
Metagenomics: A New Way to Illustrate the Crosstalk between Infectious Diseases and Host Microbiome

Abstract
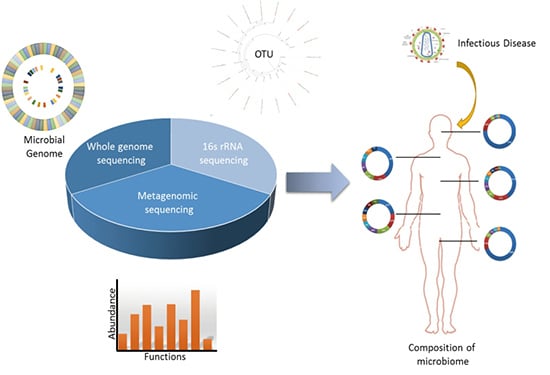
:
{kind=link}
{kind=link}
{kind=link}
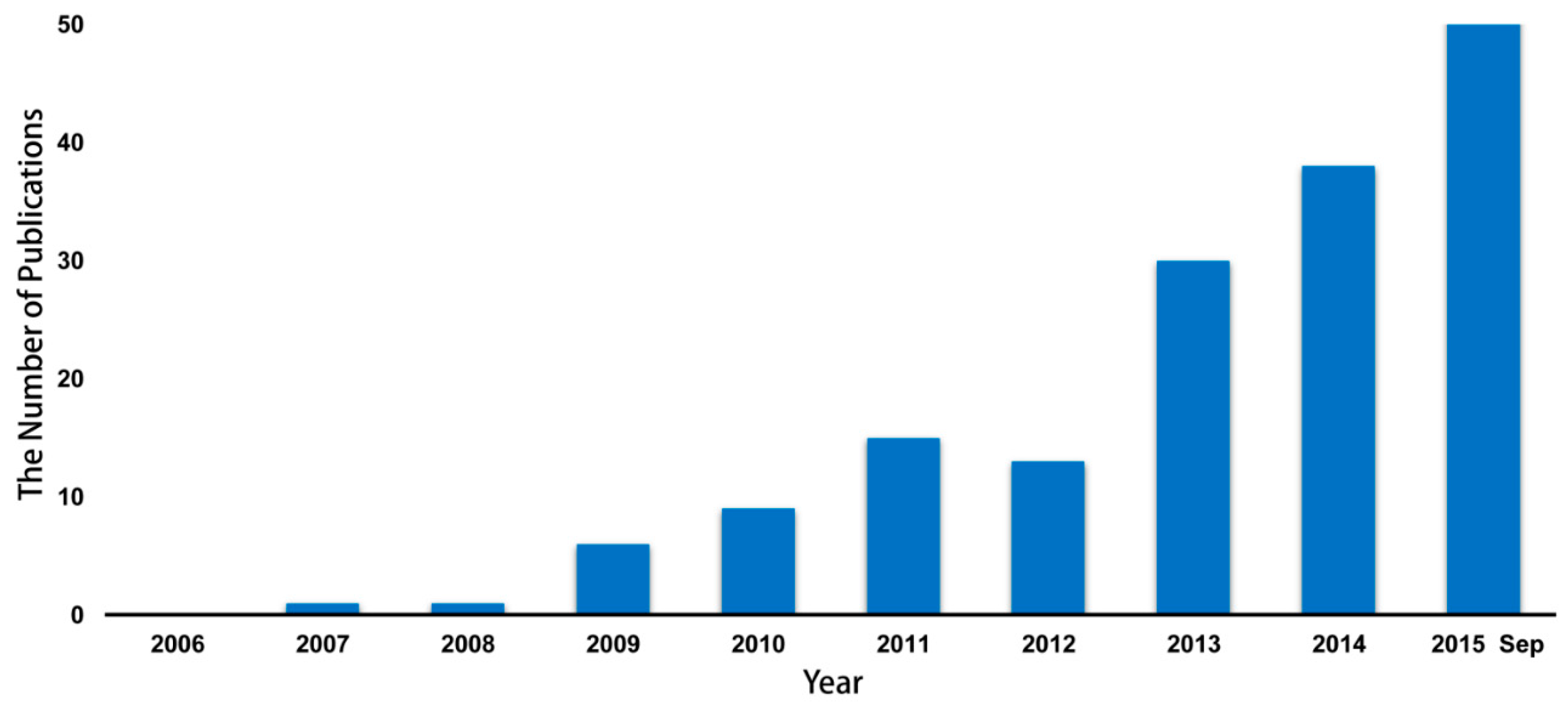
1. Introduction
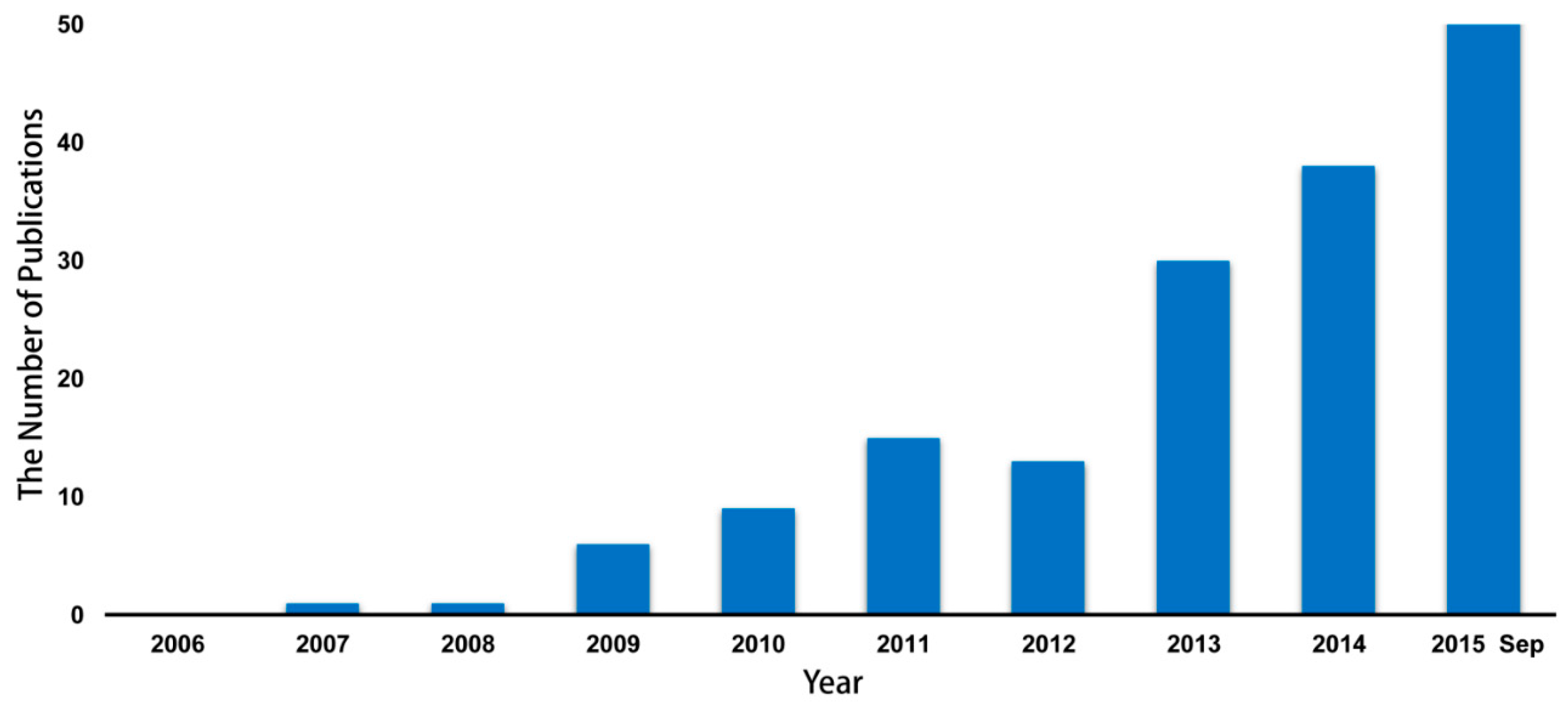
2. The Alteration of Microbiome in HIV/AIDS Patients
2.1. Alteration of the Microbiome in Gastrointestinal Tract
2.2. Alteration of the Microbiome at the Rectal Site
2.3. Alteration of the Microbiome at the Genital Tract
2.4. Alteration of the Microbiome in the Blood, Semen and Brain
2.5. Alteration of the Microbiome in Oral Cavity and Airway
3. The Alteration of Microbiome in Patients with Tuberculosis
4. The Alteration of Microbiome in Patients Infected with Influenza
5. The Alteration of Microbiome in Patients Infected HBV
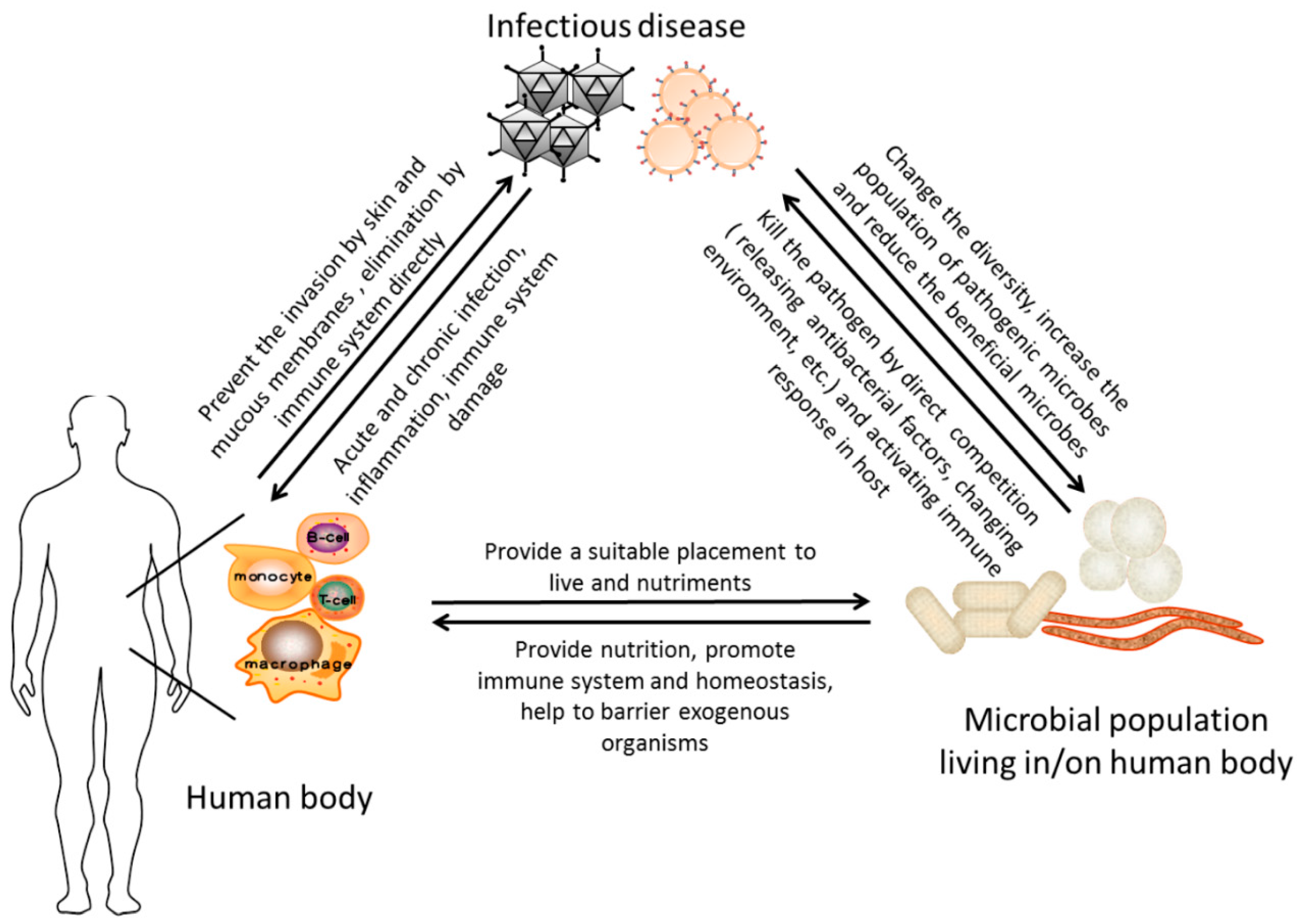
6. Conclusion
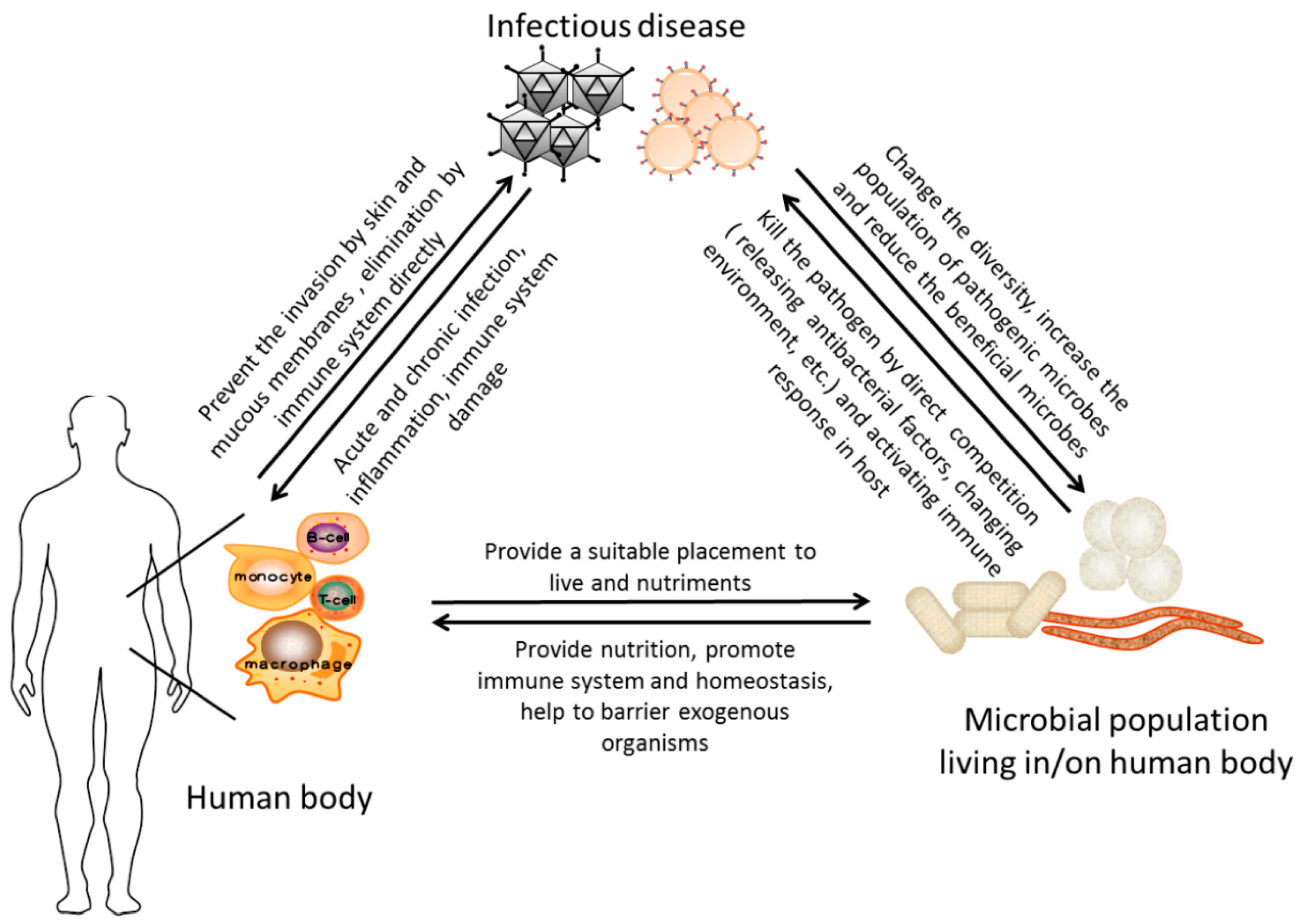
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaura, E.; Keijser, B.J.; Huse, S.M.; Crielaard, W. Defining the healthy "core microbiome" of oral microbial communities. BMC Microbiol. 2009. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.F.; Brulc, J.M.; Iovieno, A.; Bates, B.; Garoutte, A.; Miller, D.; Revanna, K.V.; Gao, X.; Antonopoulos, D.A.; Slepak, V.Z.; et al. Diversity of bacteria at healthy human conjunctiva. Investig. Ophth. Vis. Sci. 2011, 52, 5408–5413. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, K.T.; Ramette, A.; Tiedje, J.M. Toward a more robust assessment of intraspecies diversity, using fewer genetic markers. Appl. Environ. Microbiol. 2006, 72, 7286–7293. [Google Scholar] [CrossRef] [PubMed]
- Fettweis, J.M.; Serrano, M.G.; Sheth, N.U.; Mayer, C.M.; Glascock, A.L.; Brooks, J.P.; Jefferson, K.K.; Buck, G.A. Species-level classification of the vaginal microbiome. BMC Genom. 2012. [Google Scholar] [CrossRef]
- Mathieu, A.; Delmont, T.O.; Vogel, T.M.; Robe, P.; Nalin, R.; Simonet, P. Life on human surfaces: Skin metagenomics. PLoS ONE 2013. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.E.; Weinstock, G.M.; Highlander, S.K.; Worley, K.C.; Creasy, H.H.; Wortman, J.R.; Rusch, D.B.; Mitreva, M.; Sodergren, E.; Chinwalla, A.T.; et al. A catalog of reference genomes from the human microbiome. Science 2010, 328, 994–999. [Google Scholar] [PubMed]
- Dethlefsen, L.; McFall-Ngai, M.; Relman, D.A. An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature 2007, 449, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, R.D.; Adams, M.D.; White, O.; Clayton, R.A.; Kirkness, E.F.; Kerlavage, A.R.; Bult, C.J.; Tomb, J.F.; Dougherty, B.A.; Merrick, J.M.; et al. Whole-genome random sequencing and assembly of haemophilus influenza Rd. Science 1995, 269, 496–512. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, F.H.; Fak, F.; Nookaew, I.; Tremaroli, V.; Fagerberg, B.; Petranovic, D.; Backhed, F.; Nielsen, J. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat. Commun. 2012, 3, 1245. [Google Scholar] [CrossRef] [PubMed]
- Nelsons, K. Metagenomics of the Human Body; Springer: New York, NY, USA, 2010; pp. 1–14. [Google Scholar]
- Hammami, R.; Fernandez, B.; Lacroix, C.; Fliss, I. Anti-infective properties of bacteriocins: An update. Cell. Mol. Life Sci. 2013, 70, 2947–2967. [Google Scholar] [CrossRef] [PubMed]
- Leatham, M.P.; Banerjee, S.; Autieri, S.M.; Mercado-Lubo, R.; Conway, T.; Cohen, P.S. Precolonized human commensal Escherichia coli strains serve as a barrier to E. coli O157:H7 growth in the streptomycin-treated mouse intestine. Infect. Immun. 2009, 77, 2876–2886. [Google Scholar] [CrossRef] [PubMed]
