
High-Throughput Screening in Protein Engineering: Recent Advances and Future Perspectives

Abstract
:
1. Introduction
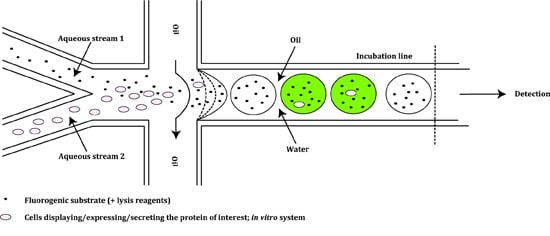
2. Nature’s Own Compartments in FACS-Based Screening Platforms
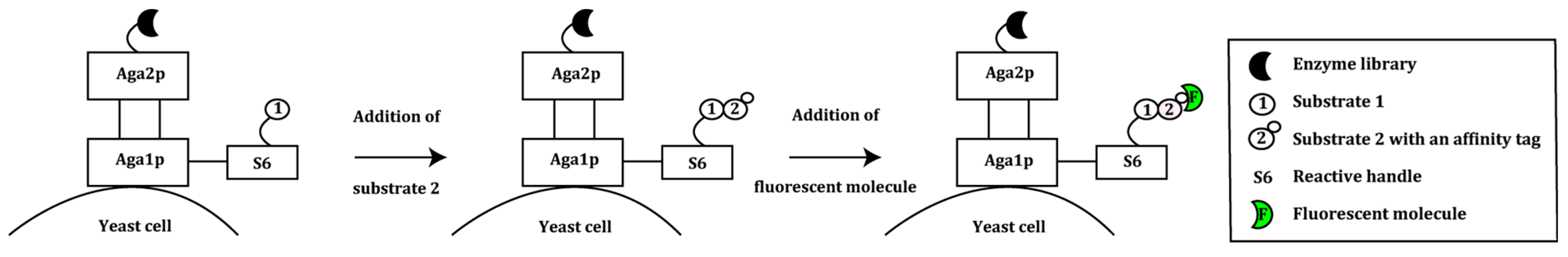
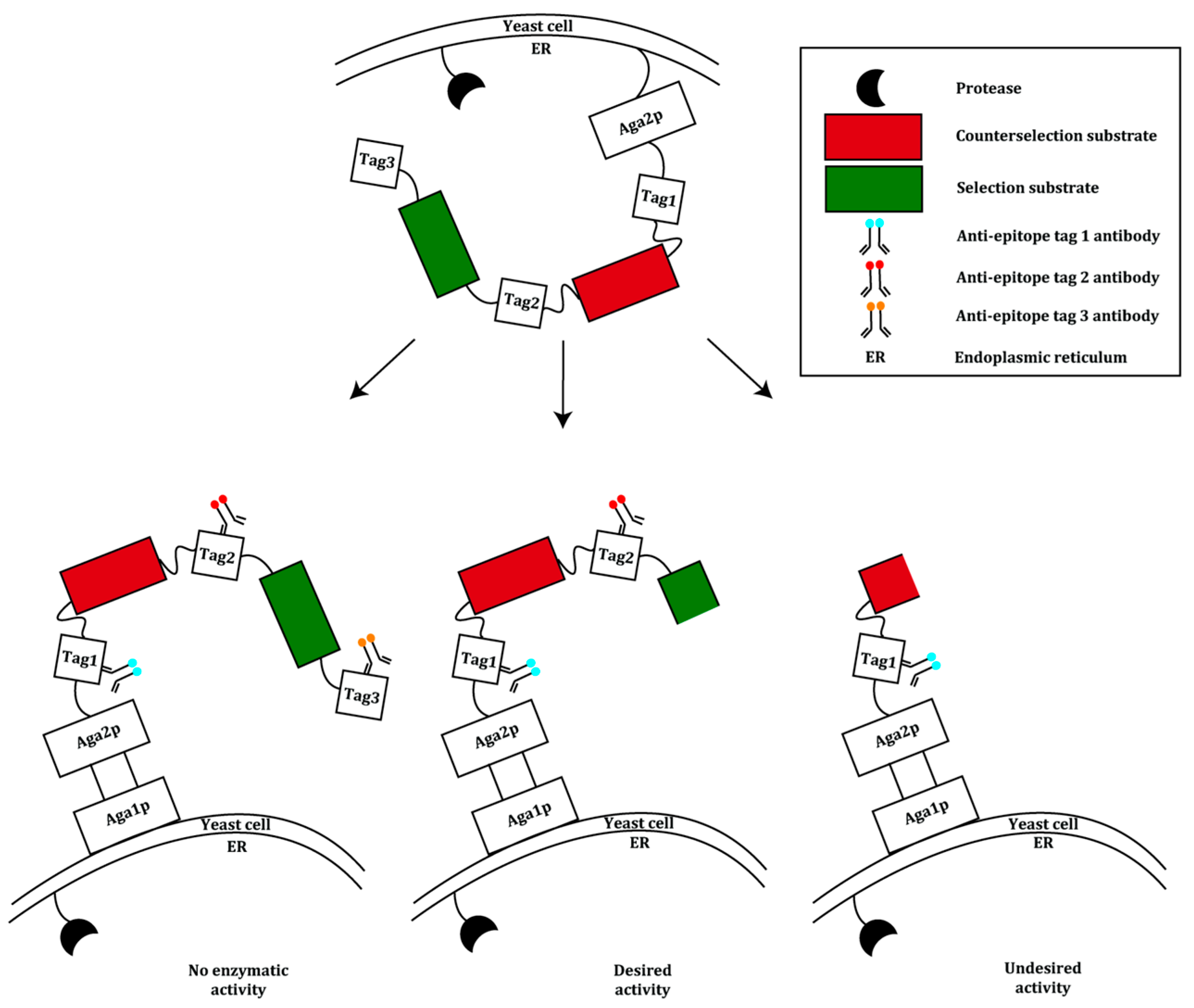
2.1. Display of Protein Variants on Eukaryotic Cells
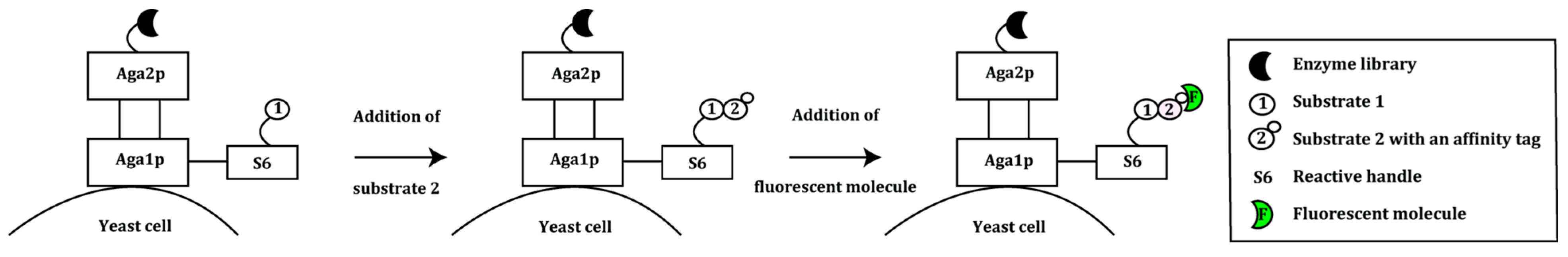
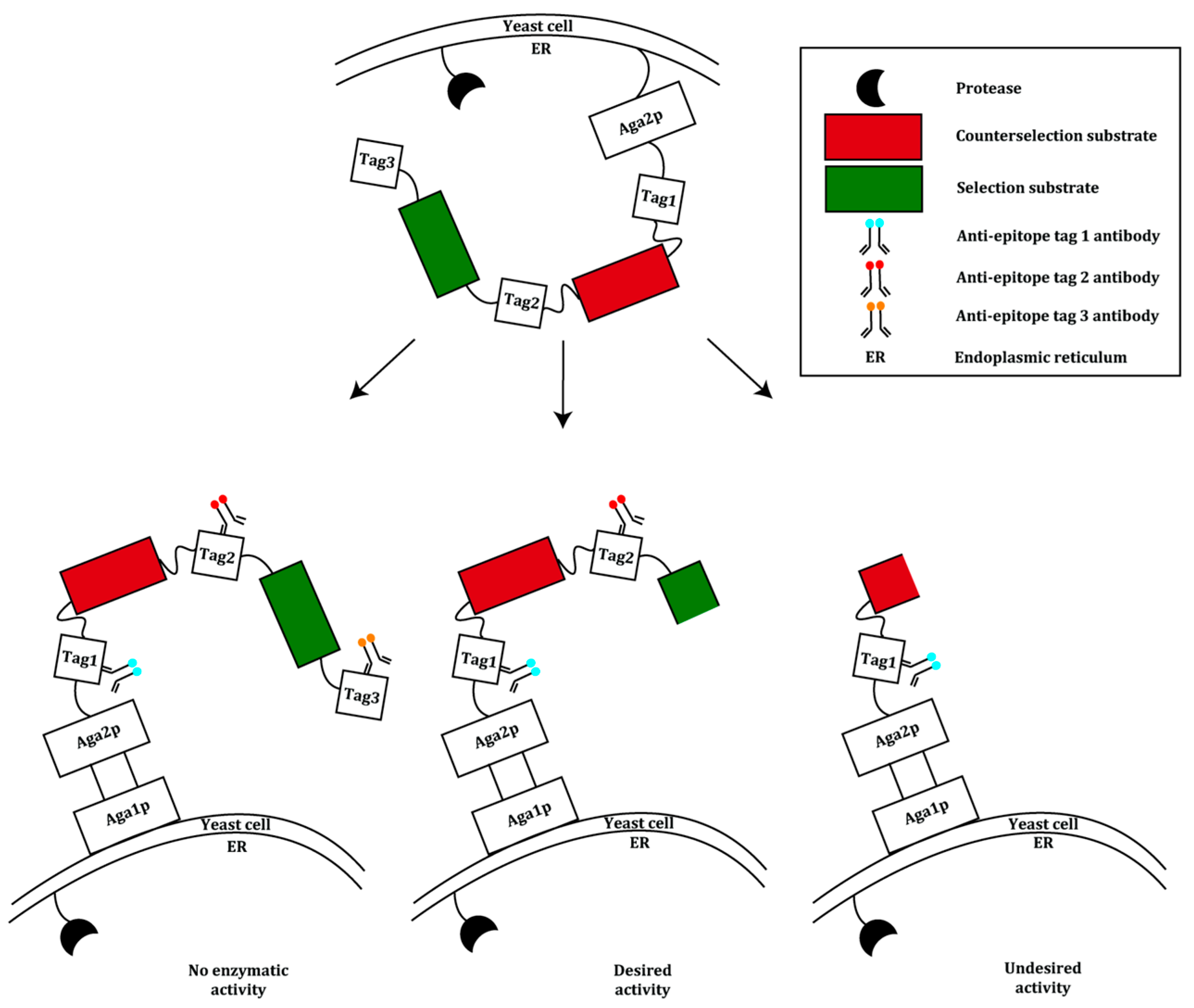
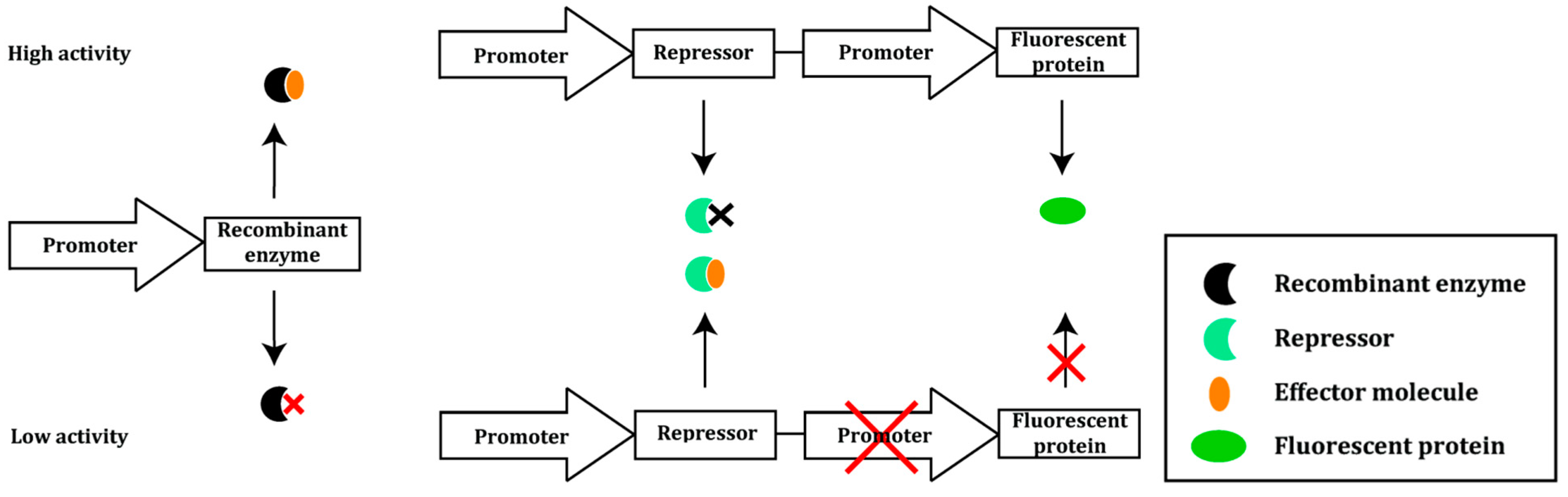
2.2. Prokaryotic Cytoplasmic Screening of Protein Variants

3. Man-Made Compartments in FACS Screening Platforms
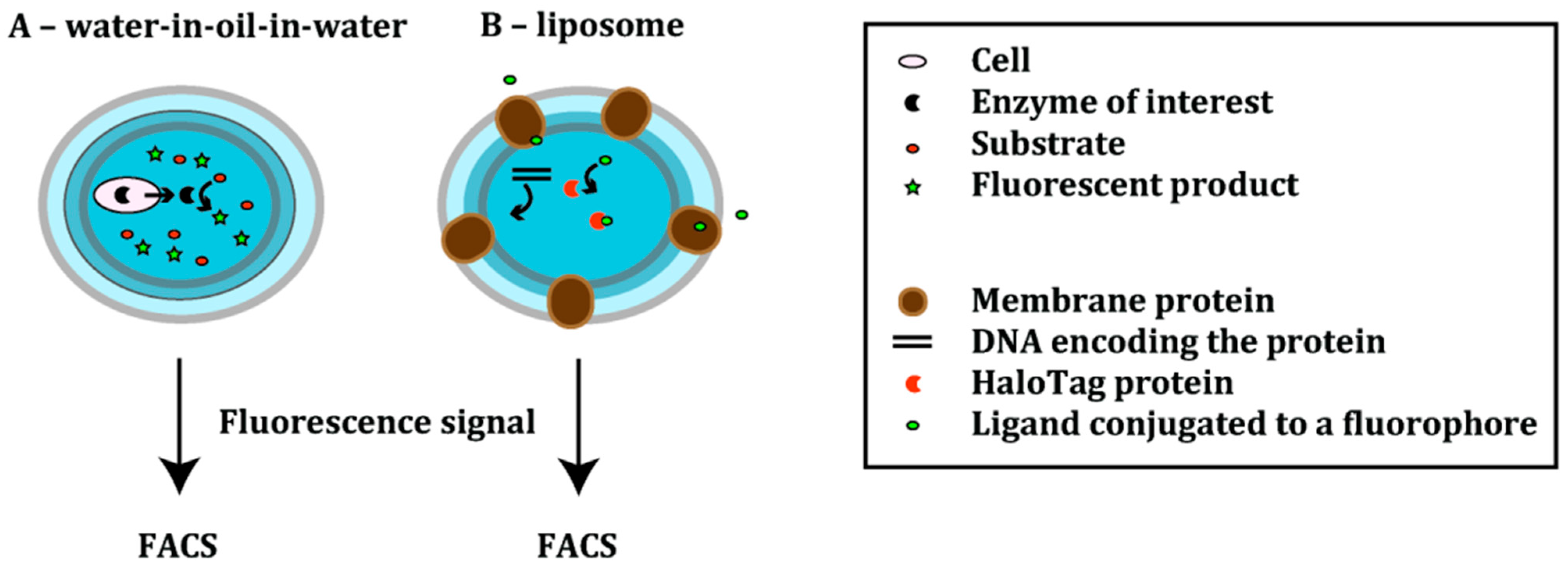
3.1. Emulsion-Based Compartments
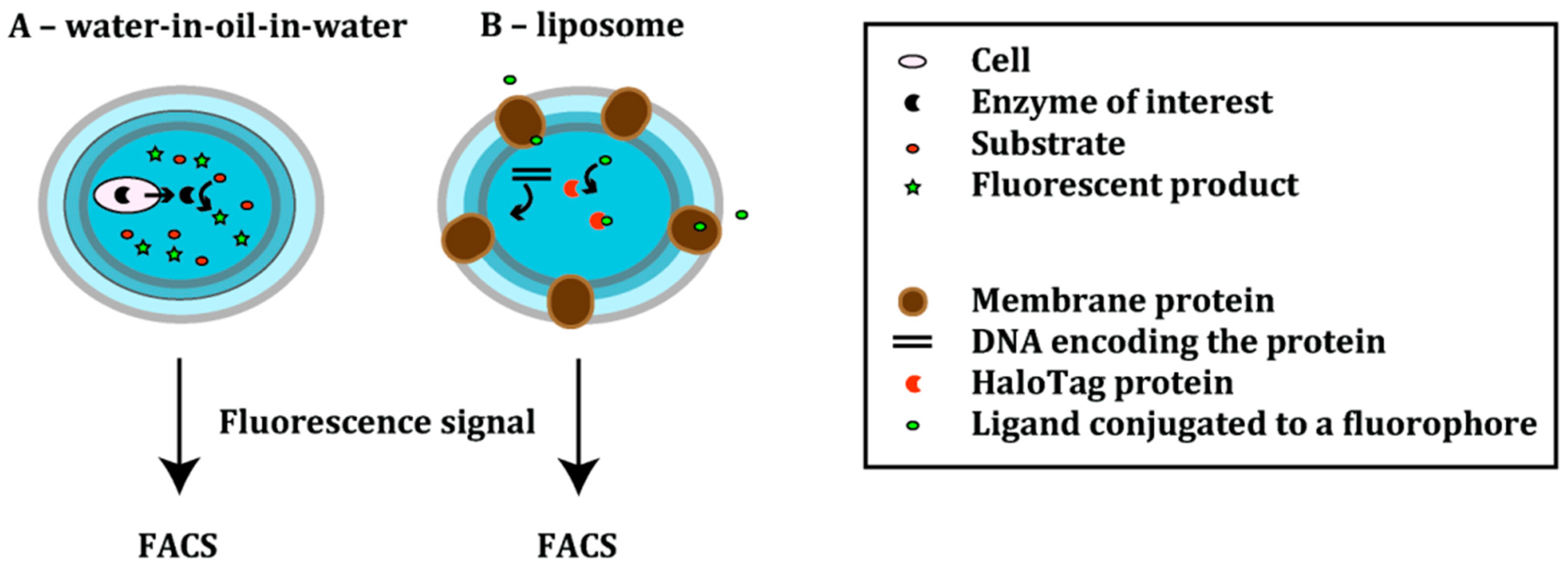
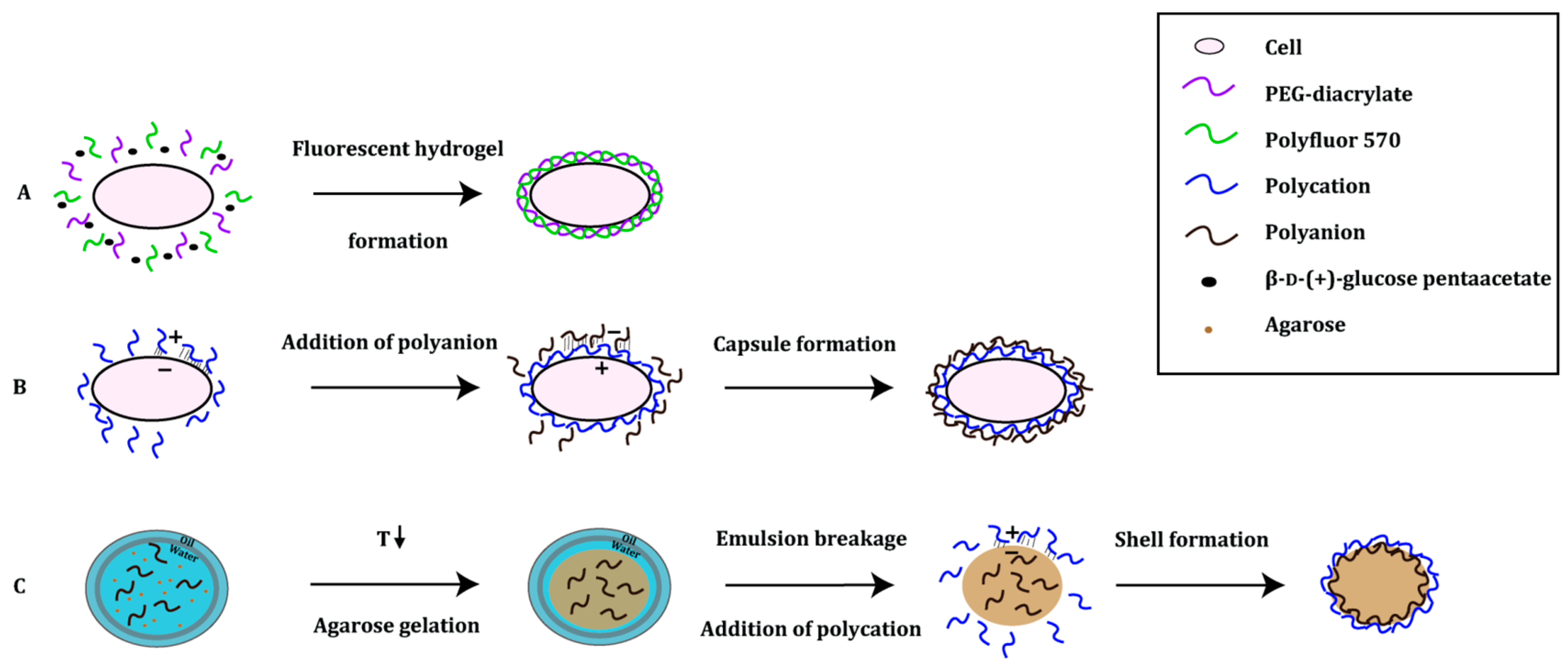
3.2. Polymer-Based Platforms


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Experiment | Compartment | Result | Throughput (Events/s) |
|---|---|---|---|---|
| Nature’s own compartments | ||||
| Antibodies (IgG) [14] | Selection and affinity maturation | Whole cells, mammalian display | Improvement in binding affinity towards human cytokine (hβNGF) | ND |
| Staphylococcus aureus sortase A (SrtA) [20] | Mutagenesis (epPCR, DNA shuffling and saturation mutagenesis) | Whole cells, yeast display | 140-fold improvement in LPETG-coupling activity | ND |
| S. aureus SrtA mutant (eSrtA [20]) [22] | Mutagenesis (epPCR, DNA shuffling and saturation mutagenesis) | Whole cells, yeast display | 51,000-fold change in specificity for LAETG instead of LPETG and a 125-fold change in specificity for LPESG instead of LPETG | ND |
| Tobacco Etch Virus protease (TEVp) [24] | Mutagenesis via epPCR | Whole cells, yeast display | 1100–5000-fold reversed substrate specificity | ~2 × 108 cells screened |
| TEVp [26] | Enrichment | Whole cells, intracellular expression | 69,000-fold enrichment for variants recognizing the natural substrate | 300 |
| TEVp [30] | Site-directed mutagenesis | Whole cells, intracellular expression | Substrate profiling | 300 |
| Pseudomonas plecoglossicida arginine deiminase (PpADI) [31] | Mutagenesis (epPCR) | Whole cells, intracellular expression | 2.8-fold increase in kcat/KM in comparison to the parent PpADI mutant M21 | 5000 |
| Man-made compartments (emulsion-based) | ||||
| Cellulase Cel5A [12] | Enrichment | Whole cells, double emulsion droplets | 12-fold enrichment of active variants form a mixture containing 5% cells expressing cellulase | 8000 to 20,000 |
| Subtilisin Carlsberg (SC) [36] | Mutagenesis (epPCR) | Whole cells, double emulsion droplets | 160% increase in resistance towards antipain dihydrochloride | 8000 |
| α-Hemolysin [37] | Mutagenesis (epPCR) | Cell-free, liposome display | 30-fold higher pore-forming activity | ND |
| Man-made compartments (polymer-based) | ||||
| Yersinia mollaretii phytase (YmPh) [45] | Mutagenesis (epPCR) | Whole cells, fur-shell | 97 U·mg−1 higher specific activity towards 4-methylumbelliferylphosphate (4-MUP) | 5000 |
| p-nitrobenzyl esterase (Bacillus licheniformis); lipase A (Bacillus subtilis); cellulase (CelA2) (metagenome library [55]) [50] | Mutagenesis (epPCR) | Whole cells, fur-shell | 7-fold increase in kcat towards pNPA; 1.3-fold increase in kcat towards β-d-(+)-glucose pentaacetate; 1.9-fold increase in kcat towards 4-methylumbelliferyl-β-d-cellobioside, respectively | 5000 |
