
Small RNAs in Plant Responses to Abiotic Stresses: Regulatory Roles and Study Methods

 , and
, and
Abstract
:
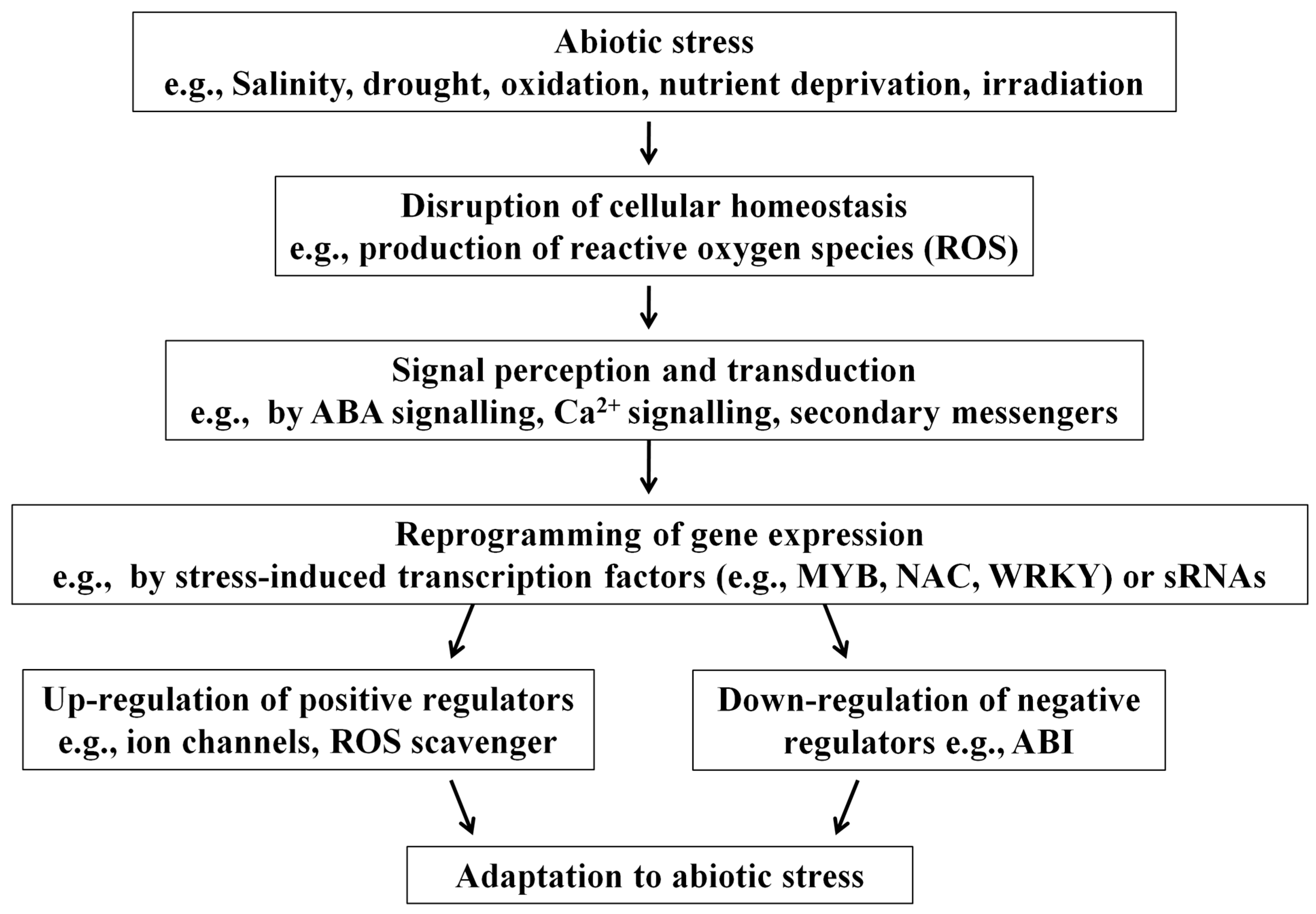
1. Introduction: The Importance of Small RNAs
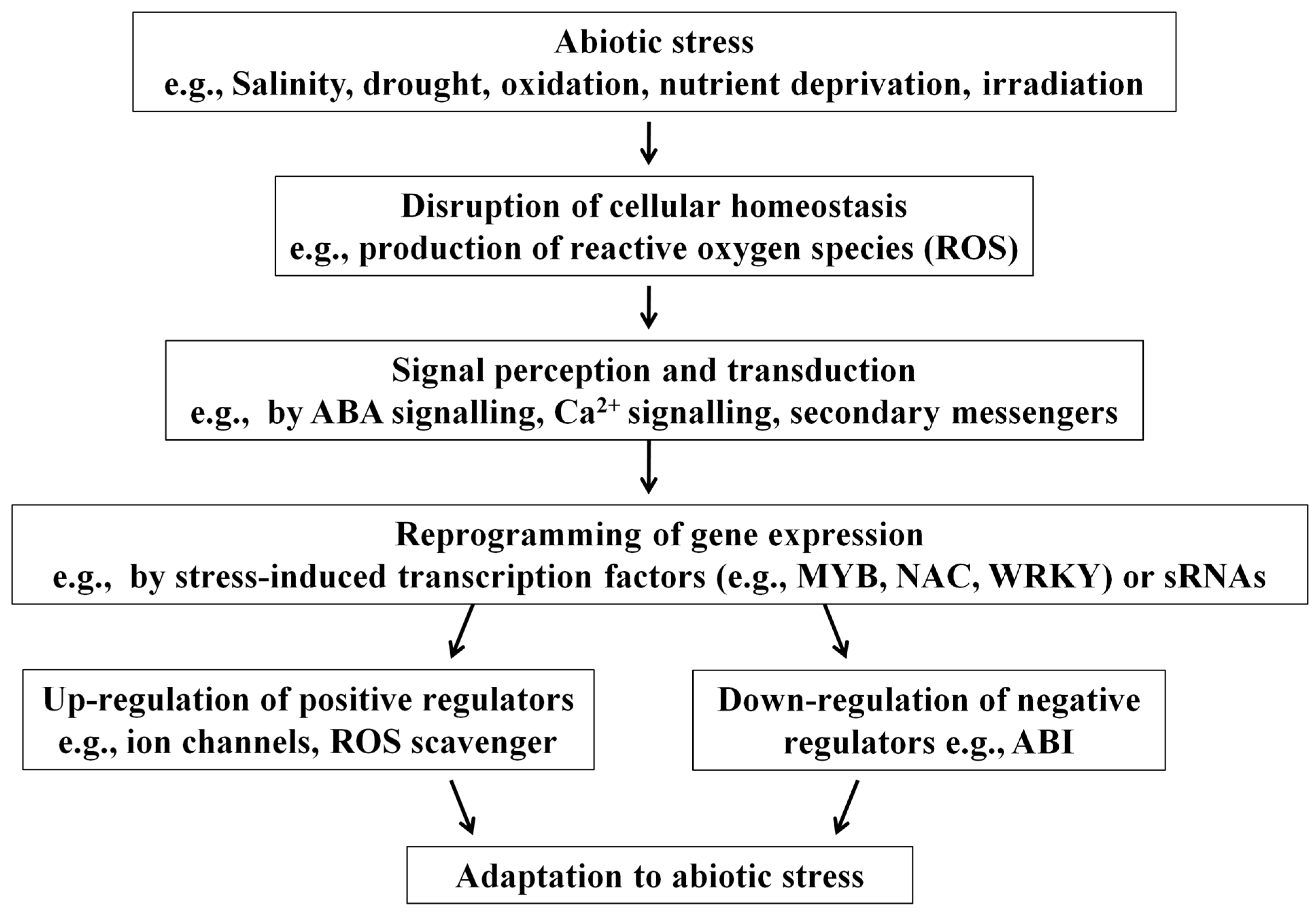
2. Mechanisms of sRNA-Mediated Genetic Regulation
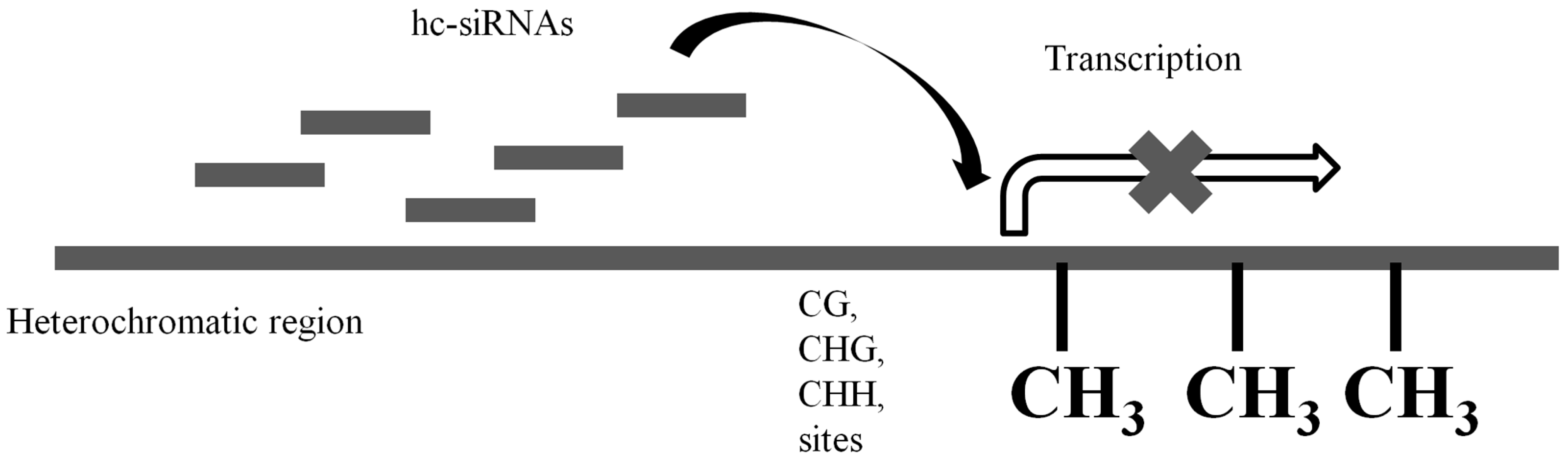
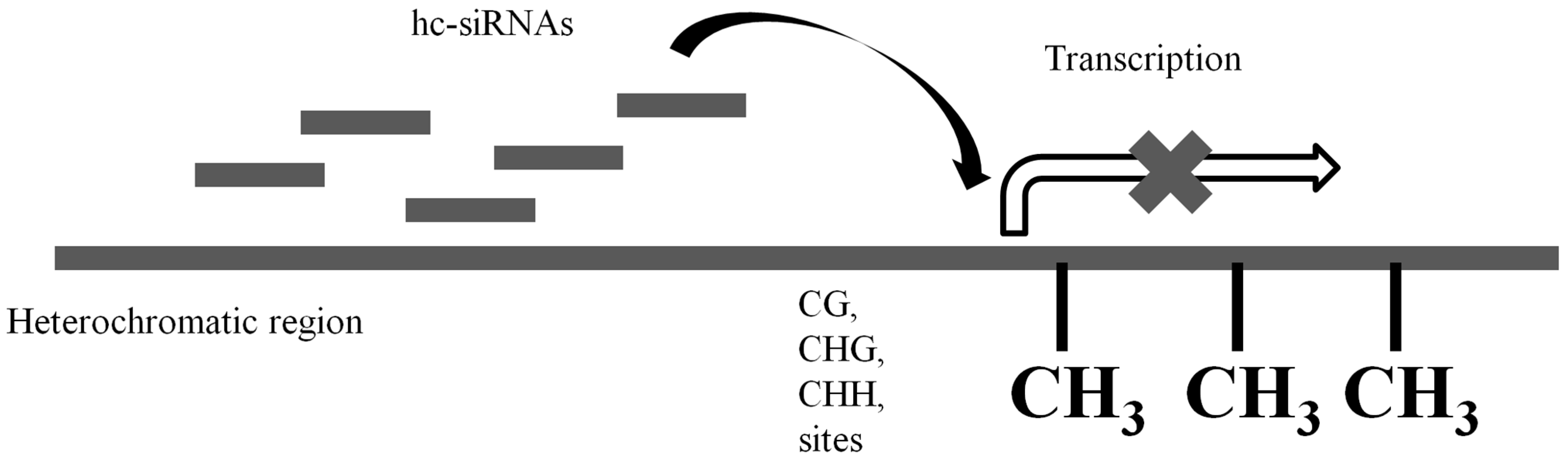
2.1. Transcriptional Gene Silencing
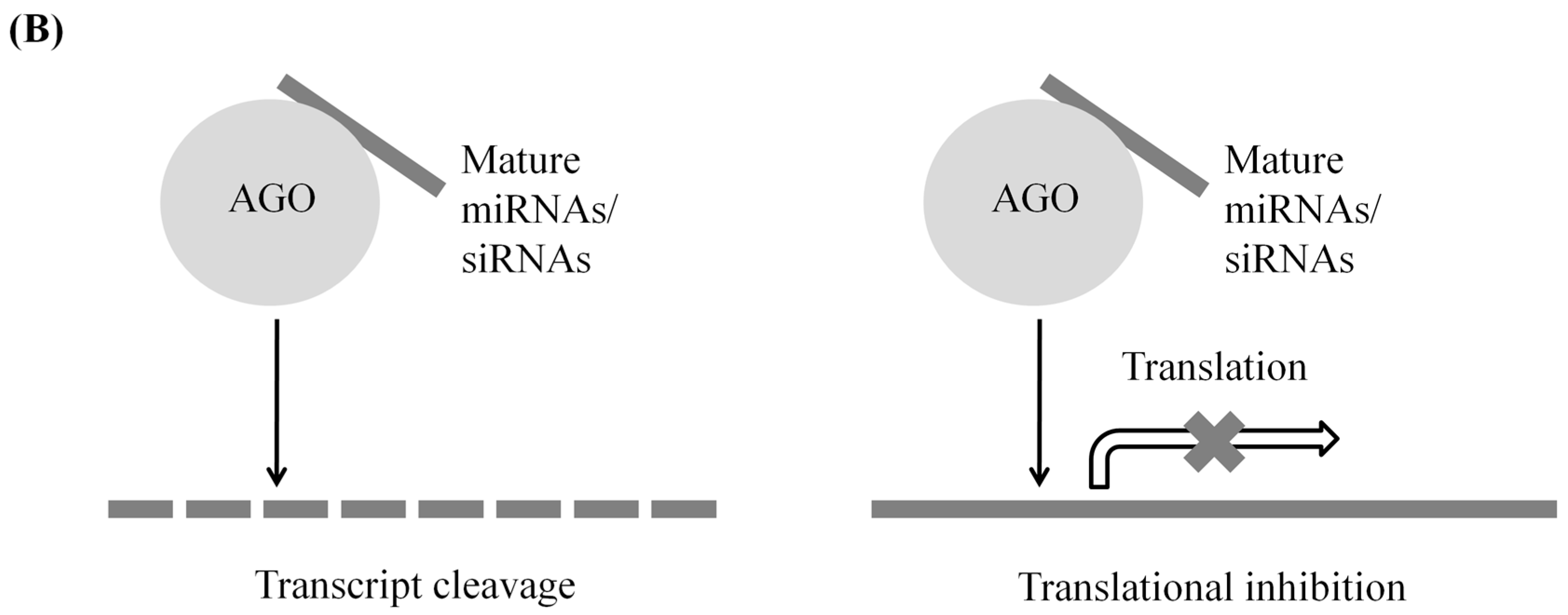
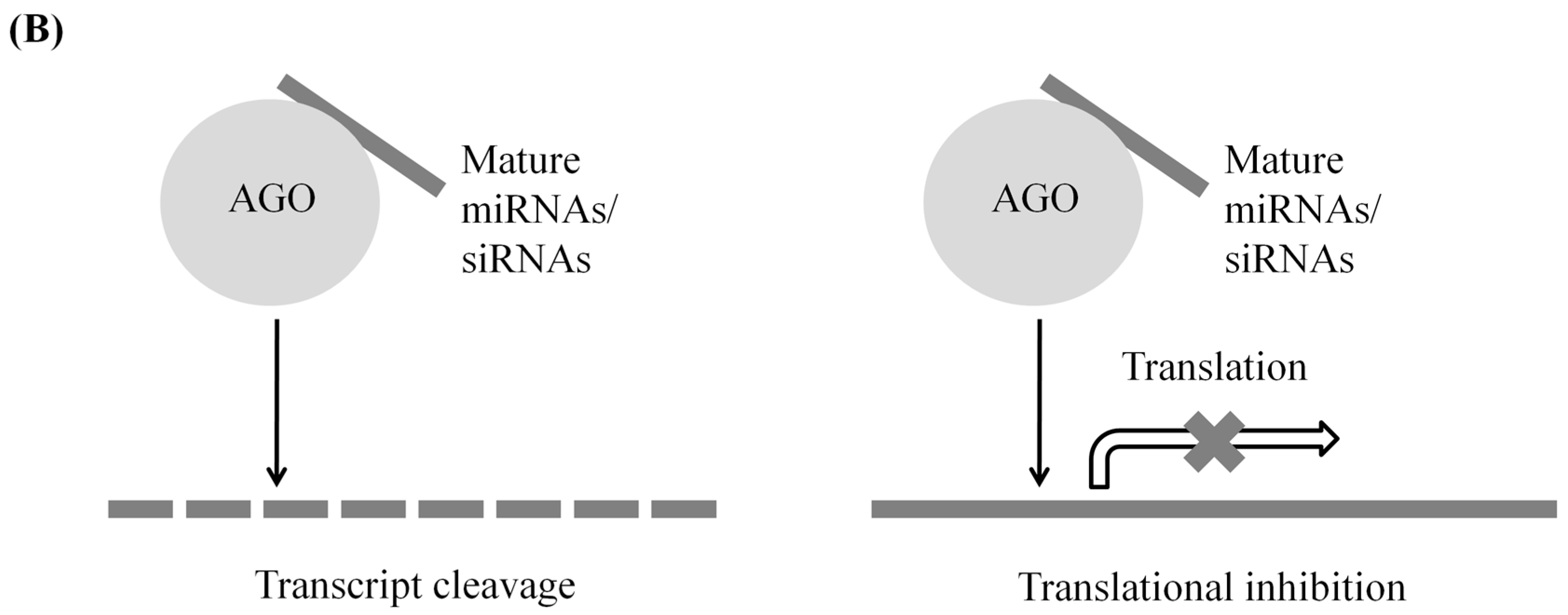
2.2. Post-Transcriptional Gene Silencing


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mechanism of Regulation | sRNA Types Participated | Origin of sRNAs | Targets of sRNAs | Modes of Action |
|---|---|---|---|---|
| Transcriptional gene silencing | hc-siRNAs | Transcripts of heterochromatic regions | Heterochromatic regions (act in cis) | RNA-directed DNA methylation |
| Post-transcriptional gene silencing | miRNAs | Short stem-loop-forming transcripts | Other transcripts (act in both cis and trans) | Transcript cleavage; translational inhibition |
| TAS-transcripts | Triggering double strand synthesis of TAS-transcripts | |||
| NAT-siRNAs | Antisense transcripts | Other transcripts in both cis and trans | Transcript cleavage; translational inhibition | |
| ta-siRNAs | TAS-loci derived transcripts | Other transcripts in both cis and trans | Transcript cleavage; translational inhibition |
2.2.1. miRNA
2.2.2. siRNA
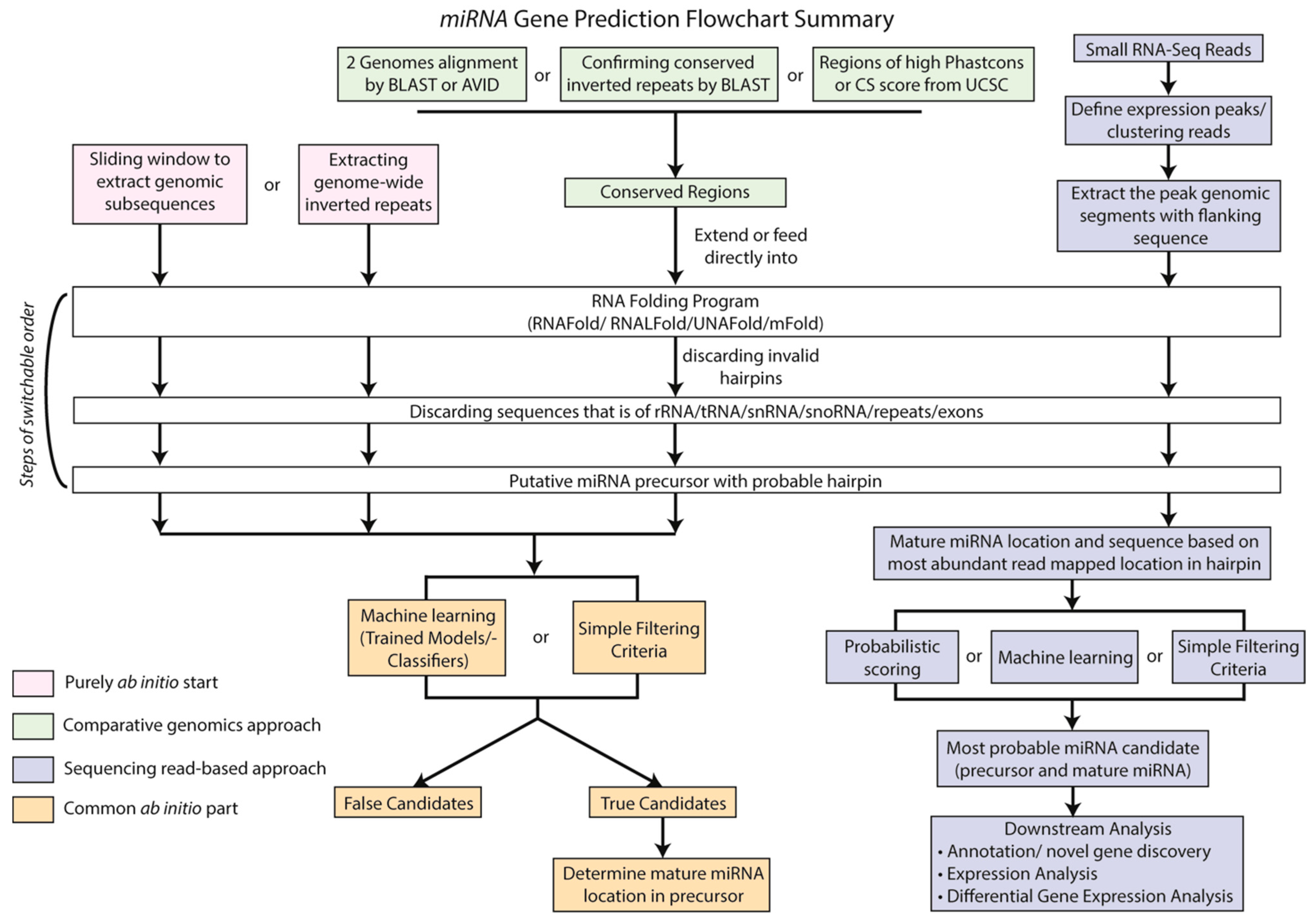
3. Computational Methods to Identify sRNAs
3.1. Computational Prediction of miRNA Gene Loci
| Tool | Application | Property | Reference |
|---|---|---|---|
| MIRFINDER | Detection of potential conserved miRNAs in Arabidopsis thaliana and Oryza satica | The use of NCBI BLAST to search for conserved short hits (~21–22 nt). The hits with flanking sequences were identified as putative hairpin precursors. | [58] |
| miRSeeker | Identification of novel miRNA candidates that are conserved in insect, nematode, or vertebrate | The use of AVID to align Drosophila melanogaster and Drosophila pseudoobscura euchromatic sequences to search for conserved sequences meeting these two criteria: 1. Having extended stem-loop structure; 2. Having nucleotide divergence from known miRNAs. | [59] |
| mirCoS | Prediction of mammalian miRNAs | Detection of known miRNAs and prediction of new miRNAs based on sequence, secondary structure and conservation by comparing human and mouse genomes. | [60] |
| miRRim | Identification of novel miRNAs in human | Detection of miRNAs with the use of a hidden Markov model. | [61] |
| miRAlign | Detection of miRNA homologs or orthologs in animals. | Detection of miRNAs based on sequence and structure alignment. The sensitivity is better than BLAST search and ERPIN search with comparable specificity. | [62] |
| microHARVESTER | Identification of plant miRNA homologs | Identification of plant miRNA homologs based on query miRNA. | [63] |
| MiRscan | Identification of vertebrate miRNA genes | Evaluation of conserved stem-loops. | [64] |
| miRDeep | Identification of miRNAs with deep sequencing data | The use of known miRNA training set obtained from Caenorhabditis elegans to deduce parameters of most probable miRNA precursors. These parameters were used to score precursor candidates using a probabilistic approach. | [65] |
| MiRCheck | Identification of miRNAs in Arabidopsis thaliana and Oryza sativa | The use of EINVERTED from EMBOSS [66] to predict genome-wide inverted repeats in both Arabidopsis thaliana and Oryza sativa to define possible hairpin regions, and the check for segments with high homology between Arabidopsis thaliana hairpins and Oryza sativa hairpins using Patscan. | [67] |
3.1.1. Choosing the Right Tools for Plant miRNA Discovery
| Tool | Property | Reference |
|---|---|---|
| miRDeep-P | Adopting miRDeep core algorithm with modified step of setting a maximal value for the MFE log-odds score to account for longer plant miRNA precursors | [70] |
| miRPlant | Implementing miRDeep* [74] with 100 and 200 nt extended genomic regions from mapped read peaks to include more bona fide miRNA precursor candidates | [71] |
| miR-PREFeR | Filtering miRNA precursor candidates with criteria suggested in [75] for annotating plant miRNAs | [76] |
| MIReNA | Filtering putative precursors with length-normalized and GC-normalized MFE to accommodate the prediction of plant miRNAs | [72] |