- Vaishnava, S.; Behrendt, C.L.; Ismail, A.S.; Eckmann, L.; Hooper, L.V. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc. Natl. Acad. Sci. USA 2008, 105, 20858–20863. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Maruya, M.; Kawamoto, S.; Sitnik, K.; Kitamura, H.; Agace, W.W.; Fagarasan, S. The sensing of environmental stimuli by follicular dendritic cells promotes immunoglobulin a generation in the gut. Immunity 2010, 33, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Strugnell, R.A.; Wijburg, O.L. The role of secretory antibodies in infection immunity. Nat. Rev. Microbiol. 2010, 8, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Kamada, N.; Chen, G.Y.; Inohara, N.; Nunez, G. Control of pathogens and pathobionts by the gut microbiota. Nat. Immunol. 2013, 14, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Kamada, N.; Sakamoto, K.; Seo, S.U.; Zeng, M.Y.; Kim, Y.G.; Cascalho, M.; Vallance, B.A.; Puente, J.L.; Nunez, G. Humoral immunity in the gut selectively targets phenotypically virulent attaching-and-effacing bacteria for intraluminal elimination. Cell Host Microbe 2015, 17, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Gophna, U.; Sommerfeld, K.; Gophna, S.; Doolittle, W.F.; Veldhuyzen van Zanten, S.J. Differences between tissue-associated intestinal microfloras of patients with Crohn’s disease and ulcerative colitis. J. Clin. Microbiol. 2006, 44, 4136–4141. [Google Scholar] [CrossRef] [PubMed]
- Knights, D.; Lassen, K.G.; Xavier, R.J. Advances in inflammatory bowel disease pathogenesis: Linking host genetics and the microbiome. Gut 2013, 62, 1505–1510. [Google Scholar] [CrossRef] [PubMed]
- Perez-Brocal, V.; Garcia-Lopez, R.; Nos, P.; Beltran, B.; Moret, I.; Moya, A. Metagenomic analysis of Crohn’s disease patients identifies changes in the virome and microbiome related to disease status and therapy, and detects potential interactions and biomarkers. Inflamm. Bowel Dis. 2015, 21, 1419–1427. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Jovel, J.; Halloran, B.; Wine, E.; Patterson, J.; Ford, G.; O’Keefe, S.; Meng, B.; Song, D.; Zhang, Y.; et al. Metagenomic analysis of microbiome in colon tissue from subjects with inflammatory bowel diseases reveals interplay of viruses and bacteria. Inflamm. Bowel Dis. 2015, 21, 1419–1427. [Google Scholar] [CrossRef] [PubMed]
- Nadal, I.; Donat, E.; Ribes-Koninckx, C.; Calabuig, M.; Sanz, Y. Imbalance in the composition of the duodenal microbiota of children with coeliac disease. J. Med. Microbiol. 2007, 56, 1669–1674. [Google Scholar] [CrossRef] [PubMed]
- Iebba, V.; Aloi, M.; Civitelli, F.; Cucchiara, S. Gut microbiota and pediatric disease. Dig. Dis. 2011, 29, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Nistal, E.; Caminero, A.; Herran, A.R.; Arias, L.; Vivas, S.; de Morales, J.M.; Calleja, S.; de Miera, L.E.; Arroyo, P.; Casqueiro, J. Differences of small intestinal bacteria populations in adults and children with/without celiac disease: Effect of age, gluten diet, and disease. Inflamm. Bowel Dis. 2012, 18, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Francavilla, R.; Ercolini, D.; Piccolo, M.; Vannini, L.; Siragusa, S.; de Filippis, F.; de Pasquale, I.; di Cagno, R.; di Toma, M.; Gozzi, G.; et al. Salivary microbiota and metabolome associated with celiac disease. Appl. Environ. Microbiol. 2014, 80, 3416–3425. [Google Scholar] [CrossRef] [PubMed]
- Clemente, J.C.; Ursell, L.K.; Parfrey, L.W.; Knight, R. The impact of the gut microbiota on human health: An integrative view. Cell 2012, 148, 1258–1270. [Google Scholar] [CrossRef] [PubMed]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Muegge, B.D.; Kuczynski, J.; Knights, D.; Clemente, J.C.; Gonzalez, A.; Fontana, L.; Henrissat, B.; Knight, R.; Gordon, J.I. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science 2011, 332, 970–974. [Google Scholar] [CrossRef] [PubMed]
- Jakobsson, H.E.; Jernberg, C.; Andersson, A.F.; Sjolund-Karlsson, M.; Jansson, J.K.; Engstrand, L. Short-term antibiotic treatment has differing long-term impacts on the human throat and gut microbiome. PLoS ONE 2010. [Google Scholar] [CrossRef] [PubMed]
- Stebbing, J.; Gazzard, B.; Douek, D.C. Where does HIV live? N. Engl. J. Med. 2004, 350, 1872–1880. [Google Scholar] [CrossRef] [PubMed]
- Mootsikapun, P. Bacteremia in adult patients with acquired immunodeficiency syndrome in the northeast of Thailand. Int. J. Infect. Dis. 2007, 11, 226–231. [Google Scholar] [CrossRef] [PubMed]
- The Antiretroviral Therapy Cohort Collaboration. Causes of death in HIV-1-infected patients treated with antiretroviral therapy, 1996–2006: Collaborative analysis of 13 HIV cohort studies. Clin. Infect. Dis. 2010, 50, 1387–1396. [Google Scholar] [CrossRef] [PubMed]