| G-protein coupled receptors (GPCRs) [54] | Mutagenesis (StEP, Slonomics® technology [53], Martinsried, Germany) | Whole cells, CHESS | ~26.8 °C increase in thermostability of NTS1 | 8000 |
| sfGFP [52] | Mutagensis (epPCR) | Whole cells, CHESS | ~19 °C increase in thermostability in 2% (w/v) SDS | 8000 |
| Pseudomonas diminuta phosphotriesterase [48] | Mutagenesis (epPCR) | Cell lysate, GSBs | 19-fold increase in kcat/KM towards tetraethyl-O-fluorescein diphosphate | ~2800 |
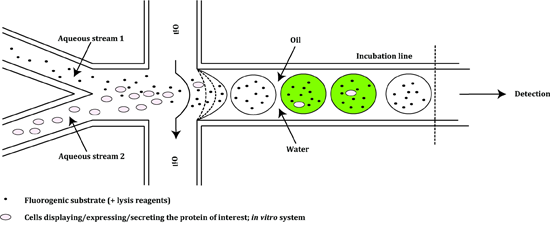
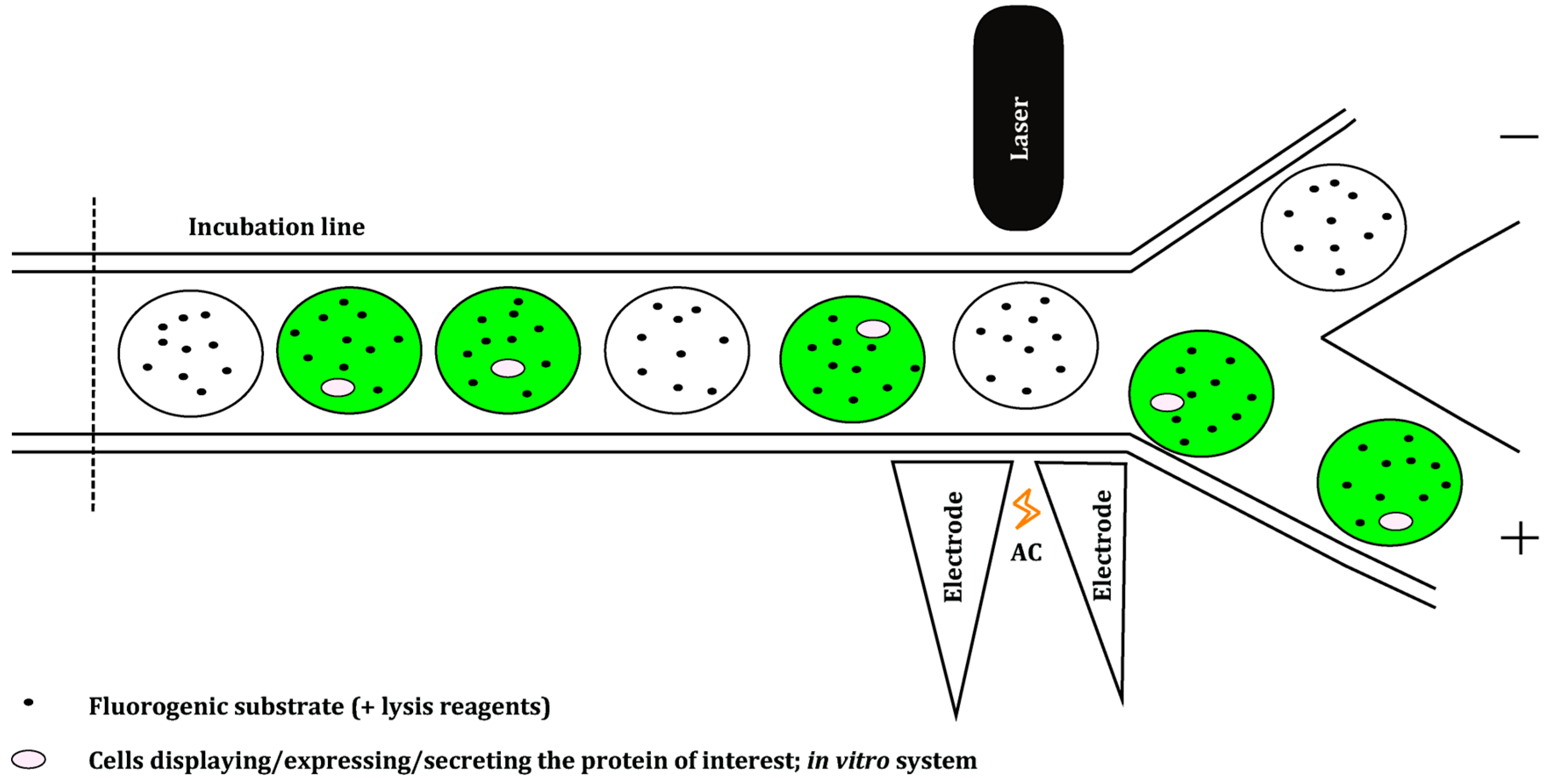
4. Microfluidics Screening Platforms


| Protein | Experiment | Compartment | Result | Throughput (Events/s) |
|---|---|---|---|---|
| Horse radish peroxidase [65] | Mutagenesis (epPCR and saturation mutagenesis) | Yeast display, drop-based microfluidic system | 7-fold increase in catalytic efficiency towards Amplex Ultrared (AUR) | 2000 |
| Pseudomonas aeruginosa arylsulfatase [66] | Mutagenesis (epPCR) | Cell lysate, drop-based microfluidic system | 6-fold increase in promiscuous hydrolytic activity towards the nonnative substrate phosphonate | 926 |
| Streptomyces sp. β-glucosidase [71] | Mutagenesis (epPCR) | Cell lysate, drop-based microfluidic system combined with high-throughput DNA sequencing | 5.3 °C increase in thermostability | >100 |
| β-galactosidase [68] | Enrichment | In vitro transcription and translation, drop-based microfluidic system | 502-fold enrichment of positive variants from a mixture of active and inactive variants | 2000 |
| Yeast strain MH34α-amylase [69] | Mutagenesis (UV irradiation) | Whole cells, drop-based microfluidic system | 2-fold increase in α-amylase production | 323 |
| Cellulases for the hydrolysis of cellulosic biomass [79] | Metagenomics | Whole cells, drop-based microfluidic system | Identification of microorganisms with 17-fold higher cellobiohydrolase activity and 7-fold higher endogluconase activity | 6667 |
5. Future Perspectives
| FACS-Based | Microfluidics-Based |
|---|---|
| The utility of FACS assays is limited to fluorophores that remain inside or on the surface of cells | The utility of microfluidics is broadened to components that are secreted |
| Water-in-oil emulsions must be converted into a water-in-oil-in-water emulsion (double emulsion) | Water-in-oil emulsions can be sorted directly |
| Pre-formed droplets are difficult to manipulate (restricted range of assays) | Pre-formed droplets are easy to manipulate: they can be divided, fused, incubated, analyzed, sorted, broken up |
| Limited control over the reaction conditions in a droplet | Much greater control over the reaction conditions in a droplet |
| Lack of control over the droplet volume, leading to polydispersity | Good control over the droplet volume, highly monodisperse |
| Requires standard cell sorters | Requires specialized instrumentation |
| Sorting speed up to 20,000 droplets/s | Sorting speed up to 2000 droplets/s |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Buchholz, K.; Kasche, V.; Bornscheuer, U.T. Biocatalysts and Enzyme Technology, 2nd ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012. [Google Scholar]
- Lutz, S.; Bornscheuer, U.T. Protein Engineering Handbook; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009; Volume 1. [Google Scholar]
