
Morphological Characters and Transcriptome Profiles Associated with Black Skin and Red Skin in Crimson Snapper (Lutjanus erythropterus)

Abstract
:1. Introduction
2. Results
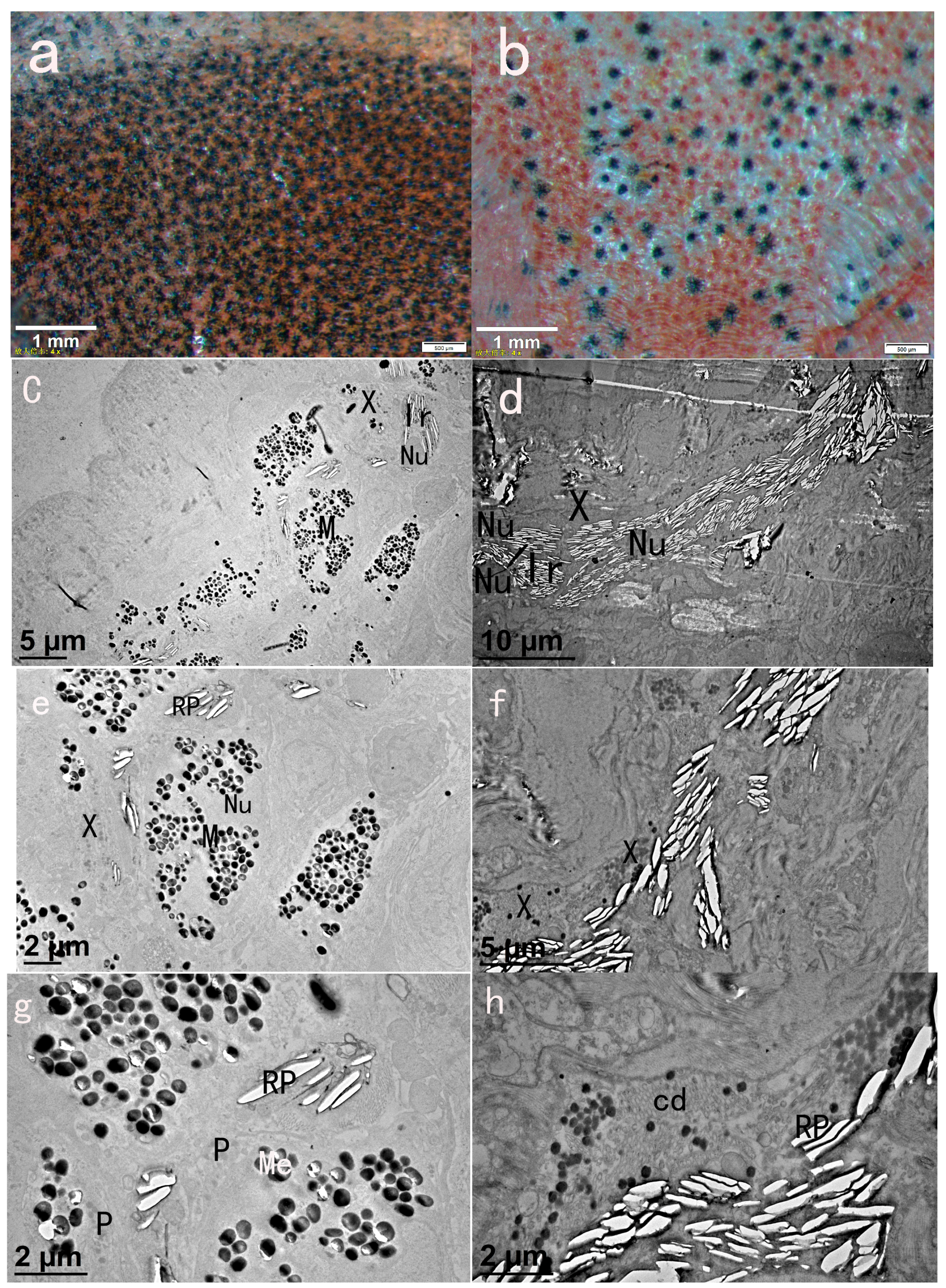
2.1. Chromatophore Distribution of Black Skin and Red Skin
2.2. Sequencing and Assembly of the Black Color Skin and Red Color Skin Transcriptomes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Back Skin | Red Skin |
|---|---|---|
| Total raw reads | 54,232,958 | 52,873,586 |
| Total clean reads | 51,438,110 | 49,531,098 |
| Total clean nucleotides (bp) | 4,629,429,900 | 4,457,798,820 |
| Q20 percentage | 94.41% | 94.35% |
| N percentage | 0.00% | 0.00% |
| GC percentage | 49.85% | 50.73% |
| Total length | 104,322,550 | 89,828,794 |
| N50 (bp) | 881 | 888 |
| Unigenes | 142,792 | 122,508 |
| Mean length (bp) | 622 | 613 |
2.3. Genes Highly Expressed in Fish Skins
| Back-Skin RPKM * | Gene Name | Red-Skin RPKM | Gene Name |
|---|---|---|---|
| 16,496.2127 | 40S ribosomal protein S25-like | 13,630.6212 | Creatine kinase M-type |
| 12,175.4483 | hypothetical protein LOC100078788 | 13,504.9082 | Fructose-bisphosphate aldolase |
| 10,046.855 | ribosomal protein L27a | 12,425.8508 | myosin light chain 3 |
| 10,041.9354 | 40S ribosomal protein S15 | 9826.1597 | cytochrome c oxidase subunit 1 |
| 9797.9735 | uncharacterized protein LOC100849392 | 8020.0354 | NADH dehydrogenase subunit 5 |
| 8880.9287 | putative ribosomal protein L14 | 6576.8346 | parvalbumin |
| 8046.257 | ribosomal protein L18 | 9305.5688 | 40S ribosomal protein S25-like |
| 914.1748 | unnamed protein product | 6265.6913 | 60S ribosomal protein L41 |
| 7404.5547 | 60S ribosomal protein L8 | 5994.7734 | ribosomal protein L27a |
| 7289.4817 | unnamed protein product | 5751.3138 | myosin light chain 2 polypeptide |
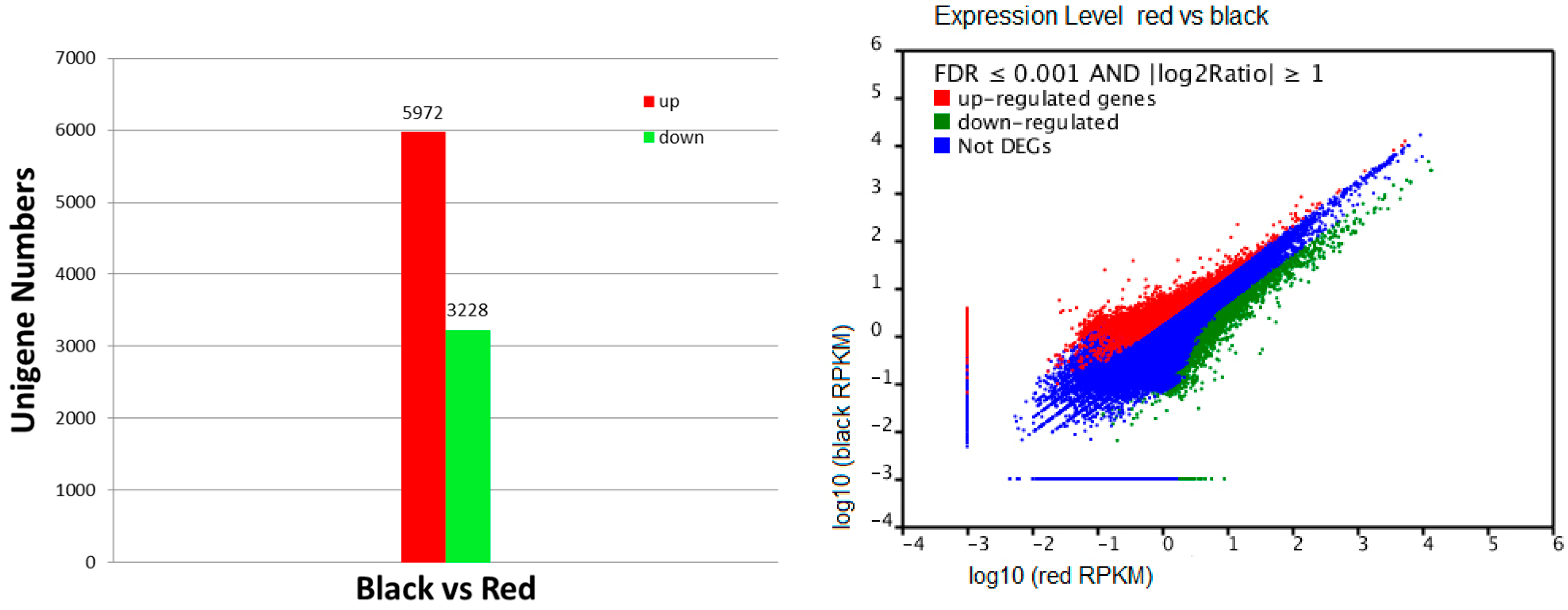
2.4. Recognition of Differentially Transcribed Genes (DTGs) in Black Skin versus Red Skin
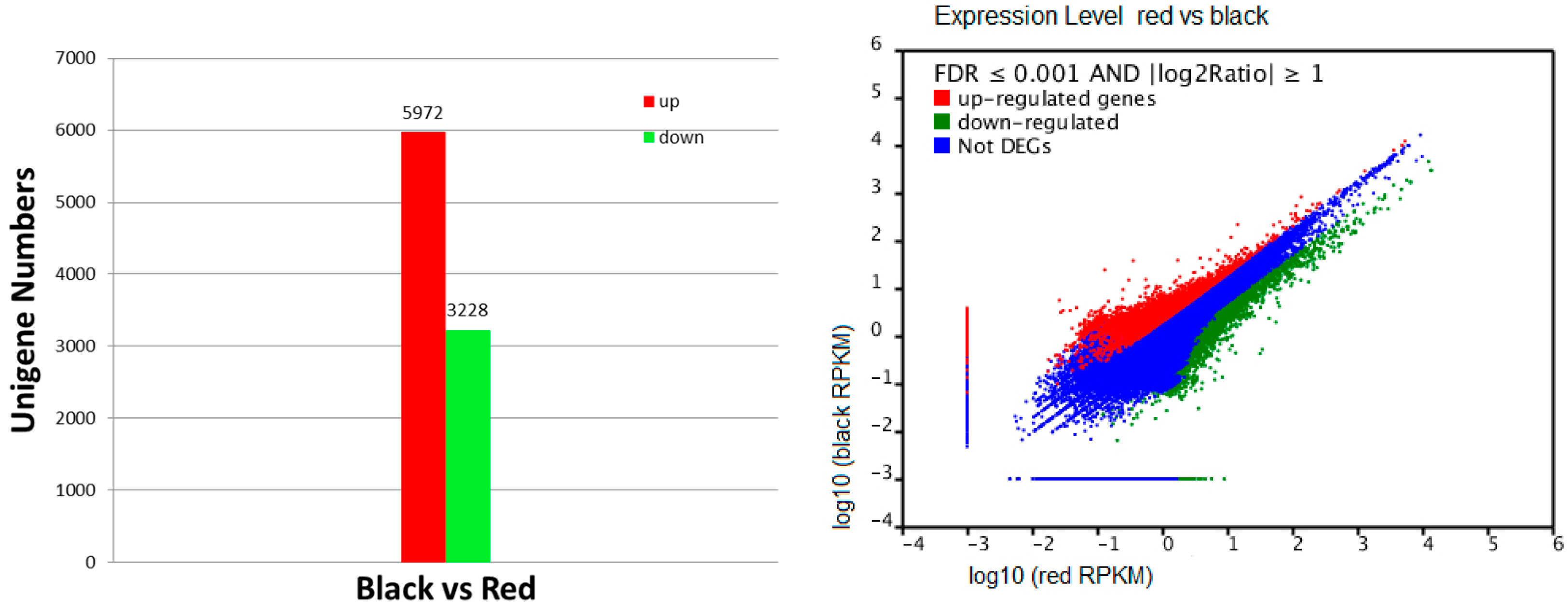
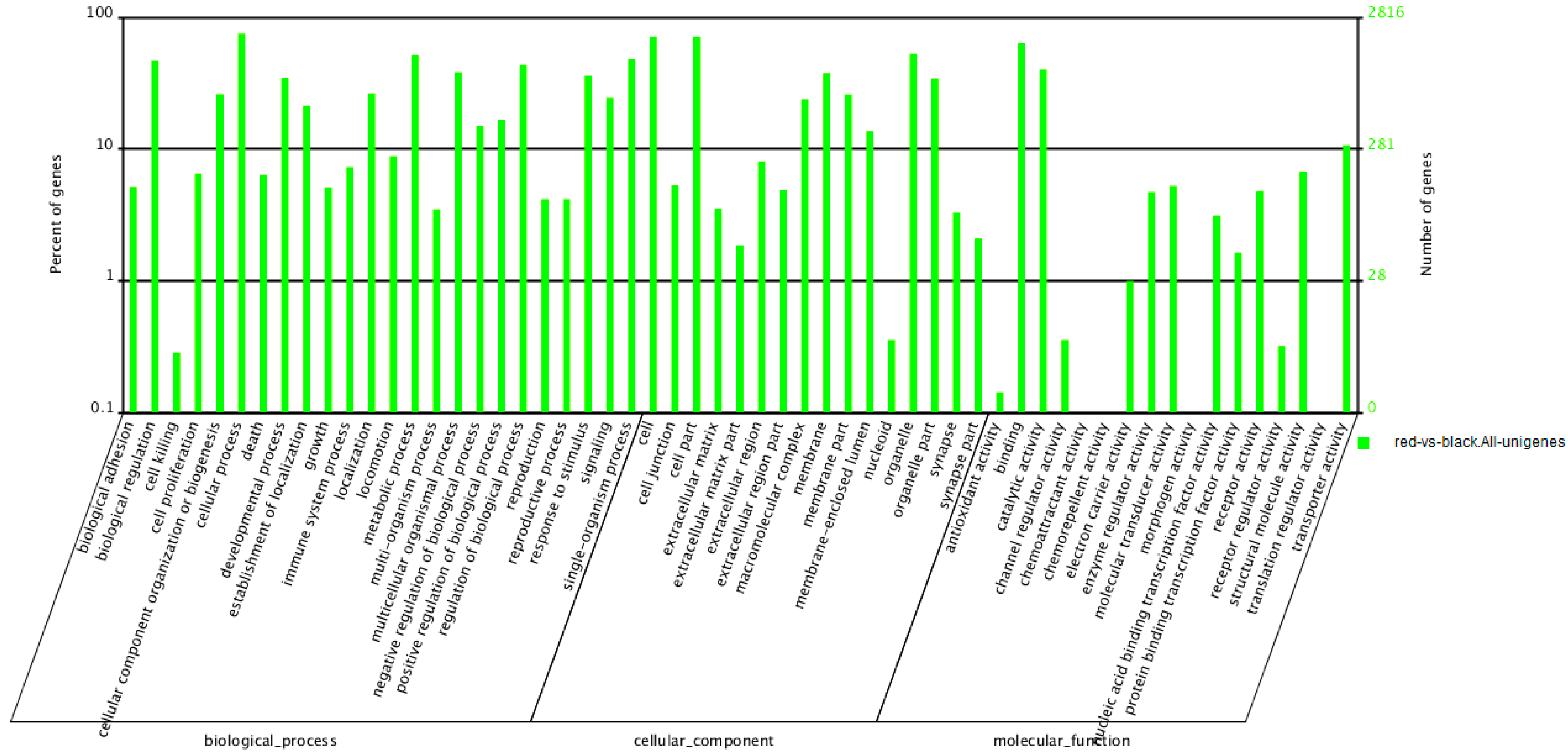
2.5. Functional Enrichment of Differentially Transcribed Genes (DTGs)

| Pathway | DTGs * with Pathway | q-Value | Pathway ID |
|---|---|---|---|
| Dilated cardiomythy | 301 | 1.04 × 10−65 | ko05414 |