| ShortStack | Defining structural miRNA parameters based on selected annotated miRNA in miRBase depending on the “miRType” specified by user, either “plant” or “animal”, subsequently filter candidates with criteria suggested in [75] | [77] |
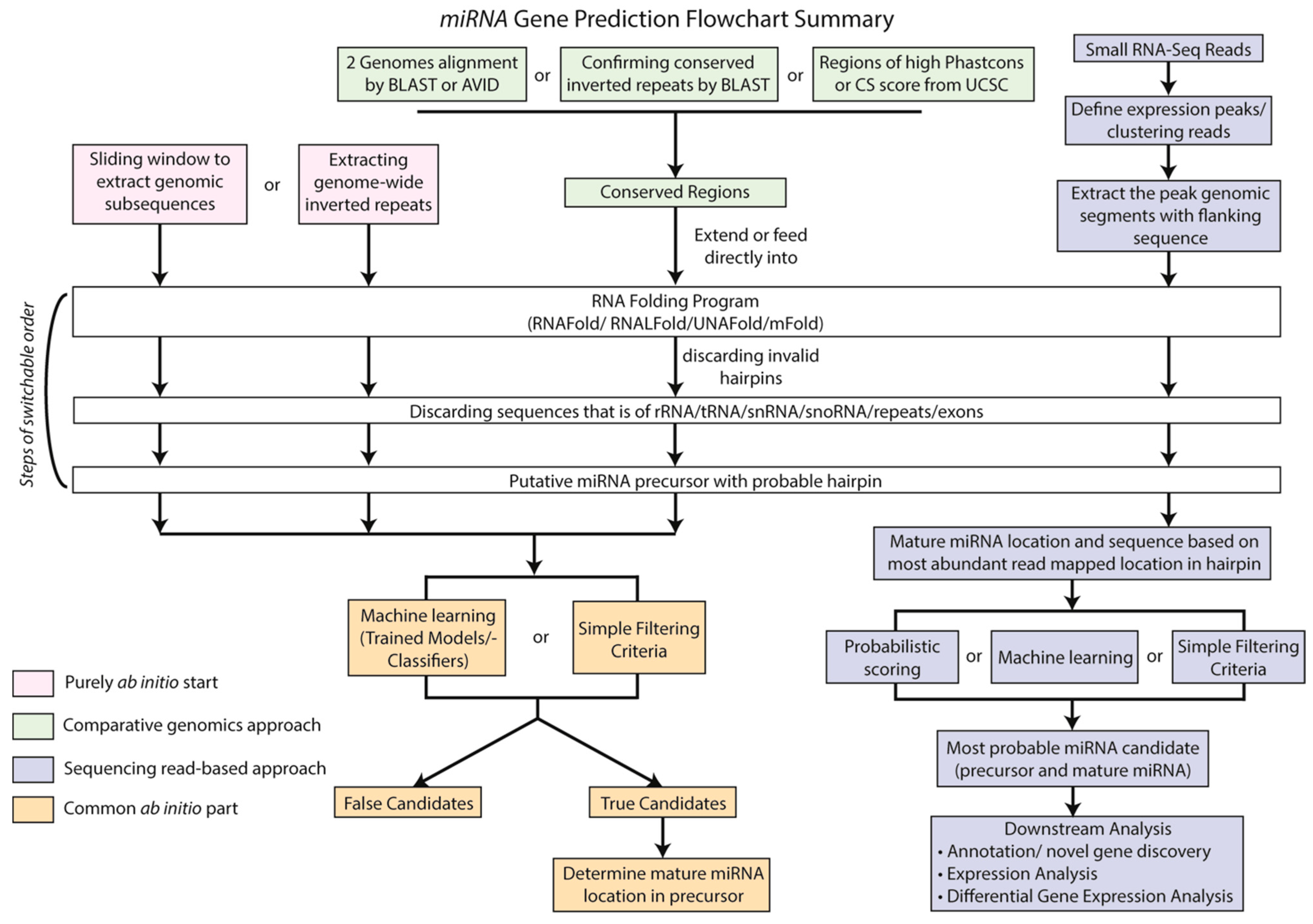
3.1.2. Computational Prediction of TAS-Like Loci
3.1.3. Common Features of Target Prediction Tools
3.1.4. Functions of Prediction Tools
3.2. Computational Prediction of sRNA Targets
3.3. High-Throughput sRNA Target Identification—Degradome
4. Experimental Validations of Predicted sRNAs
| Method | Stress | sRNA | Reference |
|---|---|---|---|
| Validation of the existence of sRNA | |||
| qRT-PCR | Salinity, copper deficiency | miR397, miR857 | [103] |
| Northern blot | Salinity, sulphur deprivation, oxidative stress, nitrogen deficiency, inorganic phosphtase deprivation, drought, irradiation, copper deficiency | miR399, miR395, miR398, miR408 | [34,90,104,105,106,107,108,109,110] |
| Validation of the target gene | |||
| 5′ RACE | Copper deficiency | miR397, miR408 | [103] |
| Transgenic plant for functional test | |||
| Arabidopsis | Inorganic phosphate deprivation | miR399 | [104] |
| Arabidopsis | Drought | miR196 | [111] |
| Creeping bentgrass | Drought, salinity | miR319 | [112] |
4.1. Validation of sRNAs Expression
4.1.1. Quantitative Detection of sRNAs by Northern Blot
4.1.2. Quantitative Detection of sRNAs by qPCR
4.1.3. In Situ Hybridization for Spatiotemporal Detection of sRNAs
4.2. Validation of sRNA Targets
4.2.1. Labeled miRNA Pull-down (LAMP) Assay System
4.2.2. RNA Ligase-Mediated Amplification of cDNA End (RLM-RACE)
4.3. Functional Validation of sRNAs
4.3.1. Reporter Assays
4.3.2. Validation of the Effect of the sRNA of Interest on the Target Gene Expression
| Purpose | Method | Advantage(s) | Disadvantage(s) |
|---|---|---|---|
| Validation of the existence of predicted sRNA | Northern blot | Quantitative, simultaneous detection of sRNA and its precursor | Optimization steps are needed to improve sensitivity and specificity. |
| qPCR | Small amount of RNA is required | Normalization by spike-in control or housekeeping genes can be unreliable. | |
| Validation of the existence of predicted sRNA | In situ hybridization | Allows tissue-specific and spatiotemporal detection | Optimization steps are needed to improve sensitivity and specificity. |
| Functional analysis of sRNA | LAMP assay | Straightforward | An in vitro approach, the pre-miRNA processing and specificity have been questioned; not popular for plants. |
| RLM-RACE | Previous knowledge of the cleaved mRNA is not required | Cannot distinguish by which type of sRNA the mRNA cleavage is mediated. | |
| Reporter assays | An in vivo approach | Transformation of the species under study is needed. |
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nakashima, K.; Yamaguchi-Shinozaki, K.; Shinozaki, K. The transcriptional regulatory network in the drought response and its crosstalk in abiotic stress responses including drought, cold, and heat. Front. Plant Sci. 2014, 5, 170. [Google Scholar] [CrossRef] [PubMed]
- Gehan, M.A.; Greenham, K.; Mockler, T.C.; McClung, C.R. Transcriptional networks—Crops, clocks, and abiotic stress. Curr. Opin. Plant Biol. 2015, 24, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Priest, H.D.; Fox, S.E.; Rowley, E.R.; Murray, J.R.; Michael, T.P.; Mockler, T.C. Analysis of global gene expression in Brachypodium distachyon reveals extensive network plasticity in response to abiotic stress. PLoS ONE 2014, 9, e87499. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, A.J.; Baulcombe, D.C. A species of small antisense RNA in posttranscriptional gene silencing in plants. Science 1999, 286, 950–952. [Google Scholar] [CrossRef] [PubMed]
- Dalmay, T.; Hamilton, A.; Mueller, E.; Baulcombe, D.C. Potato virus X amplicons in Arabidopsis mediate genetic and epigenetic gene silencing. Plant Cell 2000, 12, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Hutvagner, G.; Mlynarova, L.; Nap, J.P. Detailed characterization of the posttranscriptional gene-silencing-related small RNA in a GUS gene-silenced tobacco. RNA 2000, 6, 1445–1454. [Google Scholar] [CrossRef] [PubMed]
- Reinhart, B.J.; Weinstein, E.G.; Rhoades, M.W.; Bartel, B.; Bartel, D.P. MicroRNAs in plants. Genes Dev. 2002, 16, 1616–1626. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Llave, C.; Kasschau, K.D.; Rector, M.A.; Carrington, J.C. Endogenous and silencing-associated small RNAs in plants. Plant Cell 2002, 14, 1605–1619. [Google Scholar] [CrossRef] [PubMed]