- Reetz, M.T. Biocatalysis in organic chemistry and biotechnology: Past, present, and future. J. Am. Chem. Soc. 2013, 135, 12480–12496. [Google Scholar] [CrossRef] [PubMed]
- Martínez, R.; Schwaneberg, U. A roadmap to directed enzyme evolution and screening systems for biotechnological applications. Biol. Res. 2013, 46, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Arnold, F.H.; Volkov, A.A. Directed evolution of biocatalysts. Curr. Opin. Chem. Biol. 1999, 3, 54–59. [Google Scholar] [CrossRef]
- Boersma, Y.L.; Dröge, M.J.; Quax, W.J. Selection strategies for improved biocatalysts. FEBS J. 2007, 274, 2181–2195. [Google Scholar] [CrossRef] [PubMed]
- Leemhuis, H.; Kelly, R.M.; Dijkhuizen, L. Directed evolution of enzymes: Library screening strategies. IUBMB Life 2009, 61, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Tawfik, D.S.; Griffiths, A.D. Man-made cell-like compartments for molecular evolution. Nat. Biotechnol. 1998, 16, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, A.D.; Tawfik, D.S. Miniaturising the laboratory in emulsion droplets. Trends Biotechnol. 2006, 24, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Mayr, L.M.; Fuerst, P. The future of high-throughput screening. J. Biomol. Screen. 2008, 13, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Thorsen, T.; Maerkl, S.J.; Quake, S.R. Microfluidic large-scale integration. Science 2002, 298, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Ostafe, R.; Prodanovic, R.; Commandeur, U.; Fischer, R. Flow cytometry-based ultra-high-throughput screening assay for cellulase activity. Anal. Biochem. 2013, 435, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Beerli, R.R.; Bauer, M.; Buser, R.B.; Gwerder, M.; Muntwiler, S.; Maurer, P.; Saudan, P.; Bachmann, M.F. Isolation of human monoclonal antibodies by mammalian cell display. Proc. Natl. Acad. Sci. USA 2008, 105, 14336–14341. [Google Scholar] [CrossRef] [PubMed]
- Bowers, P.M.; Horlick, R.A.; Neben, T.Y.; Toobian, R.M.; Tomlinson, G.L.; Dalton, J.L.; Jones, H.A.; Chen, A.; Altobell, L.; Zhang, X.; et al. Coupling mammalian cell surface display with somatic hypermutation for the discovery and maturation of human antibodies. Proc. Natl. Acad. Sci. USA 2011, 108, 20455–20460. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Lin, B.; Li, J.; Qian, W.; Hou, S.; Zhang, D.; Kou, G.; Li, B.; Wang, H.; Chen, Y.; Guo, Y. Tolerability, pharmacokinetics and pharmacodynamics of CMAB007, a humanized anti-immunoglobulin E monoclonal antibody, in healthy Chinese subjects. MAbs 2012, 4, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Bowers, P.M.; Horlick, R.A.; Kehry, M.R.; Neben, T.Y.; Tomlinson, G.L.; Altobell, L.; Zhang, X.; Macomber, J.L.; Krapf, I.P.; Wu, B.F.; et al. Mammalian cell display for the discovery and optimization of antibody therapeutics. Methods 2014, 65, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Peled, J.U.; Kuang, F.L.; Iglesias-Ussel, M.D.; Roa, S.; Kalis, S.L.; Goodman, M.F.; Scharff, M.D. The biochemistry of somatic hypermutation. Annu. Rev. Immunol. 2008, 26, 481–511. [Google Scholar] [CrossRef] [PubMed]
- McConnell, A.D.; Zhang, X.; Macomber, J.L.; Chau, B.; Sheffer, J.C.; Rahmanian, S.; Hare, E.; Spasojevic, V.; Horlick, R.A.; King, D.J.; et al. A general approach to antibody thermostabilization. MAbs 2014, 6, 1274–1282. [Google Scholar] [CrossRef] [PubMed]