| Hypertrophic cardiomyopathy(HCM) | 291 | 8.36 × 10−63 | ko05410 |
| Cardiac muscle contraction | 156 | 2.02 × 10−24 | ko04260 |
| Alzheimer’s disease | 150 | 2.22 × 10−20 | ko05010 |
| Oxidative phosphorylation | 75 | 2.85 × 10−18 | ko00190 |
| Parkinson’s disease | 80 | 1.72× 10−17 | ko05012 |
| Calcium signaling pathway | 150 | 3.53 × 10−11 | ko04020 |
| Proteasome | 28 | 5.07 × 10−9 | ko03050 |
| Huntington’s disease | 115 | 1.21 × 10−7 | ko05016 |
| Viral myocarditis | 118 | 3.68 × 10−7 | ko05416 |
| Vascular smooth muscle contraction | 149 | 4.29 × 10−6 | ko04270 |
| Focal adhesion | 202 | 1.32 × 10−5 | ko04510 |
| Amoebiasis | 153 | 1.04 × 10−4 | ko05146 |
| Arrhythmogenic right ventricular cardiomyopathy | 75 | 2.47 × 10−4 | ko05412 |
| Metabolic pathways | 400 | 2.70 × 10−4 | ko01100 |
| Regulation of actin cytoskeleton | 220 | 3.05 × 10−4 | ko04810 |
| Protein digestion and absorption | 79 | 3.87 × 10−4 | ko04974 |
| Tight junction | 153 | 8.05 × 10−4 | ko04530 |
| Glycolysis/Gluconeogenesis | 34 | 8.66 × 10−4 | ko00010 |
| Salmonella infection | 120 | 1.66 × 10−3 | ko05132 |
| Phenylalanine, tyrosine and tryptophan biosynthesis | 8 | 1.66 × 10−3 | ko00400 |
| Arginine and proline metablism | 30 | 4.50 × 10−3 | ko00330 |
| Citrate cycle (TCA cycle) | 24 | 4.80 × 10−3 | ko00020 |
| Phenylalanine metabolism | 13 | 5.16 × 10−3 | ko00360 |
| Alanine, aspartate and glutamate metabolism | 21 | 9.60 × 10−3 | ko00250 |
| Tyrosine metabolism | 18 | 2.88 × 10−2 | ko00350 |
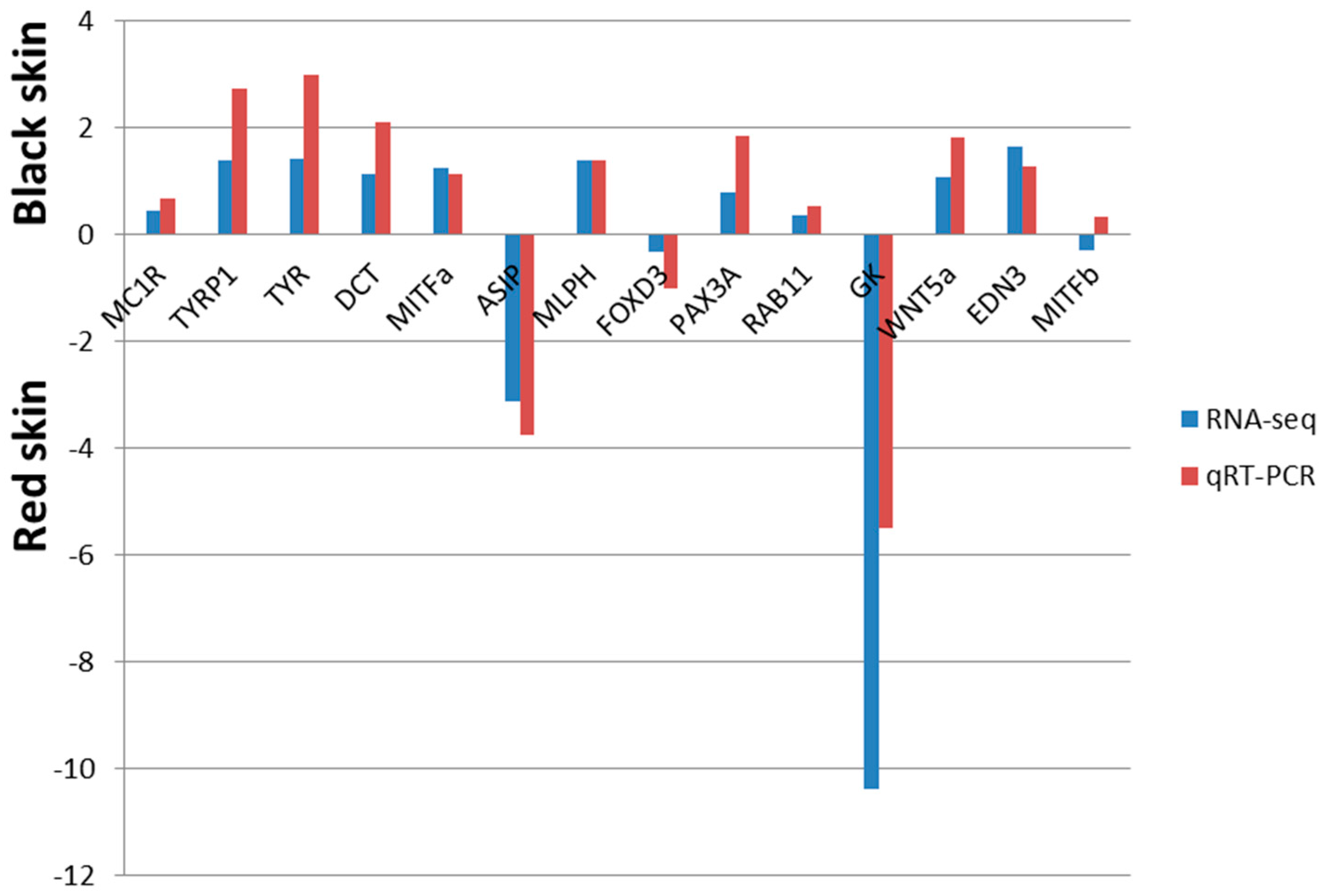
2.6. Differential Expression of Known Pigmentation Genes
2.7. Confirmation of RNA-Seq Identified DTGs by Quantiative Real-Time PCR (qRT-PCR)
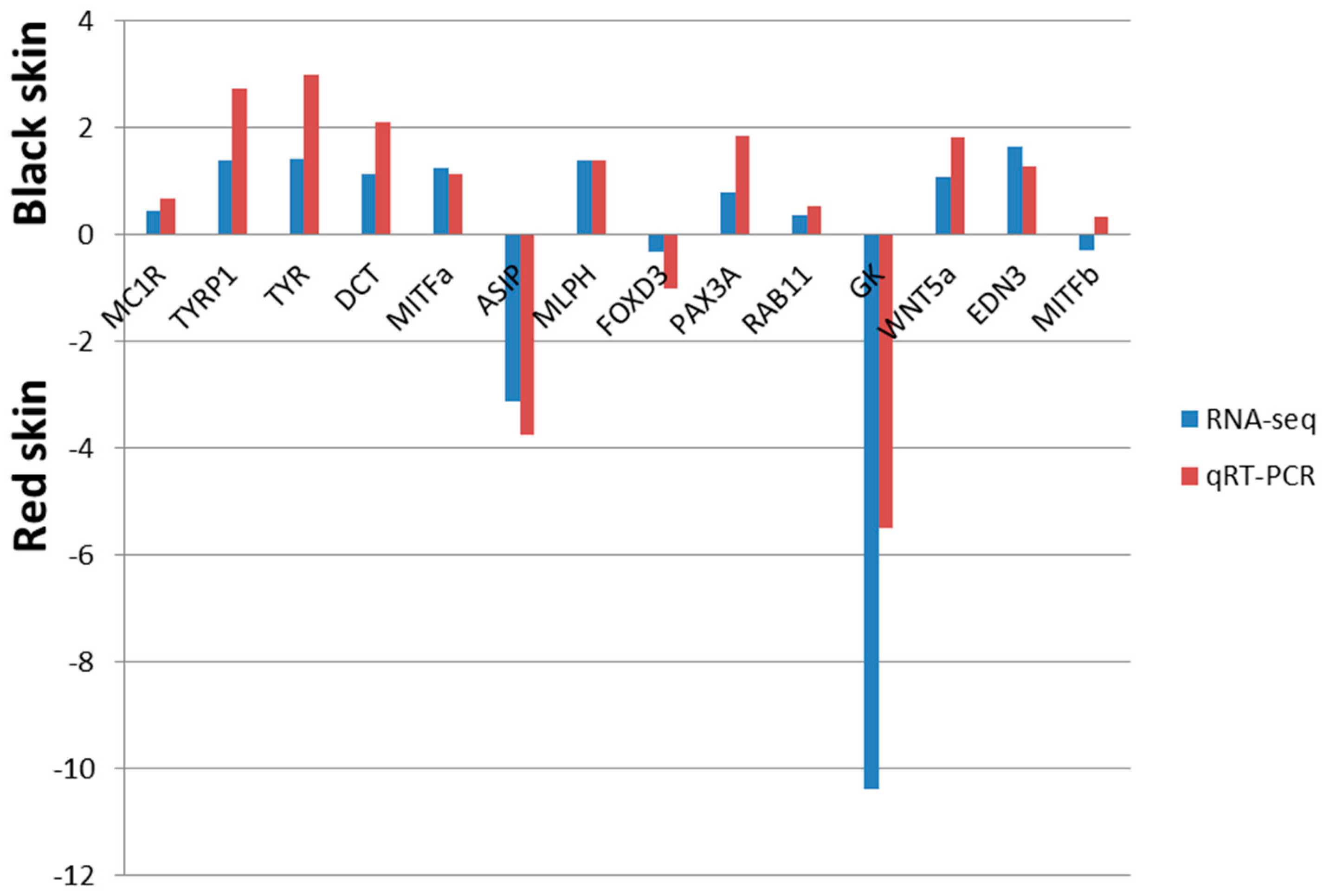
3. Discussion
4. Experimental Section
4.1. Samples for Transmission Electron Microscopy (TEM) Observation and RNA Extraction
4.2. Transcriptome Analysis
4.3. Quantitative Real-Time PCR Validate
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Roberts, R.B.; Ser, J.R.; Kocher, T.D. Sexual conflict resolved by invasion of a novel sex determiner in Lake Malawi cichlid fishes. Science 2009, 326, 998–1001. [Google Scholar] [CrossRef] [PubMed]