- Sunkar, R.; Zhu, J.K. Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell 2004, 16, 2001–2019. [Google Scholar] [CrossRef] [PubMed]
- Sunkar, R.; Girke, T.; Jain, P.K.; Zhu, J.K. Cloning and characterization of microRNAs from rice. Plant Cell 2005, 17, 1397–1411. [Google Scholar] [CrossRef] [PubMed]
- Baulcombe, D. RNA silencing in plants. Nature 2004, 431, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Kooter, J.M.; Matzke, M.A.; Meyer, P. Listening to the silent genes: Transgene silencing, gene regulation and pathogen control. Trends Plant Sci. 1999, 4, 340–347. [Google Scholar] [CrossRef]
- Mette, M.; Aufsatz, W.; van der Winden, J.; Matzke, M.; Matzke, A. Transcriptional silencing and promoter methylation triggered by double-stranded RNA. EMBO J. 2000, 19, 5194–5201. [Google Scholar] [CrossRef] [PubMed]
- Matzke, M.; Kanno, T.; Daxinger, L.; Huettel, B.; Matzke, A.J. RNA-mediated chromatin-based silencing in plants. Curr. Opin. Cell Biol. 2009, 21, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Creasey, K.M.; Zhai, J.; Borges, F.; van Ex, F.; Regulski, M.; Meyers, B.C.; Martienssen, R.A. miRNAs trigger widespread epigenetically activated siRNAs from transposons in Arabidopsis. Nature 2014, 508, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, A.; Voinnet, O.; Chappell, L.; Baulcombe, D. Two classes of short interfering RNA in RNA silencing. EMBO J. 2002, 21, 4671–4679. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Johansen, L.K.; Gustafson, A.M.; Kasschau, K.D.; Lellis, A.D.; Zilberman, D.; Jacobsen, S.E.; Carrington, J.C. Genetic and functional diversification of small RNA pathways in plants. PLoS Biol. 2004, 2, E104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zilberman, D.; Cao, X.; Jacobsen, S.E. Argonaute4 control of locus-specific siRNA accumulation and DNA and histone methylation. Science 2003, 299, 716–719. [Google Scholar] [CrossRef] [PubMed]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Shah, S.; Irshad, M. Immediate and transgenerational regulation of plant stress response through DNA methylation. J. Agric. Sci. 2015, 7, 144–151. [Google Scholar] [CrossRef]
- Boyko, A.; Blevins, T.; Yao, Y.; Golubov, A.; Bilichak, A.; Ilnytskyy, Y.; Hollander, J.; Meins, F., Jr.; Kovalchuk, I. Transgenerational adaptation of Arabidopsis to stress requires DNA methylation and the function of dicer-like proteins. PLoS ONE 2010, 5, e9514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovalchuk, O.; Burke, P.; Arkhipov, A.; Kuchma, N.; James, S.J.; Kovalchuk, I.; Pogribny, I. Genome hypermethylation in Pinus silvestris of chernobyl—A mechanism for radiation adaptation? Mutat. Res. Fundam. Mol. Mech. Mutagen. 2003, 529, 13–20. [Google Scholar] [CrossRef]
- Barakat, A.; Sriram, A.; Park, J.; Zhebentyayeva, T.; Main, D.; Abbott, A. Genome wide identification of chilling responsive microRNAs in Prunus persica. BMC Genom. 2012, 13, 481. [Google Scholar] [CrossRef] [PubMed]
- Meister, G. Argonaute proteins: Functional insights and emerging roles. Nat. Rev. Genet. 2013, 14, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Carthew, R.W.; Sontheimer, E.J. Origins and mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Wang, Z.; Li, S.; Yu, B.; Liu, J.-Y.; Chen, X. Intergenic transcription by RNA polymerase II coordinates Pol IV and Pol V in siRNA-directed transcriptional gene silencing in Arabidopsis. Genes Dev. 2009, 23, 2850–2860. [Google Scholar] [CrossRef] [PubMed]
- Dunoyer, P.; Himber, C.; Voinnet, O. Dicer-like 4 is required for RNA interference and produces the 21-nucleotide small interfering RNA component of the plant cell-to-cell silencing signal. Nat. Genet. 2005, 37, 1356–1360. [Google Scholar] [CrossRef] [PubMed]
- Gasciolli, V.; Mallory, A.C.; Bartel, D.P.; Vaucheret, H. Partially redundant functions of Arabidopsis dicer-like enzymes and a role for DCL4 in producing trans-acting siRNAs. Curr. Biol. 2005, 15, 1494–1500. [Google Scholar] [CrossRef] [PubMed]