- Gai, S.A.; Wittrup, K.D. Yeast surface display for protein engineering and characterization. Curr. Opin. Struct. Biol. 2007, 17, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.; Dorr, B.M.; Liu, D.R. A general strategy for the evolution of bond-forming enzymes using yeast display. Proc. Natl. Acad. Sci. USA 2011, 108, 11399–11404. [Google Scholar] [CrossRef] [PubMed]
- Ilangovan, U.; Ton-That, H.; Iwahara, J.; Schneewind, O.; Clubb, R.T. Structure of sortase, the transpeptidase that anchors proteins to the cell wall of Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2001, 98, 6056–6061. [Google Scholar] [CrossRef] [PubMed]
- Dorr, B.M.; Ham, H.O.; An, C.; Chaikof, E.L.; Liu, D.R. Reprogramming the specificity of sortase enzymes. Proc. Natl. Acad. Sci. USA 2014, 111, 13343–13348. [Google Scholar] [CrossRef] [PubMed]
- Kapust, R.B.; Tözsér, J.; Fox, J.D.; Anderson, D.E.; Cherry, S.; Copeland, T.D.; Waugh, D.S. Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. 2001, 14, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Gebhard, M.C.; Li, Q.; Taft, J.M.; Georgiou, G.; Iverson, B.L. Engineering of TEV protease variants by yeast ER sequestration screening (YESS) of combinatorial libraries. Proc. Natl. Acad. Sci. USA 2013, 110, 7229–7234. [Google Scholar] [CrossRef] [PubMed]
- Phan, J.; Zdanov, A.; Evdokimov, A.G.; Tropea, J.E.; Peters, H.K.; Kapust, R.B.; Li, M.; Wlodawer, A.; Waugh, D.S. Structural basis for the substrate specificity of tobacco etch virus protease. J. Biol. Chem. 2002, 277, 50564–50572. [Google Scholar] [CrossRef] [PubMed]
- Kostallas, G.; Samuelson, P. Novel fluorescence-assisted whole-cell assay for engineering and characterization of proteases and their substrates. Appl. Environ. Microbiol. 2010, 76, 7500–7508. [Google Scholar] [CrossRef] [PubMed]
- Hersch, G.L.; Baker, T.A.; Sauer, R.T. SspB delivery of substrates for ClpXP proteolysis probed by the design of improved degradation tags. Proc. Natl. Acad. Sci. USA 2004, 101, 12136–12141. [Google Scholar] [CrossRef] [PubMed]
- DeLisa, M.P.; Samuelson, P.; Palmer, T.; Georgiou, G. Genetic analysis of the twin arginine translocator secretion pathway in bacteria. J. Biol. Chem. 2002, 277, 29825–29831. [Google Scholar] [CrossRef] [PubMed]
- Karzai, A.W.; Roche, E.D.; Sauer, R.T. The SsrA–SmpB system for protein tagging, directed degradation and ribosome rescue. Nat. Struct. Biol. 2000, 7, 449–455. [Google Scholar] [PubMed]
- Kostallas, G.; Löfdahl, P.-Å.; Samuelson, P. Substrate profiling of tobacco etch virus protease using a novel fluorescence-assisted whole-cell assay. PLoS ONE 2011, 6, e16136. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Kardashliev, T.; Pitzler, C.; Shehzad, A.; Lue, H.; Bernhagen, J.; Zhu, L.; Schwaneberg, U. A competitive flow cytometry screening system for directed evolution of therapeutic enzyme. ACS Synth. Biol. 2015, 4, 768–775. [Google Scholar] [CrossRef] [PubMed]
- Caldara, M.; Dupont, G.; Leroy, F.; Goldbeter, A.; Vuyst, L.D.; Cunin, R. Arginine biosynthesis in Escherichia coli: Experimental perturbation and mathematical modeling. J. Biol. Chem. 2008, 283, 6347–6358. [Google Scholar] [CrossRef] [PubMed]