- Palella, F.J., Jr.; Baker, R.K.; Moorman, A.C.; Chmiel, J.S.; Wood, K.C.; Brooks, J.T.; Holmberg, S.D. Mortality in the highly active antiretroviral therapy era: Changing causes of death and disease in the HIV outpatient study. J. Acquir. Immune Defic. Syndr. 2006, 43, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, K.E.; Slusher, N.A.; Cabana, M.D.; Lynch, S.V. Role of the gut microbiota in defining human health. Expert Rev. Anti Infect. Ther. 2010, 8, 435–454. [Google Scholar] [CrossRef] [PubMed]
- Brenchley, J.M.; Price, D.A.; Schacker, T.W.; Asher, T.E.; Silvestri, G.; Rao, S.; Kazzaz, Z.; Bornstein, E.; Lambotte, O.; Altmann, D.; et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 2006, 12, 1365–1371. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, G.; Bellistri, G.M.; Borghi, E.; Tincati, C.; Ferramosca, S.; la Francesca, M.; Morace, G.; Gori, A.; Monforte, A.D. Microbial translocation is associated with sustained failure in CD4+ T-cell reconstitution in HIV-infected patients on long-term highly active antiretroviral therapy. AIDS 2008, 22, 2035–2038. [Google Scholar] [CrossRef] [PubMed]
- McKenna, P.; Hoffmann, C.; Minkah, N.; Aye, P.P.; Lackner, A.; Liu, Z.Z.; Lozupone, C.A.; Hamady, M.; Knight, R.; Bushman, F.D. The macaque gut microbiome in health, lentiviral infection, and chronic enterocolitis. PLoS Pathog. 2008. [Google Scholar] [CrossRef] [PubMed]
- Handley, S.A.; Thackray, L.B.; Zhao, G.; Presti, R.; Miller, A.D.; Droit, L.; Abbink, P.; Maxfield, L.F.; Kambal, A.; Duan, E.; et al. Pathogenic simian immunodeficiency virus infection is associated with expansion of the enteric virome. Cell 2012, 151, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Brenchley, J.M. Mucosal immunity in human and simian immunodeficiency lentivirus infections. Mucosal Immunol. 2013, 6, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Moeller, A.H.; Peeters, M.; Ayouba, A.; Ngole, E.M.; Esteban, A.; Hahn, B.H.; Ochman, H. Stability of the gorilla microbiome despite simian immunodeficiency virus infection. Mol. Ecol. 2015, 24, 690–697. [Google Scholar] [CrossRef] [PubMed]
- Grossman, Z.; Feinberg, M.B.; Paul, W.E. Multiple modes of cellular activation and virus transmission in HIV infection: A role for chronically and latently infected cells in sustaining viral replication. Proc. Natl. Acad. Sci. USA 1998, 95, 6314–6319. [Google Scholar] [CrossRef] [PubMed]
- Vujkovic-Cvijin, I.; Dunham, R.M.; Iwai, S.; Maher, M.C.; Albright, R.G.; Broadhurst, M.J.; Hernandez, R.D.; Lederman, M.M.; Huang, Y.; Somsouk, M.; et al. Dysbiosis of the gut microbiota is associated with HIV disease progression and tryptophan catabolism. Sci. Transl. Med. 2013. [Google Scholar] [CrossRef] [PubMed]
- Dinh, D.M.; Volpe, G.E.; Duffalo, C.; Bhalchandra, S.; Tai, A.K.; Kane, A.V.; Wanke, C.A.; Ward, H.D. Intestinal microbiota, microbial translocation, and systemic inflammation in chronic HIV infection. J. Infect. Dis. 2015, 211, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Gori, A.; Tincati, C.; Rizzardini, G.; Torti, C.; Quirino, T.; Haarman, M.; Ben Amor, K.; van Schaik, J.; Vriesema, A.; Knol, J.; et al. Early impairment of gut function and gut flora supporting a role for alteration of gastrointestinal mucosa in human immunodeficiency virus pathogenesis. J. Clin. Microbiol. 2008, 46, 757–758. [Google Scholar] [CrossRef] [PubMed]
- Gori, A.; Rizzardini, G.; van’t Land, B.; Amor, K.B.; van Schaik, J.; Torti, C.; Quirino, T.; Tincati, C.; Bandera, A.; Knol, J.; et al. Specific prebiotics modulate gut microbiota and immune activation in HAART-naive HIV-infected adults: Results of the “COPA” pilot randomized trial. Mucosal Immunol. 2011, 4, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Nowak, P.; Troseid, M.; Avershina, E.; Barqasho, B.; Neogi, U.; Holm, K.; Hov, J.R.; Noyan, K.; Vesterbacka, J.; Svard, J.; et al. Gut microbiota diversity predicts immune status in HIV-1 infection. AIDS 2015, in press. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.A.; Rhodes, M.E.; Neff, C.P.; Fontenot, A.P.; Campbell, T.B.; Palmer, B.E. HIV-induced alteration in gut microbiota: Driving factors, consequences, and effects of antiretroviral therapy. Gut Mcrobes 2014, 5, 562–570. [Google Scholar] [CrossRef] [PubMed]