- Baumberger, N.; Baulcombe, D.C. Arabidopsis argonaute1 is an RNA slicer that selectively recruits microRNAs and short interfering RNAs. Proc. Natl. Acad. Sci. USA 2005, 102, 11928–11933. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Iwasaki, S.; Watanabe, T.; Utsumi, M.; Watanabe, Y. The mechanism selecting the guide strand from small RNA duplexes is different among Argonaute proteins. Plant Cell Physiol. 2008, 49, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.S.; Xie, Q.; Fei, J.F.; Chua, N.H. MicroRNA directs mRNA cleavage of the transcription factor NAC1 to downregulate auxin signals for Arabidopsis lateral root development. Plant Cell 2005, 17, 1376–1386. [Google Scholar] [CrossRef] [PubMed]
- Llave, C.; Xie, Z.; Kasschau, K.D.; Carrington, J.C. Cleavage of scarecrow-like mRNA targets directed by a class of Arabidopsis miRNA. Science 2002, 297, 2053–2056. [Google Scholar] [CrossRef] [PubMed]
- Jagadeeswaran, G.; Saini, A.; Sunkar, R. Biotic and abiotic stress down-regulate miR398 expression in Arabidopsis. Planta 2009, 229, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Sunkar, R.; Kapoor, A.; Zhu, J.K. Posttranscriptional induction of two Cu/Zn superoxide dismutase genes in Arabidopsis is mediated by downregulation of miR398 and important for oxidative stress tolerance. Plant Cell 2006, 18, 2051–2065. [Google Scholar] [CrossRef] [PubMed]
- Bouché, N. New insights into miR398 functions in Arabidopsis. Plant Signal. Behav. 2010, 5, 684–686. [Google Scholar] [CrossRef] [PubMed]
- Peragine, A.; Yoshikawa, M.; Wu, G.; Albrecht, H.L.; Poethig, R.S. SGS3 and SGS2/SDE1/RDR6 are required for juvenile development and the production of trans-acting sirnas in Arabidopsis. Genes Dev. 2004, 18, 2368–2379. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, F.; Vaucheret, H.; Rajagopalan, R.; Lepers, C.; Gasciolli, V.; Mallory, A.C.; Hilbert, J.L.; Bartel, D.P.; Crete, P. Endogenous trans-acting siRNAs regulate the accumulation of Arabidopsis mRNAs. Mol. Cell 2004, 16, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Borsani, O.; Zhu, J.; Verslues, P.E.; Sunkar, R.; Zhu, J.K. Endogenous siRNAs derived from a pair of natural cis-antisense transcripts regulate salt tolerance in Arabidopsis. Cell 2005, 123, 1279–1291. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.J.; Gaasterland, T.; Chua, N.H. Genome-wide prediction and identification of cis-natural antisense transcripts in Arabidopsis thaliana. Genome Biol. 2005, 6, R30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Pan, X.; Cobb, G.P.; Anderson, T.A. Plant microRNA: A small regulatory molecule with big impact. Dev. Biol. 2006, 289, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Rajeswaran, R.; Aregger, M.; Zvereva, A.S.; Borah, B.K.; Gubaeva, E.G.; Pooggin, M.M. Sequencing of RDR6-dependent double-stranded RNAs reveals novel features of plant siRNA biogenesis. Nucleic Acids Res. 2012, 40, 6241–6254. [Google Scholar] [CrossRef] [PubMed]
- Zhai, J.; Jeong, D.H.; de Paoli, E.; Park, S.; Rosen, B.D.; Li, Y.; Gonzalez, A.J.; Yan, Z.; Kitto, S.L.; Grusak, M.A.; et al. MicroRNAs as master regulators of the plant NB-LRR defense gene family via the production of phased, trans-acting siRNAs. Genes Dev. 2011, 25, 2540–2553. [Google Scholar] [CrossRef] [PubMed]
- Fei, Q.; Xia, R.; Meyers, B.C. Phased, secondary, small interfering RNAs in posttranscriptional regulatory networks. Plant Cell 2013, 25, 2400–2415. [Google Scholar] [CrossRef] [PubMed]
- Allen, E.; Xie, Z.; Gustafson, A.M.; Carrington, J.C. MicroRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 2005, 121, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Axtell, M.J.; Jan, C.; Rajagopalan, R.; Bartel, D.P. A two-hit trigger for siRNA biogenesis in plants. Cell 2006, 127, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, R.; Vaucheret, H.; Trejo, J.; Bartel, D.P. A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev. 2006, 20, 3407–3425. [Google Scholar] [CrossRef] [PubMed]
- Fahlgren, N.; Montgomery, T.A.; Howell, M.D.; Allen, E.; Dvorak, S.K.; Alexander, A.L.; Carrington, J.C. Regulation of AUXIN RESPONSE FACTOR3 by TAS3 ta-siRNA affects developmental timing and patterning in Arabidopsis. Curr. Biol. 2006, 16, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, D.; Spriggs, A.; Yang, J.; Pogson, B.J.; Dennis, E.S.; Wilson, I.W. Hypoxia-responsive microRNAs and trans-acting small interfering RNAs in Arabidopsis. J. Exp. Bot. 2010, 61, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Jen, C.H.; Michalopoulos, I.; Westhead, D.R.; Meyer, P. Natural antisense transcripts with coding capacity in Arabidopsis may have a regulatory role that is not linked to double-stranded RNA degradation. Genome Biol. 2005, 6, R51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Zhou, X.; Xia, J.; Zhou, X. Identification of microRNAs and natural antisense transcript-originated endogenous siRNAs from small-RNA deep sequencing data. Methods Mol. Biol. 2012, 883, 221–227. [Google Scholar] [PubMed]
