. Hoechst Staining
4.7. Transwell Assay
4.8. Immunoblot Assay
4.9. Enzyme-Linked Immunosorbent Assay (ELISA)
4.10. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Swiderska, M.; Choromanska, B.; Dabrowska, E.; Konarzewska-Duchnowska, E.; Choromańska, K.; Szczurko, G.; Myśliwiec, P.; Dadan, J.; Ładny, J.R.; Zwierz, K. The diagnostics of colorectal cancer. Contemp. Oncol. 2014, 18, 1–6. [Google Scholar]
- Gupta, G.P.; Massagué, J. Cancer metastasis: Building a framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Fan, X.; Xu, X.; Yang, X.; Wang, X.; Liang, H.P. Evodiamine induces caspase-dependent apoptosis and S phase arrest in human colon lovo cells. Anticancer Drugs 2010, 21, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, S.; Wang, M.W. Evodiamine-induced human melanoma A375-S2 cell death was mediated by PI3K/Akt/caspase and Fas-L/NF-κB signaling pathways and augmented by ubiquitin-proteasome inhibition. Toxicol. Vitro 2010, 24, 898–904. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wu, L.J.; Tashino, S.; Onodera, S.; Ikejima, T. Reactive oxygen species and nitric oxide regulate mitochondria-dependent apoptosis and autophagy in evodiamine-treated human cervix carcinoma HeLa cells. Free Radic. Res. 2008, 42, 492–504. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Hu, C. Evodiamine: A novel anti-cancer alkaloid from Evodia rutaecarpa. Molecules 2009, 14, 1852–1859. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; He, K.; Zhang, L.; Yu, J. Crizotinib induces PUMA-dependent apoptosis in colon cancer cells. Mol. Cancer Ther. 2013, 12, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Harder, J.M.; Libby, R.T. BBC3 (PUMA) regulates developmental apoptosis but not axonal injury induced death in the retina. Mol. Neurodegener. 2011, 6, 50. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.L.; Liao, M.H.; Lee, J.W.; Shih, W.L. Induction of hepatoma cells migration by phosphoglucose isomerase/autocrine motility factor through the upregulation of matrix metalloproteinase-3. Biochem. Biophys. Res. Commun. 2004, 314, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Niinaka, Y.; Harada, K.; Fujimuro, M.; Oda, M.; Haga, A.; Hosoki, M.; Uzawa, N.; Arai, N.; Yamaguchi, S.; Yamashiro, M.; et al. Silencing of autocrine motility factor induces mesenchymal-to-epithelial transition and suppression of osteosarcoma pulmonary metastasis. Cancer Res. 2010, 70, 9483–9493. [Google Scholar] [CrossRef] [PubMed]
- Gurbuz, V.; Konac, E.; Varol, N.; Yilmaz, A.; Gurocak, S.; Menevse, S.; Sozen, S. Effects of AG490 and S3I-201 on regulation of the JAK/STAT3 signaling pathway in relation to angiogenesis in TRAIL-resistant prostate cancer cells. Oncol. Lett. 2014, 7, 755–763. [Google Scholar] [PubMed]
- Germain, D.; Frank, D.A. Targeting the cytoplasmic and nuclear functions of signal transducers and activators of transcription 3 for cancer therapy. Clin. Cancer Res. 2007, 13, 5665–5669. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Jove, R. The STATs of cancer—New molecular targets come of age. Nat. Rev. Cancer 2004, 4, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Shih, W.L.; Liao, M.H.; Yu, F.L.; Lin, P.Y.; Hsu, H.Y.; Chiu, S.J. AMF/PGI transactivates the MMP-3 gene through the activation of Src-RhoA-phosphatidylinositol 3-kinase signaling to induce hepatoma cell migration. Cancer Lett. 2008, 270, 202–217. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wilson, N.O.; Hibbert, J.M.; Stiles, J.K. STAT3 regulates MMP3 in heme-induced endothelial cell apoptosis. PLoS ONE 2013, 8, e71366. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; McCormick, J.; Connolly, M.; Balogh, E.; Veale, D.J.; Fearon, U. Hypoxia and STAT3 signalling interactions regulate pro-inflammatory pathways in rheumatoid arthritis. Ann. Rheum. Dis. 2014. [Google Scholar] [CrossRef] [PubMed]
- Liotta, L.A.; Mandler, R.; Murano, G.; Katz, D.A.; Gordon, R.K.; Chiang, P.K.; Schiffmann, E. Tumor cell autocrine motility factor. Proc. Natl. Acad. Sci. USA 1986, 83, 3302–3306. [Google Scholar] [CrossRef] [PubMed]