- McHardy, I.H.; Li, X.; Tong, M.; Ruegger, P.; Jacobs, J.; Borneman, J.; Anton, P.; Braun, J. HIV Infection is associated with compositional and functional shifts in the rectal mucosal microbiota. Microbiome 2013, 1, 26. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.A.; Li, M.; Campbell, T.B.; Flores, S.C.; Linderman, D.; Gebert, M.J.; Knight, R.; Fontenot, A.P.; Palmer, B.E. Alterations in the gut microbiota associated with HIV-1 infection. Cell Host Microbe 2013, 14, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Mutlu, E.A.; Keshavarzian, A.; Losurdo, J.; Swanson, G.; Siewe, B.; Forsyth, C.; French, A.; DeMarais, P.; Sun, Y.; Koenig, L.; et al. A compositional look at the human gastrointestinal microbiome and immune activation parameters in HIV infected subjects. PLoS Pathog. 2014. [Google Scholar] [CrossRef] [PubMed]
- Dillon, S.M.; Lee, E.J.; Kotter, C.V.; Austin, G.L.; Dong, Z.; Hecht, D.K.; Gianella, S.; Siewe, B.; Smith, D.M.; Landay, A.L.; et al. An altered intestinal mucosal microbiome in HIV-1 infection is associated with mucosal and systemic immune activation and endotoxemia. Mucosal Immunol. 2014, 7, 983–994. [Google Scholar] [CrossRef] [PubMed]
- Burgener, A.; McGowan, I.; Klatt, N.R. HIV and mucosal barrier interactions: Consequences for transmission and pathogenesis. Curr. Opin. Immunol. 2015, 36, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Hladik, F.; McElrath, M.J. Setting the stage: Host invasion by HIV. Nat. Rev. Immunol. 2008, 8, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Haase, A.T. Perils at mucosal front lines for HIV and SIV and their hosts. Nat. Rev. Immunol. 2005, 5, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, K.T.; Marcus, J.L.; Nieri, G.; Philip, S.S.; Klausner, J.D. Rectal gonorrhea and chlamydia reinfection is associated with increased risk of HIV seroconversion. J. Acquir. Immune Defic. Syndr. 2010, 53, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Kane, C.T.; Diawara, S.; Ndiaye, H.D.; Diallo, P.A.N.; Wade, A.S.; Diallo, A.G.; Belec, L.; Mboup, S. Concentrated and linked epidemics of both HSV-2 and HIV-1/HIV-2 infections in senegal: Public health impacts of the spread of HIV. Int. J. STD AIDS 2009, 20, 793–796. [Google Scholar] [CrossRef] [PubMed]
- Larsson, P.G.; Bergstrom, M.; Forsum, U.; Jacobsson, B.; Strand, A.; Wolner-Hanssen, P. Bacterial vaginosis transmission, role in genital tract infection and pregnancy outcome: An enigma. Apmis 2005, 113, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, C.M.; Balkus, J.; Agnew, K.J.; Cohn, S.; Luque, A.; Lawler, R.; Coombs, R.W.; Hitti, J.E. Bacterial vaginosis, not HIV, is primarily responsible for increased vaginal concentrations of proinflammatory cytokines. AIDS Res. Hum. Retrovir. 2008, 24, 667–671. [Google Scholar] [CrossRef]
- Schellenberg, J.J.; Card, C.M.; Ball, T.B.; Mungai, J.N.; Irungu, E.; Kimani, J.; Jaoko, W.; Wachihi, C.; Fowke, K.R.; Plummer, F.A. Bacterial vaginosis, HIV serostatus and T-cell subset distribution in a cohort of East African commercial sex workers: Retrospective analysis. AIDS 2012, 26, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Hummelen, R.; Fernandes, A.D.; Macklaim, J.M.; Dickson, R.J.; Changalucha, J.; Gloor, G.B.; Reid, G. Deep sequencing of the vaginal microbiota of women with HIV. PLoS ONE 2010. [Google Scholar] [CrossRef] [PubMed]
- Salas, J.T.; Chang, T.L. Microbiome in human immunodeficiency virus infection. Clin. Lab. Med. 2014, 34, 733–745. [Google Scholar] [CrossRef] [PubMed]
- Cone, R.A. Vaginal microbiota and sexually transmitted infections that may influence transmission of cell-associated HIV. J. Infect. Dis. 2014, 210, S616–S621. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.D.; Donovan, B.; Weber, K.M.; Cohen, M.; Ravel, J.; Gajer, P.; Gilbert, D.; Burgad, D.; Spear, G.T. The vaginal microbiota over an 8- to 10-year period in a cohort of HIV-infected and HIV-uninfected women. PLoS ONE 2015. [Google Scholar] [CrossRef] [PubMed]