- Jin, H.; Vacic, V.; Girke, T.; Lonardi, S.; Zhu, J.K. Small RNAs and the regulation of cis-natural antisense transcripts in Arabidopsis. BMC Mol. Biol. 2008, 9, 6. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Sunkar, R.; Jin, H.; Zhu, J.K.; Zhang, W. Genome-wide identification and analysis of small RNAs originated from natural antisense transcripts in Oryza sativa. Genome Res. 2009, 19, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Zubko, E.; Meyer, P. A natural antisense transcript of the Petunia hybrida Sho gene suggests a role for an antisense mechanism in cytokinin regulation. Plant J. 2007, 52, 1131–1139. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xia, J.; Lii, Y.E.; Barrera-Figueroa, B.E.; Zhou, X.; Gao, S.; Lu, L.; Niu, D.; Chen, Z.; Leung, C. Genome-wide analysis of plant NAT-siRNAs reveals insights into their distribution, biogenesis and function. Genome Biol. 2012, 13, R20. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, M.; Miura, S.; Nei, M. Origins and evolution of microRNA genes in plant species. Genome Biol. Evol. 2012, 4, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Merchan, F.; Boualem, A.; Crespi, M.; Frugier, F. Plant polycistronic precursors containing non-homologous microRNAs target transcripts encoding functionally related proteins. Genome Biol. 2009, 10, R136. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, E.; Wuyts, J.; Rouze, P.; van de Peer, Y. Detection of 91 potential conserved plant microRNAs in Arabidopsis thaliana and Oryza sativa identifies important target genes. Proc. Natl. Acad. Sci. USA 2004, 101, 11511–11516. [Google Scholar] [CrossRef] [PubMed]
- Lai, E.C.; Tomancak, P.; Williams, R.W.; Rubin, G.M. Computational identification of Drosophila microRNA genes. Genome Biol. 2003, 4, R42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, Y.; Engstrom, P.G.; Lenhard, B. Mammalian microRNA prediction through a support vector machine model of sequence and structure. PLoS ONE 2007, 2, e946. [Google Scholar] [CrossRef] [PubMed]
- Terai, G.; Komori, T.; Asai, K.; Kin, T. miRRim: A novel system to find conserved miRNAs with high sensitivity and specificity. RNA 2007, 13, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, J.; Li, F.; Gu, J.; He, T.; Zhang, X.; Li, Y. MicroRNA identification based on sequence and structure alignment. Bioinformatics 2005, 21, 3610–3614. [Google Scholar] [CrossRef] [PubMed]
- Dezulian, T.; Remmert, M.; Palatnik, J.F.; Weigel, D.; Huson, D.H. Identification of plant microRNA homologs. Bioinformatics 2006, 22, 359–360. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.P.; Glasner, M.E.; Yekta, S.; Burge, C.B.; Bartel, D.P. Vertebrate microRNA genes. Science 2003, 299, 1540–1540. [Google Scholar] [CrossRef] [PubMed]
- Sewer, A.; Paul, N.; Landgraf, P.; Aravin, A.; Pfeffer, S.; Brownstein, M.J.; Tuschl, T.; van Nimwegen, E.; Zavolan, M. Identification of clustered microRNAs using an ab initio prediction method. BMC Bioinform. 2005, 6, 267. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. Emboss: The European molecular biology open software suite. Trends Genet. 2000, 16, 276–277. [Google Scholar]
- Jones-Rhoades, M.W.; Bartel, D.P. Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol. Cell 2004, 14, 787–799. [Google Scholar] [CrossRef] [PubMed]
- Axtell, M.J.; Westholm, J.O.; Lai, E.C. Vive la difference—Biogenesis ans evolution of microRNAs in plants and animals. Genome Biol. 2011, 12, 221. [Google Scholar] [CrossRef] [PubMed]
- Thakur, V.; Wanchana, S.; Xu, M.; Bruskiewich, R.; Quick, W.P.; Mosig, A.; Zhu, X.G. Characterization of statistical features for plant microRNA prediction. BMC Genom. 2011, 12, 108. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, L. miRDeep-P: A computational tool for analyzing the microRNA transcriptome in plants. Bioinformatics 2011, 27, 2614–2615. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Lai, J.; Sajjanhar, A.; Lehman, M.L.; Nelson, C.C. miRPlant: An integrated tool for identification of plant miRNA from RNA sequencing data. BMC Bioinform. 2014, 15, 275. [Google Scholar] [CrossRef] [PubMed]