- Parichy, D.M. Evolution of danio pigment pattern development. Heredity 2006, 97, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wachholtz, M.; Wang, J.; Liao, X.; Lu, G. Analysis of the skin transcriptome in two oujiang color varieties of common carp. PLoS ONE 2014, 9, e90074. [Google Scholar] [CrossRef] [PubMed]
- Parichy, D.M. Pigment patterns: Fish in stripes and spots. Curr. Biol. 2003, 13, 947–950. [Google Scholar] [CrossRef]
- Guo, H.; Huang, B.; Qi, F.; Zhang, S. Distribution and ultrastructure of pigment cells in the skins of normal and albino adult turbot, Scophthalmus maximus. Chin. J. Oceanol. Limnol. 2007, 25, 199–208. [Google Scholar] [CrossRef]
- Hawkes, J.W. The structure of fish skin. Cell Tissue Res. 1974, 149, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Kuriyama, T.; Miyaji, K.; Sugimoto, M.; Hasegawa, M. Ultrastructure of the dermal chromatophores in a lizard (Scincidae: Plestiodon latiscutatus) with conspicuous body and tail coloration. Zool. Sci. 2006, 23, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Zarnescu, O. Ultrastructure of the skin melanophores and iridophores in paddlefish, Polyodon spathula. Micron 2007, 38, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Failde, L.D.; Bermudez, R.; Vigliano, F.; Coscelli, G.A.; Quiroga, M.I. Morphological, immunohistochemical and ultrastructural characterization of the skin of turbot (Psetta maxima L.). Tissue Cell 2014, 46, 334–342. [Google Scholar] [CrossRef]
- Newton, R.A.; Smit, S.E.; Barnes, C.C.; Pedley, J.; Parsons, P.G.; Sturm, R.A. Activation of the cAMP pathway by variant human MC1R alleles expressed in HEK and in melanoma cells. Peptides 2005, 26, 1818–1824. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, H.E. Genetics, development and evolution of adaptive pigmentation in vertebrates. Heredity 2006, 97, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, H.E.; Hirschmann, R.J.; Bundey, R.A.; Insel, P.A.; Crossland, J.P. A single amino acid mutation contributes to adaptive beach mouse color pattern. Science 2006, 313, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Rieder, S.; Taourit, S.; Mariat, D.; Langlois, B.; Guerin, G. Mutations in the agouti (ASIP), the extension (MC1R), and the brown (TYRP1) loci and their association to coat color phenotypes in horses (Equus