- Dodevski, I.; Markou, G.C.; Sarkar, C.A. Conceptual and methodological advances in cell-free directed evolution. Curr. Opin. Struct. Biol. 2015, 33, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bernath, K.; Hai, M.; Mastrobattista, E.; Griffiths, A.D.; Magdassi, S.; Tawfik, D.S. In vitro compartmentalization by double emulsions: Sorting and gene enrichment by fluorescence activated cell sorting. Anal. Biochem. 2004, 325, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Prodanovic, R.; Ostafe, R.; Blanusa, M.; Schwaneberg, U. Vanadium bromoperoxidase-coupled fluorescent assay for flow cytometry sorting of glucose oxidase gene libraries in double emulsions. Anal. Bioanal. Chem. 2012, 404, 1439–1447. [Google Scholar] [CrossRef] [PubMed]
- Tu, R.; Martinez, R.; Prodanovic, R.; Klein, M.; Schwaneberg, U. A flow cytometry-based screening system for directed evolution of proteases. J. Biomol. Screen. 2011, 16, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Matsuura, T.; Sunami, T.; Nishikawa, T.; Kazuta, Y.; Yomo, T. Liposome display for in vitro selection and evolution of membrane proteins. Nat. Protoc. 2014, 9, 1578–1591. [Google Scholar] [CrossRef] [PubMed]
- Pautot, S.; Frisken, B.J.; Weitz, D.A. Production of unilamellar vesicles using an inverted emulsion. Langmuir 2003, 19, 2870–2879. [Google Scholar] [CrossRef]
- Yamada, A.; Yamanaka, T.; Hamada, T.; Hase, M.; Yoshikawa, K.; Baigl, D. Spontaneous transfer of phospholipid-coated oil-in-oil and water-in-oil micro-droplets through an oil/water interface. Langmuir 2006, 22, 9824–9828. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Hobaugh, M.R.; Shustak, C.; Cheley, S.; Bayley, H.; Gouaux, J.E. Structure of staphylococcal α-hemolysin, a heptameric transmembrane pore. Science 1996, 274, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Kuruma, Y.; Suzuki, T.; Ono, S.; Yoshida, M.; Ueda, T. Functional analysis of membranous Fo-a subunit of F1Fo-ATP synthase by in vitro protein synthesis. Biochem. J. 2012, 442, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Periasamy, A.; Shadiac, N.; Amalraj, A.; Garajová, S.; Nagarajan, Y.; Waters, S.; Mertens, H.D.T.; Hrmova, M. Cell-free protein synthesis of membrane (1,3)-β-d-glucan (curdlan) synthase: co-translational insertion in liposomes and reconstitution in nanodiscs. Biochim. Biophys. Acta 2013, 1828, 743–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soga, H.; Fujii, S.; Yomo, T.; Kato, Y.; Watanabe, H.; Matsuura, T. In vitro membrane protein synthesis inside cell-sized vesicles reveals the dependence of membrane protein integration on vesicle volume. ACS Synth. Biol. 2014, 3, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Städler, B.; Price, A.D.; Chandrawati, R.; Hosta-Rigau, L.; Zelikin, A.N.; Caruso, F. Polymer hydrogel capsules: En route toward synthetic cellular systems. Nanoscale 2009, 1, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Pitzler, C.; Wirtz, G.; Vojcic, L.; Hiltl, S.; Böker, A.; Martinez, R.; Schwaneberg, U. A fluorescent hydrogel-based flow cytometry high-throughput screening platform for hydrolytic enzymes. Chem. Biol. 2014, 21, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Diaspro, A.; Silvano, D.; Krol, S.; Cavalleri, O.; Gliozzi, A. Single living cell encapsulation in nano-organized polyelectrolyte shells. Langmuir 2002, 18, 5047–5050. [Google Scholar] [CrossRef]