- Mathelier, A.; Carbone, A. MIReNA: Finding microRNAs with high accuracy and no learning at genome scale and from deep sequencing data. Bioinformatics 2010, 26, 2226–2234. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, M.R.; Chen, W.; Adamidi, C.; Maaskola, J.; Einspanier, R.; Knespel, S.; Rajewsky, N. Discovering microRNAs from deep sequencing data using miRDeep. Nat. Biotechnol. 2008, 26, 407–415. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Lai, J.; Lehman, M.L.; Nelson, C.C. miRDeep*: An integrated application tool for miRNA identification from RNA sequencing data. Nucleic Acids Res. 2013, 41, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Meyers, B.C.; Axtell, M.J.; Bartel, B.; Bartel, D.P.; Baulcombe, D.; Bowman, J.L.; Cao, X.; Carrington, J.C.; Chen, X.; Green, P.J.; et al. Criteria for annotation of plant microRNAs. Plant Cell 2008, 20, 3186–3190. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Sun, Y. miR-PREFeR: An accurate, fast and easy-to-use plant miRNA prediction tool using small RNA-seq data. Bioinformatics 2014, 30, 2837–2839. [Google Scholar] [CrossRef] [PubMed]
- Axtell, M.J. ShortStack: Comprehensive annotation and quantification of small RNA genes. RNA 2013, 19, 740–751. [Google Scholar] [CrossRef]
- Chen, H.-M.; Li, Y.-H.; Wu, S.-H. Bioinformatic prediction and experimental validation of a microRNA-directed tandem trans-acting siRNA cascade in Arabidopsis. Proc. Natl. Acad. Sci. USA 2007, 104, 3318–3323. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Zhao, P.X. pssRNAMiner: A plant short small RNA regulatory cascade analysis server. Nucleic Acids Res. 2008, 36, W114–W118. [Google Scholar] [CrossRef] [PubMed]
- Howell, M.D.; Fahlgren, N.; Chapman, E.J.; Cumbie, J.S.; Sullivan, C.M.; Givan, S.A.; Kasschau, K.D.; Carrington, J.C. Genome-wide analysis of the RNA-dependent RNA polymerase6/dicer-like4 pathway in Arabidopsis reveals dependency on miRNA-and tasiRNA-directed targeting. Plant Cell 2007, 19, 926–942. [Google Scholar] [CrossRef] [PubMed]
- De Paoli, E.; Dorantes-Acosta, A.; Zhai, J.; Accerbi, M.; Jeong, D.-H.; Park, S.; Meyers, B.C.; Jorgensen, R.A.; Green, P.J. Distinct extremely abundant siRNAs associated with cosuppression in petunia. RNA 2009, 15, 1965–1970. [Google Scholar] [CrossRef] [PubMed]
- McGinnis, S.; Madden, T.L. BLAST: At the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 2004, 32, W20–W25. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.F.; Waterman, M.S. Identification of common molecular subsequences. J. Mol. Biol. 1981, 147, 195–197. [Google Scholar] [CrossRef]
- Fahlgren, N.; Howell, M.D.; Kasschau, K.D.; Chapman, E.J.; Sullivan, C.M.; Cumbie, J.S.; Givan, S.A.; Law, T.F.; Grant, S.R.; Dangl, J.L. High-throughput sequencing of Arabidopsis microRNAs: Evidence for frequent birth and death of miRNA genes. PLoS ONE 2007, 2, e219. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. miRU: An automated plant miRNA target prediction server. Nucleic Acids Res. 2005, 33, W701–W704. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Orban, R.; Baker, B. SoMART: A web server for plant miRNA, tasiRNA and target gene analysis. Plant J. 2012, 70, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Numnark, S.; Mhuantong, W.; Ingsriswang, S.; Wichadakul, D. C-mii: A tool for plant miRNA and target identification. BMC Genom. 2012, 13, S16. [Google Scholar] [CrossRef] [PubMed]
- Katara, P.; Gautam, B.; Kuntal, H.; Sharma, V. Prediction of miRNA targets, affected proteins and their homologs in Glycine max. Bioinformation 2010, 5, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, G.; Wang, J.; Fang, J. Identification of trans-acting siRNAs and their regulatory cascades in grapevine. Bioinformatics 2012, 28, 2561–2568. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Ai, Q.; Yu, D. Uncovering miRNAs involved in crosstalk between nutrient deficienfies in Arabidopsis. Sci. Rep. 2015, 5, 11813. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, E.; He, Y.; Billiau, K.; van de Peer, Y. TAPIR, a web server for the prediction of plant microRNA targets, including target mimics. Bioinformatics 2010, 26, 1566–1568. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server. Nucleic Acids Res. 2011, 39, W155–W159. [Google Scholar] [CrossRef] [PubMed]
- Milev, I.; Yahubyan, G.; Minkov, I.; Baev, V. miRTour: Plant miRNA and target prediction tool. Bioinformation 2011, 6, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Enright, A.J.; John, B.; Gaul, U.; Tuschl, T.; Sander, C.; Marks, D.S. MicroRNA targets in Drosophila. Genome Biol. 2004, 5, R1. [Google Scholar] [CrossRef] [PubMed]