caballus). Mamm. Genome 2001, 12, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Gratten, J.; Beraldi, D.; Lowder, B.V.; McRae, A.F.; Visscher, P.M.; Pemberton, J.M.; Slate, J. Compelling evidence that a single nucleotide substitution in TYRP1 is responsible for coat-colour polymorphism in a free-living population of Soay sheep. Proc. Biol. Sci. R. Soc. 2007, 274, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Dunn, K.J.; Brady, M.; Ochsenbauer-Jambor, C.; Snyder, S.; Incao, A.; Pavan, W.J. WNT1 and WNT3a promote expansion of melanocytes through distinct modes of action. Pigment Cell Res. Spons. Eur. Soc. Pigment Cell Res. Int. Pigment Cell Soc. 2005, 18, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Sefc, K.M.; Brown, A.C.; Clotfelter, E.D. Carotenoid-based coloration in cichlid fishes. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2014, 173C, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Higdon, C.W.; Mitra, R.D.; Johnson, S.L. Gene expression analysis of zebrafish melanocytes, iridophores, and retinal pigmented epithelium reveals indicators of biological function and developmental origin. PLoS ONE 2013, 8, e67801. [Google Scholar] [CrossRef] [PubMed]
- Ng, A.; Uribe, R.A.; Yieh, L.; Nuckels, R.; Gross, J.M. Zebrafish mutations in gart and paics identify crucial roles for de novo purine synthesis in vertebrate pigmentation and ocular development. Development 2009, 136, 2601–2611. [Google Scholar] [CrossRef] [PubMed]
- Fan, R.; Xie, J.; Bai, J.; Wang, H.; Tian, X.; Bai, R.; Jia, X.; Yang, L.; Song, Y.; Herrid, M.; et al. Skin transcriptome profiles associated with coat color in sheep. BMC Genom. 2013, 14, 389. [Google Scholar] [CrossRef] [PubMed]
- Croucher, P.J.; Brewer, M.S.; Winchell, C.J.; Oxford, G.S.; Gillespie, R.G. De novo characterization of the gene-rich transcriptomes of two color-polymorphic spiders, Theridion grallator and T. californicum (Araneae: Theridiidae), with special reference to pigment genes. BMC Genom. 2013, 14, 862. [Google Scholar] [CrossRef] [PubMed]