- Hillberg, A.L.; Tabrizian, M. Biorecognition through layer-by-layer polyelectrolyte assembly: In-situ hybridization on living cells. Biomacromolecules 2006, 7, 2742–2750. [Google Scholar] [CrossRef] [PubMed]
- Fischlechner, M.; Schaerli, Y.; Mohamed, M.F.; Patil, S.; Abell, C.; Hollfelder, F. Evolution of enzyme catalysts caged in biomimetic gel-shell beads. Nat. Chem. 2014, 6, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Fenton, H.J.H. LXXIII.—Oxidation of tartaric acid in presence of iron. J. Chem. Soc. Trans. 1894, 65, 899–910. [Google Scholar] [CrossRef]
- Lülsdorf, N.; Pitzler, C.; Biggel, M.; Martinez, R.; Vojcic, L.; Schwaneberg, U. A flow cytometer-based whole cell screening toolbox for directed hydrolase evolution through fluorescent hydrogels. Chem. Commun. 2015, 51, 8679–8682. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.J.; Plückthun, A. Direct molecular evolution of detergent-stable G protein-coupled receptors using polymer encapsulated cells. J. Mol. Biol. 2013, 425, 662–677. [Google Scholar] [CrossRef] [PubMed]
- Yong, K.J.; Scott, D.J. Rapid directed evolution of stabilized proteins with cellular high-throughput encapsulation solubilization and screening (CHESS). Biotechnol. Bioeng. 2015, 112, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Schlinkmann, K.M.; Hillenbrand, M.; Rittner, A.; Künz, M.; Strohner, R.; Plückthun, A. Maximizing detergent stability and functional expression of a GPCR by exhaustive recombination and evolution. J. Mol. Biol. 2012, 422, 414–428. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.J.; Kummer, L.; Egloff, P.; Bathgate, R.A.D.; Plückthun, A. Improving the apo-state detergent stability of NTS1 with CHESS for pharmacological and structural studies. BBA Biomembr. 2014, 1838, 2817–2824. [Google Scholar] [CrossRef] [PubMed]
- Ilmberger, N.; Meske, D.; Juergensen, J.; Schulte, M.; Barthen, P.; Rabausch, U.; Angelov, A.; Mientus, M.; Liebl, W.; Schmitz, R.A.; et al. Metagenomic cellulases highly tolerant towards the presence of ionic liquids—Linking thermostability and halotolerance. Appl. Microbiol. Biotechnol. 2012, 95, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-G.; Xu, Z.-R.; Wang, J.-H. Manipulation of droplets in microfluidic systems. TrAC Trends Anal. Chem. 2010, 29, 141–157. [Google Scholar] [CrossRef]
- Zinchenko, A.; Devenish, S.R.A.; Kintses, B.; Colin, P.-Y.; Fischlechner, M.; Hollfelder, F. One in a million: Flow cytometric sorting of single cell-lysate assays in monodisperse picoliter double emulsion droplets for directed evolution. Anal. Chem. 2014, 86, 2526–2533. [Google Scholar] [CrossRef] [PubMed]
- Drott, J.; Rosengren, L.; Lindström, K.; Laurell, T. Pore morphology influence on catalytic turn-over for enzyme activated porous silicon matrices. Thin Solid Films 1998, 330, 161–166. [Google Scholar] [CrossRef]
- Beneyton, T.; Coldren, F.; Baret, J.-C.; Griffiths, A.D.; Taly, V. CotA laccase: High-throughput manipulation and analysis of recombinant enzyme libraries expressed in E. coli using droplet-based microfluidics. Analyst 2014, 139, 3314–3323. [Google Scholar] [CrossRef] [PubMed]
- Teh, S.-Y.; Lin, R.; Hung, L.-H.; Lee, A.P. Droplet microfluidics. Lab Chip 2008, 8, 198–220. [Google Scholar] [CrossRef] [PubMed]