- Krüger, J.; Rehmsmeier, M. RNAhybrid: MicroRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006, 34, W451–W454. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Zhuang, Z.; Zhao, P.X. Computational analysis of miRNA targets in plants: Current status and challenges. Brief. Bioinform. 2011, 12, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, P.K.; Moturu, T.R.; Pandey, P.; Baldwin, I.T.; Pandey, S.P. A comparison of performance of plant miRNA target prediction tools and the characterization of features for genome-wide target prediction. BMC Genom. 2014, 15, 348. [Google Scholar] [CrossRef] [PubMed]
- Addo-Quaye, C.; Eshoo, T.W.; Bartel, D.P.; Axtell, M.J. Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr. Biol. 2008, 18, 758–762. [Google Scholar] [CrossRef] [PubMed]
- German, M.A.; Luo, S.; Schroth, G.; Meyers, B.C.; Green, P.J. Construction of parallel analysis of RNA ends (PARE) libraries for the study of cleaved miRNA targets and the RNA degradome. Nat. Protoc. 2009, 4, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Zhai, J.; Arikit, S.; Simon, S.A.; Kingham, B.F.; Meyers, B.C. Rapid construction of parallel analysis of RNA end (PARE) libraries for Illumina sequencing. Methods 2014, 67, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.-X.; Liu, Y.-F.; Hu, X.-Y.; Zhang, W.-K.; Ma, B.; Chen, S.-Y.; Zhang, J.-S. Identification of miRNAs and their target genes in developing soybean seeds by deep sequencing. BMC Plant Biol. 2011, 11, 5. [Google Scholar] [CrossRef] [PubMed]
- Addo-Quaye, C.; Miller, W.; Axtell, M.J. Cleaveland: A pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 2009, 25, 130–131. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Ghany, S.E.; Pilon, M. MicroRNA-mediated systemic down-regulation of copper protein expression in response to low copper availability in Arabidopsis. J. Biol. Chem. 2008, 283, 15932–15945. [Google Scholar] [CrossRef] [PubMed]
- Fujii, H.; Chiou, T.-J.; Lin, S.-I.; Aung, K.; Zhu, J.-K. A miRNA involved in phosphate-starvation response in Arabidopsis. Curr. Biol. 2005, 15, 2038–2043. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Wang, W.-X.; Ren, L.; Chen, Q.-J.; Mendu, V.; Willcut, B.; Dinkins, R.; Tang, X.; Tang, G. Differential and dynamic regulation of miR398 in response to ABA and salt stress in Populus tremula and Arabidopsis thaliana. Plant Mol. Biol. 2009, 71, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Jagadeeswaran, G.; Li, Y.F.; Sunkar, R. Redox signaling mediates the expression of a sulfate-deprivation-inducible microRNA395 in Arabidopsis. Plant J. 2014, 77, 85–96. [Google Scholar] [CrossRef]
- Zhao, M.; Tai, H.; Sun, S.; Zhang, F.; Xu, Y.; Li, W.-X. Cloning and characterization of maize miRNAs involved in responses to nitrogen deficiency. PLoS ONE 2012, 7, e29669. [Google Scholar] [CrossRef]
- Hackenberg, M.; Gustafson, P.; Langridge, P.; Shi, B.J. Differential expression of microRNAs and other small RNAs in barley between water and drought conditions. Plant Biotechnol. J. 2015, 13, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, C.G.; Yoshimoto, N.; Maruyama-Nakashita, A.; Tsuchiya, Y.N.; Saito, K.; Takahashi, H.; Dalmay, T. Sulphur starvation induces the expression of microRNA-395 and one of its target genes but in different cell types. Plant J. 2009, 57, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, H.; Abdel-Ghany, S.E.; Cohu, C.M.; Kobayashi, Y.; Shikanai, T.; Pilon, M. Regulation of copper homeostasis by micro-RNA in Arabidopsis. J. Biol. Chem. 2007, 282, 16369–16378. [Google Scholar] [CrossRef] [PubMed]
- Li, W.X.; Oono, Y.; Zhu, J.; He, X.J.; Wu, J.M.; Iida, K.; Lu, X.Y.; Cui, X.; Jin, H.; Zhu, J.K. The Arabidopsis NYFA5 transcription factor is regulated transcriptionally and posttranscriptionally to promote drought resistance. Plant Cell 2008, 20, 2238–2251. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Li, D.; Li, Z.; Hu, Q.; Yang, C.; Zhu, L.; Luo, H. Constitutive expression of a miR319 gene alters plant development and enhances salt and drought tolerance in transgenic creeping bentgrass. Plant Physiol. 2013, 161, 1375–1391. [Google Scholar] [CrossRef] [PubMed]
- Pall, G.S.; Hamilton, A.J. Improved northern blot method for enhanced detection of small RNA. Nat. Protoc. 2008, 3, 1077–1084. [Google Scholar] [CrossRef] [PubMed]