- Yim, K.M.; Brewer, M.S.; Miller, C.T.; Gillespie, R.G. Comparative transcriptomics of maturity-associated color change in Hawaiian spiders. J. Hered. 2014, 105 (Suppl. S1), 771–781. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, P.; Hansson, B.; Kraaijeveld, K.; de Knijff, P.; Svensson, E.I.; Wellenreuther, M. De novo transcriptome of Ischnura elegans provides insights into sensory biology, colour and vision genes. BMC Genom. 2014, 15, 808. [Google Scholar] [CrossRef] [PubMed]
- Hou, R.; Bao, Z.M.; Wang, S.; Su, H.L.; Li, Y.; Du, H.X.; Hu, J.J.; Wang, S.; Hu, X.L. Transcriptome Sequencing and de Novo Analysis for Yesso Scallop (Patinopecten yessoensis) Using 454 GS FLX. PLoS ONE 2011, 6, e21560. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhang, S.; Xu, J.; Feng, J.; Mahboob, S.; Al-Ghanim, K.A.; Sun, X.; Xu, P. Comparative transcriptome analysis reveals the genetic basis of skin color variation in common carp. PLoS ONE 2014, 9, e108200. [Google Scholar] [CrossRef] [PubMed]
- Henning, F.; Jones, J.C.; Franchini, P.; Meyer, A. Transcriptomics of morphological color change in polychromatic Midas cichlids. BMC Genom. 2013, 14, 171. [Google Scholar] [CrossRef] [PubMed]
- Russ, G.R.; Alcala, A.C. Effects of intense fishing pressure on an assemblage of coral reef fishes. Mar. Ecol. Prog. Ser. 1989, 56, 13–27. [Google Scholar] [CrossRef]
- Wang, Z.; Guo, Y.; Tan, W.; Li, L.; Tang, E.; Liu, C.; Liu, Y. DNA barcoding, phylogenetic relationships and speciation of snappers (genus Lutjanus). Sci. China Life Sci. 2010, 53, 1025–1030. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Wang, Z.; Liu, C.; Liu, L.; Liu, Y. Phylogenetic relationships of South China Sea snappers (genus Lutjanus; family Lutjanidae) based on mitochondrial DNA sequences. Mar. Biotechnol. 2007, 9, 682–688. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.L.; Cribb, T.H. Phylogenetic relationships of some common Indo-Pacific snappers (Perciformes: Lutjanidae) based on mitochondrial DNA sequences, with comments on the taxonomic position of the Caesioninae. Mol. Phylogenet. Evolut. 2007, 44, 450–460. [Google Scholar] [CrossRef] [PubMed]
- FishBase. Available online: http://www.fishbase.org/home.htm (accessed on 5 June 2015).
- Djurdjevic, I.; Kreft, M.E.; Susnik-Bajec, S. Comparison of pigment cell ultrastructure and organisation in the dermis of marble trout and brown trout, and first description of erythrophore ultrastructure in salmonids. J. Anat. 2015, 227, 583–595. [Google Scholar] [CrossRef] [PubMed]