- Benning, L.; Golub, E.T.; Anastos, K.; French, A.L.; Cohen, M.; Gilbert, D.; Gillevet, P.; Munyazesa, E.; Landay, A.L.; Sikaroodi, M.; et al. Comparison of lower genital tract microbiota in HIV-infected and uninfected women from Rwanda and the US. PLoS ONE 2014. [Google Scholar] [CrossRef] [PubMed]
- Ameur, A.; Meiring, T.L.; Bunikis, I.; Haggqvist, S.; Lindau, C.; Lindberg, J.H.; Gustavsson, I.; Mbulawa, Z.Z.; Williamson, A.L.; Gyllensten, U. Comprehensive profiling of the vaginal microbiome in HIV positive women using massive parallel semiconductor sequencing. Sci. Rep. 2014. [Google Scholar] [CrossRef] [PubMed]
- Li, S.K.; Leung, R.K.; Guo, H.X.; Wei, J.F.; Wang, J.H.; Kwong, K.T.; Lee, S.S.; Zhang, C.; Tsui, S.K. Detection and identification of plasma bacterial and viral elements in HIV/AIDS patients in comparison to healthy adults. Clin. Microbiol. Infect. 2012, 18, 1126–1133. [Google Scholar] [CrossRef] [PubMed]
- Merlini, E.; Bai, F.; Bellistri, G.M.; Tincati, C.; d’Arminio Monforte, A.; Marchetti, G. Evidence for polymicrobic flora translocating in peripheral blood of HIV-infected patients with poor immune response to antiretroviral therapy. PLoS ONE 2011. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Deng, X.; Linsuwanon, P.; Bangsberg, D.; Bwana, M.B.; Hunt, P.; Martin, J.N.; Deeks, S.G.; Delwart, E. AIDS alters the commensal plasma virome. J. Virol. 2013, 87, 10912–10915. [Google Scholar] [CrossRef] [PubMed]
- Branton, W.G.; Ellestad, K.K.; Maingat, F.; Wheatley, B.M.; Rud, E.; Warren, R.L.; Holt, R.A.; Surette, M.G.; Power, C. Brain microbial populations in HIV/AIDS: α-proteobacteria predominate independent of host immune status. PLoS ONE 2013. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.M.; Osborne, B.J.; Hungate, B.A.; Shahabi, K.; Huibner, S.; Lester, R.; Dwan, M.G.; Kovacs, C.; Contente-Cuomo, T.L.; Benko, E.; et al. The semen microbiome and its relationship with local immunology and viral load in HIV infection. PLoS Pathog. 2014. [Google Scholar] [CrossRef] [PubMed]
- Dang, A.T.; Cotton, S.; Sankaran-Walters, S.; Li, C.S.; Lee, C.Y.M.; Dandekar, S.; Paster, B.J.; George, M.D. Evidence of an increased pathogenic footprint in the lingual microbiome of untreated HIV infected patients. BMC Microbiol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.; Cota-Gomez, A.; Palmer, B.E.; Linderman, D.J.; Charlson, E.S.; Sodergren, E.; Mitreva, M.; Abubucker, S.; Martin, J.; Yao, G.H.; et al. Widespread colonization of the lung by Tropheryma whipplei in HIV infection. Am. J. Respir. Crit. Care 2013, 187, 1110–1117. [Google Scholar] [CrossRef] [PubMed]
- Iwai, S.; Fei, M.; Huang, D.; Fong, S.; Subramanian, A.; Grieco, K.; Lynch, S.V.; Huang, L. Oral and airway microbiota in HIV-infected pneumonia patients. J. Clin. Microbiol. 2012, 50, 2995–3002. [Google Scholar] [CrossRef] [PubMed]
- Iwai, S.; Huang, D.; Fong, S.; Jarlsberg, L.G.; Worodria, W.; Yoo, S.; Cattamanchi, A.; Davis, J.L.; Kaswabuli, S.; Segal, M.; et al. The lung microbiome of Ugandan HIV-infected pneumonia patients is compositionally and functionally distinct from that of San Franciscan patients. PLoS ONE 2014. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.M.; Schloss, P.D.; Venkataraman, A.; Twigg Iii, H.; Jablonski, K.A.; Bushman, F.D.; Campbell, T.B.; Charlson, E.S.; Collman, R.G.; Crothers, K.; et al. Multi-center comparison of lung and oral microbiomes of HIV-infected and HIV-uninfected individuals. Am. J. Respir. Crit. Care Med. 2015. [Google Scholar] [CrossRef] [PubMed]
- Kistler, J.O.; Arirachakaran, P.; Poovorawan, Y.; Dahlen, G.; Wade, W.G. The oral microbiome in human immunodeficiency virus (HIV)-positive individuals. J. Med. Microbiol. 2015, 64, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Lucht, L.; Tipton, L.; Rogers, M.B.; Fitch, A.; Kessinger, C.; Camp, D.; Kingsley, L.; Leo, N.; Greenblatt, R.M.; et al. Topographic diversity of the respiratory tract mycobiome and alteration in HIV and lung disease. Am. J. Respir. Crit. Care Med. 2015, 191, 932–942. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Morris, A.; Huang, L.; Beck, J.M.; Twigg III, H.L.; von Mutius, E.; Ghedin, E. The microbiome and the lung. Ann. Am. Thorac. Soc. 2014, 11, S227–S232. [Google Scholar] [CrossRef] [PubMed]