- Anna, S.L.; Bontoux, N.; Stone, H.A. Formation of dispersions using “flow focusing” in microchannels. Appl. Phys. Lett. 2003, 82, 364–366. [Google Scholar] [CrossRef]
- Utada, A.S.; Lorenceau, E.; Link, D.R.; Kaplan, P.D.; Stone, H.A.; Weitz, D.A. Monodisperse double emulsions generated from a microcapillary device. Science 2005, 308, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Ismagilov, R.F. Millisecond kinetics on a microfluidic chip using nanoliters of reagents. J. Am. Chem. Soc. 2003, 125, 14613–14619. [Google Scholar] [CrossRef] [PubMed]
- Baret, J.-C.; Miller, O.J.; Taly, V.; Ryckelynck, M.; El-Harrak, A.; Frenz, L.; Rick, C.; Samuels, M.L.; Hutchison, J.B.; Agresti, J.J.; et al. Fluorescence-activated droplet sorting (FADS): Efficient microfluidic cell sorting based on enzymatic activity. Lab Chip 2009, 9, 1850–1858. [Google Scholar] [CrossRef] [PubMed]
- Agresti, J.J.; Antipov, E.; Abate, A.R.; Ahn, K.; Rowat, A.C.; Baret, J.-C.; Marquez, M.; Klibanov, A.M.; Griffiths, A.D.; Weitz, D.A. Ultrahigh-throughput screening in drop-based microfluidics for directed evolution. Proc. Natl. Acad. Sci. USA 2010, 107, 4004–4009. [Google Scholar] [CrossRef] [PubMed]
- Kintses, B.; Hein, C.; Mohamed, M.F.; Fischlechner, M.; Courtois, F.; Lainé, C.; Hollfelder, F. Picoliter cell lysate assays in microfluidic droplet compartments for directed enzyme evolution. Chem. Biol. 2012, 19, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Abate, A.R.; Hung, T.; Mary, P.; Agresti, J.J.; Weitz, D.A. High-throughput injection with microfluidics using picoinjectors. Proc. Natl. Acad. Sci. USA 2010, 107, 19163–19166. [Google Scholar] [CrossRef] [PubMed]
- Fallah-Araghi, A.; Baret, J.-C.; Ryckelynck, M.; Griffiths, A.D. A completely in vitro ultrahigh-throughput droplet-based microfluidic screening system for protein engineering and directed evolution. Lab Chip 2012, 12, 882–891. [Google Scholar] [CrossRef] [PubMed]
- Sjostrom, S.L.; Bai, Y.; Huang, M.; Liu, Z.; Nielsen, J.; Joensson, H.N.; Andersson Svahn, H. High-throughput screening for industrial enzyme production hosts by droplet microfluidics. Lab Chip 2014, 14, 806–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostafe, R.; Prodanovic, R.; Ung, W.L.; Weitz, D.A.; Fischer, R. A high-throughput cellulase screening system based on droplet microfluidics. Biomicrofluidics 2014, 8, 41102. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.A.; Tran, T.M.; Abate, A.R. Dissecting enzyme function with microfluidic-based deep mutational scanning. Proc. Natl. Acad. Sci. USA 2015. [Google Scholar] [CrossRef] [PubMed]
- Nurumbetov, G.; Ballard, N.; Bon, S.A.F. A simple microfluidic device for fabrication of double emulsion droplets and polymer microcapsules. Polym. Chem. 2012, 3, 1043–1047. [Google Scholar] [CrossRef]
- Fidalgo, L.M.; Whyte, G.; Bratton, D.; Kaminski, C.F.; Abell, C.; Huck, W.T.S. From microdroplets to microfluidics: Selective emulsion separation in microfluidic devices. Angew. Chem. Int. Ed. 2008, 47, 2042–2045. [Google Scholar] [CrossRef] [PubMed]
- Jacquier, H.; Birgy, A.; Le Nagard, H.; Mechulam, Y.; Schmitt, E.; Glodt, J.; Bercot, B.; Petit, E.; Poulain, J.; Barnaud, G.; et al. Capturing the mutational landscape of the β-lactamase TEM-1. Proc. Natl. Acad. Sci. USA 2013, 110, 13067–13072. [Google Scholar] [CrossRef] [PubMed]