- Várallyay, É.; Burgyán, J.; Havelda, Z. MicroRNA detection by northern blotting using locked nucleic acid probes. Nat. Protoc. 2008, 3, 190–196. [Google Scholar]
- Kim, S.W.; Li, Z.; Moore, P.S.; Monaghan, A.P.; Chang, Y.; Nichols, M.; John, B. A sensitive non-radioactive northern blot method to detect small RNAs. Nucleic Acids Res. 2010, 38, e98. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.J.; Zhu, J.; Yang, M.; Zhang, Z.Y.; Tie, Y.; Jiang, H.; Sun, Z.X.; Zheng, X.F. A novel method to monitor the expression of microRNAs. Mol. Biotechnol. 2006, 32, 197–204. [Google Scholar] [CrossRef]
- Roberts, T.C.; Coenen-Stass, A.M.; Wood, M.J. Assessment of RT-qPCR normalization strategies for accurate quantification of extracellular microRNAs in Murine Serum. PLoS ONE 2014, 9, e89237. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Ridzon, D.A.; Broomer, A.J.; Zhou, Z.; Lee, D.H.; Nguyen, J.T.; Barbisin, M.; Xu, N.L.; Mahuvakar, V.R.; Andersen, M.R. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005, 33, e179. [Google Scholar] [CrossRef] [PubMed]
- Moreau, V.; Voirin, E.; Paris, C.; Kotera, M.; Nothisen, M.; Rémy, J.-S.; Behr, J.-P.; Erbacher, P.; Lenne-Samuel, N. Zip Nucleic Acids: New high affinity oligonucleotides as potent primers for PCR and reverse transcription. Nucleic Acids Res. 2009, 19, e130. [Google Scholar] [CrossRef] [PubMed]
- Paris, C.; Moreau, V.; Deglane, G.; Voirin, E.; Erbacher, P.; Lenne-Samuel, N. Zip nucleic acids are potent hydrolysis probes for quantitative PCR. Nucleic Acids Res. 2010, 38, e95. [Google Scholar] [CrossRef] [PubMed]
- Javelle, M.; Timmermans, M.C. In situ localization of small RNAs in plants by using LNA probes. Nat. Protoc. 2012, 7, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Begheldo, M.; Ditengou, F.; Cimoli, G.; Trevisan, S.; Quaggiotti, S.; Nonis, A.; Palme, K.; Ruperti, B. Whole-mount in situ detection of microRNAs on Arabidopsis tissues using Zip Nucleic Acid probes. Anal. Biochem. 2013, 434, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Hsu, R.J.; Yang, H.J.; Tsai, H.J. Labeled microRNA pull-down assay system: An experimental approach for high-throughput identification of microRNA-target mRNAs. Nucleic Acids Res 2009, 37, e77. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Tuo, W.; Lian, H.; Liu, Q.; Zhu, X.Q.; Gao, H. Strategies to identify microRNA targets: New advances. New Biotechnol. 2010, 27, 734–738. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, D.E.; Martin, M.M.; Feldman, D.S.; Terry, A.V., Jr.; Nuovo, G.J.; Elton, T.S. Experimental validation of miRNA targets. Methods 2008, 44, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Ghildiyal, M.; Zamore, P.D. Small silencing RNAs: An expanding universe. Nat. Rev. Genet. 2009, 10, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Alonso, J.M.; Stepanova, A.N.; Leisse, T.J.; Kim, C.J.; Chen, H.; Shinn, P.; Stevenson, D.K.; Zimmerman, J.; Barajas, P.; Cheuk, R. Genome-wide insertional mutagenesis of Arabidopsis thaliana. Science 2003, 301, 653–657. [Google Scholar] [CrossRef] [PubMed]
- Eamens, A.L.; Agius, C.; Smith, N.A.; Waterhouse, P.M.; Wang, M.B. Efficient silencing of endogenous microRNAs using artificial microRNAs in Arabidopsis thaliana. Mol. Plant 2010, 4, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Franco-Zorrilla, J.M.; Valli, A.; Todesco, M.; Mateos, I.; Puga, M.I.; Rubio-Somoza, I.; Leyva, A.; Weigel, D.; García, J.A.; Paz-Ares, J. Target mimicry provides a new mechanism for regulation of microRNA activity. Nat. Genet. 2007, 39, 1033–1037. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ku, Y.-S.; Wong, J.W.-H.; Mui, Z.; Liu, X.; Hui, J.H.-L.; Chan, T.-F.; Lam, H.-M. Small RNAs in Plant Responses to Abiotic Stresses: Regulatory Roles and Study Methods. Int. J. Mol. Sci. 2015, 16, 24532-24554. https://doi.org/10.3390/ijms161024532
Ku Y-S, Wong JW-H, Mui Z, Liu X, Hui JH-L, Chan T-F, Lam H-M. Small RNAs in Plant Responses to Abiotic Stresses: Regulatory Roles and Study Methods. International Journal of Molecular Sciences. 2015; 16(10):24532-24554. https://doi.org/10.3390/ijms161024532
Chicago/Turabian StyleKu, Yee-Shan, Johanna Wing-Hang Wong, Zeta Mui, Xuan Liu, Jerome Ho-Lam Hui, Ting-Fung Chan, and Hon-Ming Lam. 2015. "Small RNAs in Plant Responses to Abiotic Stresses: Regulatory Roles and Study Methods" International Journal of Molecular Sciences 16, no. 10: 24532-24554. https://doi.org/10.3390/ijms161024532