- Perry, S.; de Jong, B.C.; Solnick, J.V.; de la Luz Sanchez, M.; Yang, S.; Lin, P.L.; Hansen, L.M.; Talat, N.; Hill, P.C.; Hussain, R.; et al. Infection with Helicobacter pylori is associated with protection against tuberculosis. PLoS ONE 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Z.; Zhou, Y.; Li, H.; Zhang, Y.; Zhang, S.; Tang, S.; Guo, X. Complex sputum microbial composition in patients with pulmonary tuberculosis. BMC Microbiol. 2012, 12, 276. [Google Scholar] [CrossRef] [PubMed]
- Cheung, M.K.; Lam, W.Y.; Fung, W.Y.; Law, P.T.; Au, C.H.; Nong, W.; Kam, K.M.; Kwan, H.S.; Tsui, S.K. Sputum microbiota in tuberculosis as revealed by 16s rRNA pyrosequencing. PLoS ONE 2013. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liu, W.; He, L.; Huang, F.; Chen, J.; Cui, P.; Shen, Y.; Zhao, J.; Wang, W.; Zhang, Y.; et al. Sputum microbiota associated with new, recurrent and treatment failure tuberculosis. PLoS ONE 2013. [Google Scholar] [CrossRef] [PubMed]
- Botero, L.E.; Delgado-Serrano, L.; Cepeda, M.L.; Bustos, J.R.; Anzola, J.M.; Del Portillo, P.; Robledo, J.; Zambrano, M.M. Respiratory tract clinical sample selection for microbiota analysis in patients with pulmonary tuberculosis. Microbiome 2014, 2, 29. [Google Scholar] [CrossRef] [PubMed]
- Winglee, K.; Eloe-Fadrosh, E.; Gupta, S.; Guo, H.D.; Fraser, C.; Bishai, W. Aerosol Mycobacterium tuberculosis infection causes rapid loss of diversity in gut microbiota. PLoS ONE 2014. [Google Scholar] [CrossRef] [PubMed]
- Morens, D.M.; Taubenberger, J.K.; Fauci, A.S. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: Implications for pandemic influenza preparedness. J. Infect. Dis. 2008, 198, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Greninger, A.L.; Chen, E.C.; Sittler, T.; Scheinerman, A.; Roubinian, N.; Yu, G.X.; Kim, E.; Pillai, D.R.; Guyard, C.; Mazzulli, T.; et al. A metagenomic analysis of pandemic influenza A (2009 H1N1) infection in patients from North America. PLoS ONE 2010. [Google Scholar] [CrossRef] [PubMed]
- Palacios, G.; Hornig, M.; Cisterna, D.; Savji, N.; Bussetti, A.V.; Kapoor, V.; Hui, J.; Tokarz, R.; Briese, T.; Baumeister, E.; et al. Streptococcus pneumoniae coinfection is correlated with the severity of H1N1 pandemic influenza. PLoS ONE 2009. [Google Scholar] [CrossRef] [PubMed]
- Leung, R.K.; Zhou, J.W.; Guan, W.; Li, S.K.; Yang, Z.F.; Tsui, S.K. Modulation of potential respiratory pathogens by pH1N1 viral infection. Clin. Microbiol. Infect. 2013, 19, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Chaban, B.; Albert, A.; Links, M.G.; Gardy, J.; Tang, P.; Hill, J.E. Characterization of the upper respiratory tract microbiomes of patients with pandemic H1N1 influenza. PLoS ONE 2013. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Yong, D.; Lee, K.; Cho, Y.J.; Chun, J. Profiling bacterial community in upper respiratory tracts. BMC Infect. Dis. 2014, 14, 583. [Google Scholar] [CrossRef] [PubMed]
- Norman, K.; Pirlich, M. Gastrointestinal tract in liver disease: Which organ is sick? Curr. Opin. Clin. Nutr. Metab. Care 2008, 11, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Riordan, S.M.; Williams, R. Gut flora and hepatic encephalopathy in patients with cirrhosis. N. Engl. J. Med. 2010, 362, 1140–1142. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Tsao, G.; Wiest, R. Gut microflora in the pathogenesis of the complications of cirrhosis. Best Pract. Res. Clin. Gastroenterol. 2004, 18, 353–372. [Google Scholar] [CrossRef] [PubMed]