- Fowler, D.M.; Fields, S. Deep mutational scanning: A new style of protein science. Nat. Methods 2014, 11, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Kuruma, Y.; Ying, B.-W.; Umekage, S.; Ueda, T. Cell-free translation systems for protein engineering. FEBS J. 2006, 273, 4133–4140. [Google Scholar] [CrossRef] [PubMed]
- Najah, M.; Mayot, E.; Mahendra-Wijaya, I.P.; Griffiths, A.D.; Ladame, S.; Drevelle, A. New glycosidase substrates for droplet-based microfluidic screening. Anal. Chem. 2013, 85, 9807–9814. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, P.; Eck, J. Metagenomics and industrial applications. Nat. Rev. Microbiol. 2005, 3, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Najah, M.; Calbrix, R.; Mahendra-Wijaya, I.P.; Beneyton, T.; Griffiths, A.D.; Drevelle, A. Droplet-based microfluidics platform for ultra-high-throughput bioprospecting of cellulolytic microorganisms. Chem. Biol. 2014, 21, 1722–1732. [Google Scholar] [CrossRef] [PubMed]
- Davids, T.; Schmidt, M.; Böttcher, D.; Bornscheuer, U.T. Strategies for the discovery and engineering of enzymes for biocatalysis. Curr. Opin. Chem. Biol. 2013, 17, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Bornscheuer, U.T.; Huisman, G.W.; Kazlauskas, R.J.; Lutz, S.; Moore, J.C.; Robins, K. Engineering the third wave of biocatalysis. Nature 2012, 485, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Steiner, K.; Schwab, H. Recent advances in rational approaches for enzyme engineering. Comput. Struct. Biotechnol. J. 2012, 2, e201209010. [Google Scholar] [CrossRef] [PubMed]
- Kries, H.; Blomberg, R.; Hilvert, D. De novo enzymes by computational design. Curr. Opin. Chem. Biol. 2013, 17, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Deal, K.S.; Easley, C.J. Self-regulated, droplet-based sample chopper for microfluidic absorbance detection. Anal. Chem. 2012, 84, 1510–1516. [Google Scholar] [CrossRef] [PubMed]
- Cecchini, M.P.; Hong, J.; Lim, C.; Choo, J.; Albrecht, T.; deMello, A.J.; Edel, J.B. Ultrafast surface enhanced resonance Raman scattering detection in droplet-based microfluidic systems. Anal. Chem. 2011, 83, 3076–3081. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.A.; Zheng, G.; Mukherjee, N.; Yang, C. On-chip continuous monitoring of motile microorganisms on an ePetri platform. Lab Chip 2012, 12, 2385–2390. [Google Scholar] [CrossRef] [PubMed]
- Sciambi, A.; Abate, A.R. Accurate microfluidic sorting of droplets at 30 kHz. Lab Chip 2014, 15, 47–51. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wójcik, M.; Telzerow, A.; Quax, W.J.; Boersma, Y.L. High-Throughput Screening in Protein Engineering: Recent Advances and Future Perspectives. Int. J. Mol. Sci. 2015, 16, 24918-24945. https://doi.org/10.3390/ijms161024918
Wójcik M, Telzerow A, Quax WJ, Boersma YL. High-Throughput Screening in Protein Engineering: Recent Advances and Future Perspectives. International Journal of Molecular Sciences. 2015; 16(10):24918-24945. https://doi.org/10.3390/ijms161024918
Chicago/Turabian StyleWójcik, Magdalena, Aline Telzerow, Wim J. Quax, and Ykelien L. Boersma. 2015. "High-Throughput Screening in Protein Engineering: Recent Advances and Future Perspectives" International Journal of Molecular Sciences 16, no. 10: 24918-24945. https://doi.org/10.3390/ijms161024918