- Ensemble. Available online: http://asia.ensembl.org/index.html?redirect=no (accessed on 25 June 2015).
- Li, X.M.; Song, Y.N.; Xiao, G.B.; Zhu, B.H.; Xu, G.C.; Sun, M.Y.; Xiao, J.; Mahboob, S.; Al-Ghanim, K.A.; Sun, X.W.; et al. Gene Expression Variations of Red-White Skin Coloration in Common Carp (Cyprinus carpio). Int. J. Mol. Sci. 2015, 16, 21310–21329. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Oliver, E.R.; Saunders, T.L.; Tarle, S.A.; Glaser, T. Ribosomal protein L24 defect in belly spot and tail (Bst), a mouse Minute. Development 2004, 131, 3907–3920. [Google Scholar] [CrossRef] [PubMed]
- Skarnes, W.C.; Rosen, B.; West, A.P.; Koutsourakis, M.; Bushell, W.; Iyer, V.; Mujica, A.O.; Thomas, M.; Harrow, J.; Cox, T. A conditional knockout resource for the genome-wide study of mouse gene function. Nature 2011, 474, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Curran, K.; Lister, J.A.; Kunkel, G.R.; Prendergast, A.; Parichy, D.M.; Raible, D.W. Interplay between Foxd3 and MITF regulates cell fate plasticity in the zebrafish neural crest. Dev. Biol. 2010, 344, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Lan, F.; Zheng, Z.; Xie, F.; Han, J.; Dong, L.; Xie, Y.; Zheng, F. Akt2 kinase suppresses glyceraldehyde-3-phosphate dehydrogenase (GAPDH)-mediated apoptosis in ovarian cancer cells via phosphorylating GAPDH at threonine 237 and decreasing its nuclear translocation. J. Biol. Chem. 2011, 286, 42211–42220. [Google Scholar] [CrossRef] [PubMed]
- Hirata, M.; Nakamura, K.I.; Kanemaru, T.; Shibata, Y.; Kondo, S. Pigment cell organization in the hypodermis of zebrafish. Dev. Dyn. 2003, 227, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, N.; Taketo, M.M.; Mori, M.; Korinek, V.; Kozmik, Z. Spatial and temporal regulation of Wnt/β-catenin signaling is essential for development of the retinal pigment epithelium. Dev. Biol. 2009, 334, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Squarzoni, P.; Parveen, F.; Zanetti, L.; Ristoratore, F.; Spagnuolo, A. FGF/MAPK/Ets signaling renders pigment cell precursors competent to respond to Wnt signal by directly controlling Ci-Tcf transcription. Development 2011, 138, 1421–1432. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Xu, W.; Yang, K.; Lian, X.; Yang, T. Immunolocalization of Wnt5a during the hair cycle and its role in hair shaft growth in mice. Acta Histochem. 2011, 113, 608–612. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.P.; Bradley, A.; McMahon, A.P.; Jones, S. A Wnt5a pathway underlies outgrowth of multiple structures in the vertebrate embryo. Development 1999, 126, 1211–1223. [Google Scholar] [PubMed]
- Braasch, I.; Schartl, M.; Volff, J.-N. Evolution of pigment synthesis pathways by gene and genome duplication in fish. BMC Evolut. Biol. 2007, 7, 74. [Google Scholar] [CrossRef] [PubMed]
- Postlethwait, J.; Amores, A.; Cresko, W.; Singer, A.; Yan, Y.L. Subfunction partitioning, the teleost radiation and the annotation of the human genome. Trends Genet. TIG 2004, 20, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Lekmine, F.; Chang, C.K.; Sethakorn, N.; Das Gupta, T.K.; Salti, G.I. Role of microphthalmia transcription factor (MITF) in melanoma differentiation. Biochem. Biophys. Res. Commun. 2007, 354, 830–835. [Google Scholar] [CrossRef] [PubMed]
- Levy, C.; Khaled, M.; Fisher, D.E. MITF: Master regulator of melanocyte development and melanoma oncogene. Trends Mol. Med. 2006, 12, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Voisey, J.; Box, N.F.; Daal, A.V. A polymorphism study of the human Agouti gene and its association with MC1R. Pigment Cell Res. 2001, 14, 264–267. [Google Scholar] [CrossRef] [PubMed]
- Dooley, C.M.; Schwarz, H.; Mueller, K.P.; Mongera, A.; Konantz, M.; Neuhauss, S.C.F.; Nusslein-Volhard, C.; Geisler, R. Slc45a2 and V-ATPase are regulators of melanosomal pH homeostasis in zebrafish, providing a mechanism for human pigment evolution and disease. Pigment Cell Melanoma Res. 2013, 26, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Lamason, R.L.; Mohideen, M.A.; Mest, J.R.; Wong, A.C.; Norton, H.L.; Aros, M.C.; Jurynec, M.J.; Mao, X.; Humphreville, V.R.; Humbert, J.E.; et al. SLC24A5, a putative cation exchanger, affects pigmentation in zebrafish and humans. Science 2005, 310, 1782–1786. [Google Scholar] [CrossRef] [PubMed]