- Kan, S.F.; Yu, C.H.; Pu, H.F.; Hsu, J.M.; Chen, M.J.; Wang, P.S. Anti-proliferative effects of evodiamine on human prostate cancer cell lines DU145 and PC3. J. Cell. Biochem. 2007, 101, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.L.; Li, G.; Moon, D.C.; Lee, C.S.; Woo, M.H.; Lee, E.S.; Jahng, Y.; Chang, H.W.; Lee, S.H.; Son, J.K. Cytotoxicity and DNA topoisomerase inhibitory activity of constituents isolated from the fruits of Evodia officinalis. Arch. Pharm. Res. 2006, 29, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.J.; Kim, E.J.; Kim, S.; Jung, E.M.; Park, J.W.; Jeong, S.H.; Park, S.E.; Yoo, Y.H.; Kwon, T.K. Caspase-dependent and caspase-independent apoptosis induced by evodiamine in human leukemic U937 cells. Mol. Cancer Ther. 2006, 5, 2398–2407. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Wang, X.F.; Zhou, Q.M.; Zhang, T.L.; Lu, Y.Y.; Zhang, H.; Su, S.B. Evodiamine induces apoptosis and inhibits metastasis in MDAMB-231 human breast cancer cells in vitro and in vivo. Oncol. Rep. 2013, 30, 685–694. [Google Scholar] [PubMed]
- Yang, J.; Cai, X.; Lu, W.; Hu, C.; Xu, X.; Yu, Q.; Cao, P. Evodiamine inhibits STAT3 signaling by inducing phosphatase shatterproof 1 in hepatocellular carcinoma cells. Cancer Lett. 2013, 328, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J.; Liloglou, T.; Rogers, S.N.; Brown, J.S.; Vaughan, E.D.; Lowe, D.; Field, J.K.; Risk, J.M. Promoter methylation of P16, RARβ, E-cadherin, cyclin A1 and cytoglobin in oral cancer: Quantitative evaluation using pyrosequencing. Br. J. Cancer 2006, 94, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Faik, P.; Walker, J.I.; Redmill, A.A.; Morgan, M.J. Mouse glucose-6-phosphate isomerase and neuroleukin have identical 3′ sequences. Nature 1988, 332, 455–457. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Takehana, K.; Date, M.; Shinozaki, T.; Raz, A. Tumor cell autocrine motility factor is the neuroleukin/phosphohexose isomerase polypeptide. Cancer Res. 1996, 56, 2960–2963. [Google Scholar] [PubMed]
- Turkson, J.; Jove, R. STAT proteins: Novel molecular targets for cancer drug discovery. Oncogene 2000, 19, 6613–6626. [Google Scholar] [CrossRef] [PubMed]
- Turkson, J.; Bowman, T.; Garcia, R.; Caldenhoven, E.; de Groot, R.P.; Jove, R. STAT3 activation by Src induces specific gene regulation and is required for cell transformation. Mol. Cell. Biol. 1998, 18, 2545–2552. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Han, Z.C. STAT3: A critical transcription activator in angiogenesis. Med. Res. Rev. 2008, 28, 185–200. [Google Scholar] [CrossRef] [PubMed]
- Devarajan, E.; Huang, S. STAT3 as a central regulator of tumor metastases. Curr. Mol. Med. 2009, 9, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Shih, W.L.; Liao, M.H.; Lin, P.Y.; Chang, C.I.; Cheng, H.L.; Yu, F.L.; Lee, J.W. PI 3-kinase/Akt and STAT3 are required for the prevention of TGF-β-induced Hep3B cell apoptosis by autocrine motility factor/phosphoglucose isomerase. Cancer Lett. 2010, 290, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.Y.; Park, S.H.; Min, H.Y.; Park, H.J.; Lee, S.K. Anti-proliferative effects of evodiamine in human lung cancer cells. J. Cancer Prev. 2014, 19, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.C.; Yu, C.H.; Wang, S.W.; Pu, H.F.; Kan, S.F.; Lin, L.C.; Chi, C.W.; Ho, L.L.; Lee, C.H.; Wang, P.S. Anti-proliferative effects of evodiamine on human thyroid cancer cell line ARO. J. Cell. Biochem. 2010, 110, 1495–1503. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, L.-C.; Li, J.; Liao, K.; Luo, N.; Shi, Q.-Q.; Feng, Z.-Q.; Chen, D.-L. Evodiamine Induces Apoptosis and Inhibits Migration of HCT-116 Human Colorectal Cancer Cells. Int. J. Mol. Sci. 2015, 16, 27411-27421. https://doi.org/10.3390/ijms161126031
Zhao L-C, Li J, Liao K, Luo N, Shi Q-Q, Feng Z-Q, Chen D-L. Evodiamine Induces Apoptosis and Inhibits Migration of HCT-116 Human Colorectal Cancer Cells. International Journal of Molecular Sciences. 2015; 16(11):27411-27421. https://doi.org/10.3390/ijms161126031
Chicago/Turabian StyleZhao, Lv-Cui, Jing Li, Ke Liao, Nian Luo, Qing-Qiang Shi, Zi-Qiang Feng, and Di-Long Chen. 2015. "Evodiamine Induces Apoptosis and Inhibits Migration of HCT-116 Human Colorectal Cancer Cells" International Journal of Molecular Sciences 16, no. 11: 27411-27421. https://doi.org/10.3390/ijms161126031