- Law, J.; Jovel, J.; Patterson, J.; Ford, G.; O’Keefe, S.; Wang, W.; Meng, B.; Song, D.; Zhang, Y.; Tian, Z.; et al. Identification of hepatotropic viruses from plasma using deep sequencing: A next generation diagnostic tool. PLoS ONE 2013. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, Z.J.; Guo, R.Y.; Chen, N.; Lu, H.F.; Huang, S.A.; Wang, J.; Li, L.J. Correlation between gastrointestinal fungi and varying degrees of chronic hepatitis B virus infection. Diagn. Microbiol. Infect. Dis. 2011, 70, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Yan, X.; Zou, D.; Yang, Z.; Wang, X.; Liu, W.; Wang, S.; Li, X.; Han, J.; Huang, L.; et al. Abnormal fecal microbiota community and functions in patients with hepatitis B liver cirrhosis as revealed by a metagenomic approach. BMC Gastroenterol. 2013, 13, 175. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, F.; Lu, H.; Wang, B.; Chen, Y.; Lei, D.; Wang, Y.; Zhu, B.; Li, L. Characterization of fecal microbial communities in patients with liver cirrhosis. J. Hepatol. 2011, 54, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Riordan, S.M.; Williams, R. The intestinal flora and bacterial infection in cirrhosis. J. Hepatol. 2006, 45, 744–757. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Lee, S.; Lee, H.; Song, Y.M.; Lee, K.; Han, M.J.; Sung, J.; Ko, G. Association of the vaginal microbiota with human papillomavirus infection in a korean twin cohort. PLoS ONE 2013. [Google Scholar] [CrossRef] [PubMed]
- De Jong, H.K.; Parry, C.M.; van der Poll, T.; Wiersinga, W.J. Host-pathogen interaction in invasive salmonellosis. PLoS Pathog. 2012. [Google Scholar] [CrossRef] [PubMed]
- Khoruts, A.; Dicksved, J.; Jansson, J.K.; Sadowsky, M.J. Changes in the composition of the human fecal microbiome after bacteriotherapy for recurrent clostridium difficile-associated diarrhea. J. Clin. Gastroenterol. 2010, 44, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, C.; Hill, D.A.; Minkah, N.; Kirn, T.; Troy, A.; Artis, D.; Bushman, F. Community-wide response of the gut microbiota to enteropathogenic Citrobacter rodentium infection revealed by deep sequencing. Infect. Immun. 2009, 77, 4668–4678. [Google Scholar] [CrossRef] [PubMed]
- Buffie, C.G.; Bucci, V.; Stein, R.R.; McKenney, P.T.; Ling, L.; Gobourne, A.; No, D.; Liu, H.; Kinnebrew, M.; Viale, A.; et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 2015, 517, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Deatherage Kaiser, B.L.; Li, J.; Sanford, J.A.; Kim, Y.M.; Kronewitter, S.R.; Jones, M.B.; Peterson, C.T.; Peterson, S.N.; Frank, B.C.; Purvine, S.O.; et al. A multi-omic view of host-pathogen-commensal interplay in Salmonella-mediated intestinal infection. PLoS ONE 2013. [Google Scholar] [CrossRef] [PubMed]
- Lindsay, B.; Oundo, J.; Hossain, M.A.; Antonio, M.; Tamboura, B.; Walker, A.W.; Paulson, J.N.; Parkhill, J.; Omore, R.; Faruque, A.S. Microbiota that affect risk for shigellosis in children in low-income countries. Emerg. Infect. Dis. 2015, 21, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, M.; Zhang, C.; Du, H.; Wei, G.; Pang, X.; Zhou, H.; Liu, B.; Zhao, L. Pattern extraction of structural responses of gut microbiota to rotavirus infection via multivariate statistical analysis of clone library data. FEMS Microbiol. Ecol. 2009, 70, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Huda, M.N.; Lewis, Z.; Kalanetra, K.M.; Rashid, M.; Ahmad, S.M.; Raqib, R.; Qadri, F.; Underwood, M.A.; Mills, D.A.; Stephensen, C.B. Stool microbiota and vaccine responses of infants. Pediatrics 2014, 134, e362–e372. [Google Scholar] [CrossRef] [PubMed]
- Irvine, S.L.; Hummelen, R.; Hekmat, S. Probiotic yogurt consumption may improve gastrointestinal symptoms, productivity, and nutritional intake of people living with human immunodeficiency virus in mwanza, tanzania. Nutr. Res. 2011, 31, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.F.; Zhang, C.X.; Qian, G.R.; Hu, X.J.; Zhang, H.; Chen, C.L.; Liang, W.F.; Gao, H.; Yang, Y.M.; Li, L.J. An analysis of microbiota-targeted therapies in patients with avian influenza virus subtype H7N9 infection. BMC Infect. Dis. 2014. [Google Scholar] [CrossRef] [PubMed]
- Klatt, N.R.; Canary, L.A.; Sun, X.; Vinton, C.L.; Funderburg, N.T.; Morcock, D.R.; Quinones, M.; Deming, C.B.; Perkins, M.; Hazuda, D.J.; et al. Probiotic/prebiotic supplementation of antiretrovirals improves gastrointestinal immunity in SIV-infected macaques. J. Clin. Investig. 2013, 123, 903–907. [Google Scholar] [CrossRef] [PubMed]
- Cunningham-Rundles, S.; Ahrne, S.; Johann-Liang, R.; Abuav, R.; Dunn-Navarra, A.M.; Grassey, C.; Bengmark, S.; Cervia, J.S. Effect of probiotic bacteria on microbial host defense, growth, and immune function in human immunodeficiency virus type-1 infection. Nutrients 2011, 3, 1042–1070. [Google Scholar] [CrossRef] [PubMed]