- Tsetskhladze, Z.R.; Canfield, V.A.; Ang, K.C.; Wentzel, S.M.; Reid, K.P.; Berg, A.S.; Johnson, S.L.; Kawakami, K.; Cheng, K.C. Functional assessment of human coding mutations affecting skin pigmentation using zebrafish. PLoS ONE 2012, 7, e47398. [Google Scholar] [CrossRef] [PubMed]
- Hutton, S.M.; Spritz, R.A. Comprehensive analysis of oculocutaneous albinism among non-Hispanic caucasians shows that OCA1 is the most prevalent OCA type. J. Investig. Dermatol. 2008, 128, 2442–2450. [Google Scholar] [CrossRef] [PubMed]
- Watt, B.; van Niel, G.; Raposo, G.; Marks, M.S. PMEL: A pigment cell-specific model for functional amyloid formation. Pigment Cell Melanoma Res. 2013, 26, 300–315. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, A.K.; Cech, J.N.; Peichel, C.L. Molecular and developmental contributions to divergent pigment patterns in marine and freshwater sticklebacks. Evolut. Dev. 2012, 14, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Ng’oma, E.; Groth, M.; Ripa, R.; Platzer, M.; Cellerino, A. Transcriptome profiling of natural dichromatism in the annual fishes Nothobranchius furzeri and Nothobranchius kadleci. BMC Genom. 2014, 15, 754. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L.; et al. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 3, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.-P.; Wang, Z.-D.; Guo, Y.-S.; Liu, L.; Yu, J.; Zhang, S.; Liu, S.-J.; Liu, C.-W. Morphological Characters and Transcriptome Profiles Associated with Black Skin and Red Skin in Crimson Snapper (Lutjanus erythropterus). Int. J. Mol. Sci. 2015, 16, 26991-27004. https://doi.org/10.3390/ijms161126005
Zhang Y-P, Wang Z-D, Guo Y-S, Liu L, Yu J, Zhang S, Liu S-J, Liu C-W. Morphological Characters and Transcriptome Profiles Associated with Black Skin and Red Skin in Crimson Snapper (Lutjanus erythropterus). International Journal of Molecular Sciences. 2015; 16(11):26991-27004. https://doi.org/10.3390/ijms161126005
Chicago/Turabian StyleZhang, Yan-Ping, Zhong-Duo Wang, Yu-Song Guo, Li Liu, Juan Yu, Shun Zhang, Shao-Jun Liu, and Chu-Wu Liu. 2015. "Morphological Characters and Transcriptome Profiles Associated with Black Skin and Red Skin in Crimson Snapper (Lutjanus erythropterus)" International Journal of Molecular Sciences 16, no. 11: 26991-27004. https://doi.org/10.3390/ijms161126005