- Schunter, M.; Chu, H.; Hayes, T.L.; McConnell, D.; Crawford, S.S.; Luciw, P.A.; Bengmark, S.; Asmuth, D.M.; Brown, J.; Bevins, C.L.; et al. Randomized pilot trial of a synbiotic dietary supplement in chronic HIV-1 infection. BMC Complement. Altern. Med. 2012. [Google Scholar] [CrossRef] [PubMed]
- Corona, A.W.; Norden, D.M.; Skendelas, J.P.; Huang, Y.; O’Connor, J.C.; Lawson, M.; Dantzer, R.; Kelley, K.W.; Godbout, J.P. Indoleamine 2,3-dioxygenase inhibition attenuates lipopolysaccharide induced persistent microglial activation and depressive-like complications in fractalkine receptor (CX3CR1)-deficient mice. Brain Behav. Immun. 2013, 31, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Dunham, R.M.; Gordon, S.N.; Vaccari, M.; Piatak, M.; Huang, Y.; Deeks, S.G.; Lifson, J.; Franchini, G.; McCune, J.M. Preclinical evaluation of HIV eradication strategies in the simian immunodeficiency virus-infected rhesus macaque: A pilot study testing inhibition of indoleamine 2,3-dioxygenase. AIDS Res. Hum. Retrovir. 2013, 29, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Vyboh, K.; Jenabian, M.A.; Mehraj, V.; Routy, J.P. HIV and the gut microbiota, partners in crime: Breaking the vicious cycle to unearth new therapeutic targets. J. Immunol. Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Cahn, P.; Pozniak, A.L.; Mingrone, H.; Shuldyakov, A.; Brites, C.; Andrade-Villanueva, J.F.; Richmond, G.; Buendia, C.B.; Fourie, J.; Ramgopal, M.; et al. Dolutegravir versus raltegravir in antiretroviral-experienced, integrase-inhibitor-naive adults with HIV: Week 48 results from the randomised, double-blind, non-inferiority SAILING study. Lancet 2013, 382, 700–708. [Google Scholar] [CrossRef]
- Asmuth, D.M.; Ma, Z.M.; Albanese, A.; Sandler, N.G.; Devaraj, S.; Knight, T.H.; Flynn, N.M.; Yotter, T.; Garcia, J.C.; Tsuchida, E.; et al. Oral serum-derived bovine immunoglobulin improves duodenal immune reconstitution and absorption function in patients with HIV enteropathy. AIDS 2013, 27, 2207–2217. [Google Scholar] [CrossRef] [PubMed]
- Nwosu, F.C.; Avershina, E.; Wilson, R.; Rudi, K. Gut microbiota in HIV infection: Implication for disease progression and management. Gastroenterol. Res. Pract. 2014. [Google Scholar] [CrossRef] [PubMed]
- Angiuoli, S.V.; White, J.R.; Matalka, M.; White, O.; Fricke, W.F. Resources and costs for microbial sequence analysis evaluated using virtual machines and cloud computing. PLoS ONE 2011. [Google Scholar] [CrossRef] [PubMed]
- Human Microbiome Project Consortium. A framework for human microbiome research. Nature 2012, 486, 215–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korem, T.; Zeevi, D.; Suez, J.; Weinberger, A.; Avnit-Sagi, T.; Pompan-Lotan, M.; Matot, E.; Jona, G.; Harmelin, A.; Cohen, N.; et al. Growth dynamics of gut microbiota in health and disease inferred from single metagenomic samples. Science 2015, 349, 1101–1106. [Google Scholar] [CrossRef] [PubMed]
- Dubourg, G.; Lagier, J.C.; Armougom, F.; Robert, C.; Hamad, I.; Brouqui, P.; Raoult, D. The gut microbiota of a patient with resistant tuberculosis is more comprehensively studied by culturomics than by metagenomics. Eur. J. Clin. Microbiol. 2013, 32, 637–645. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Lun, C.-Y.; Tsui, S.K.-W. Metagenomics: A New Way to Illustrate the Crosstalk between Infectious Diseases and Host Microbiome. Int. J. Mol. Sci. 2015, 16, 26263-26279. https://doi.org/10.3390/ijms161125957
Zhang Y, Lun C-Y, Tsui SK-W. Metagenomics: A New Way to Illustrate the Crosstalk between Infectious Diseases and Host Microbiome. International Journal of Molecular Sciences. 2015; 16(11):26263-26279. https://doi.org/10.3390/ijms161125957
Chicago/Turabian StyleZhang, Yinfeng, Cheuk-Yin Lun, and Stephen Kwok-Wing Tsui. 2015. "Metagenomics: A New Way to Illustrate the Crosstalk between Infectious Diseases and Host Microbiome" International Journal of Molecular Sciences 16, no. 11: 26263-26279. https://doi.org/10.3390/ijms161125957