
Alkyl Chain Growth on a Transition Metal Center: How Does Iron Compare to Ruthenium and Osmium?


Abstract
:
1. Introduction

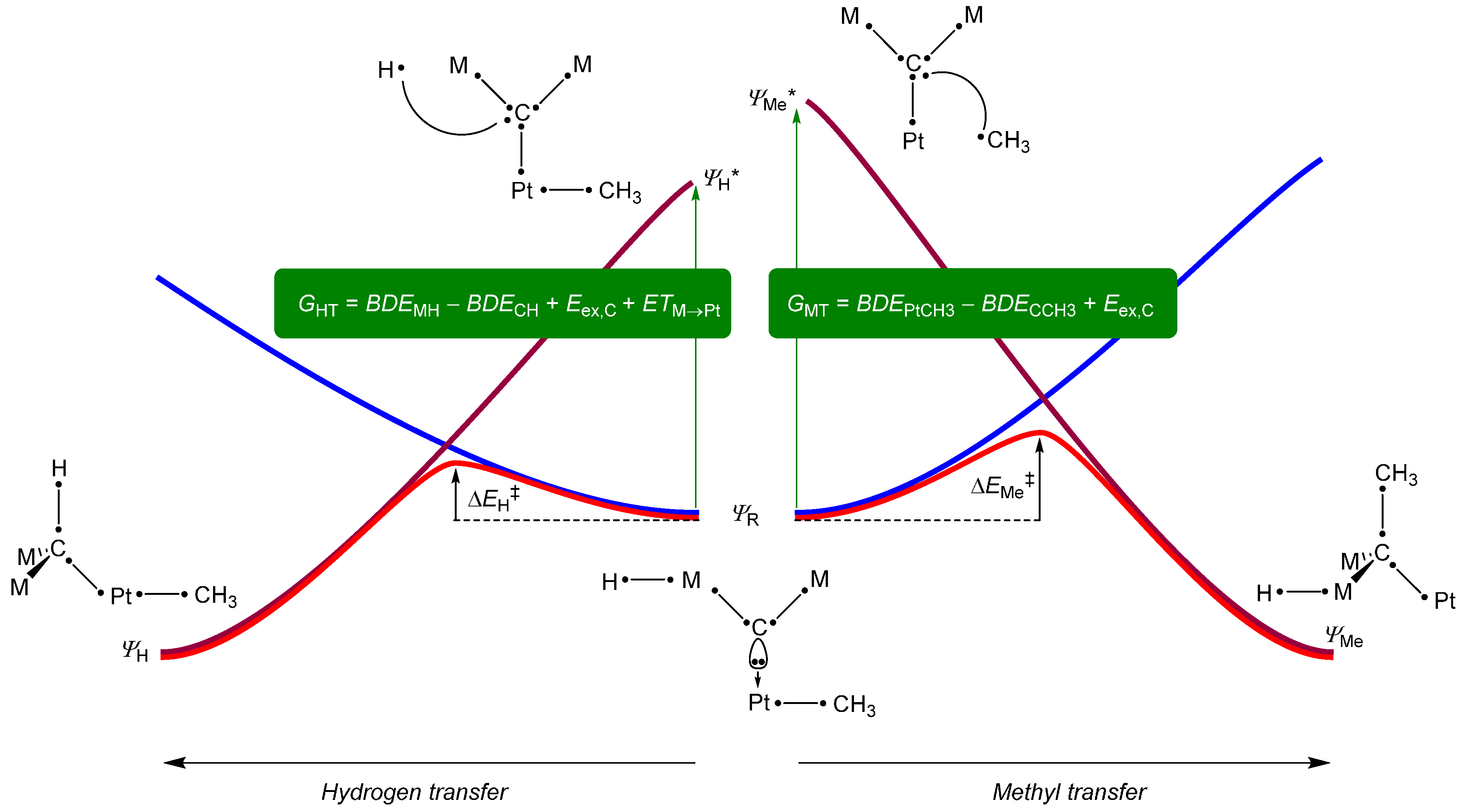
2. Results and Discussion
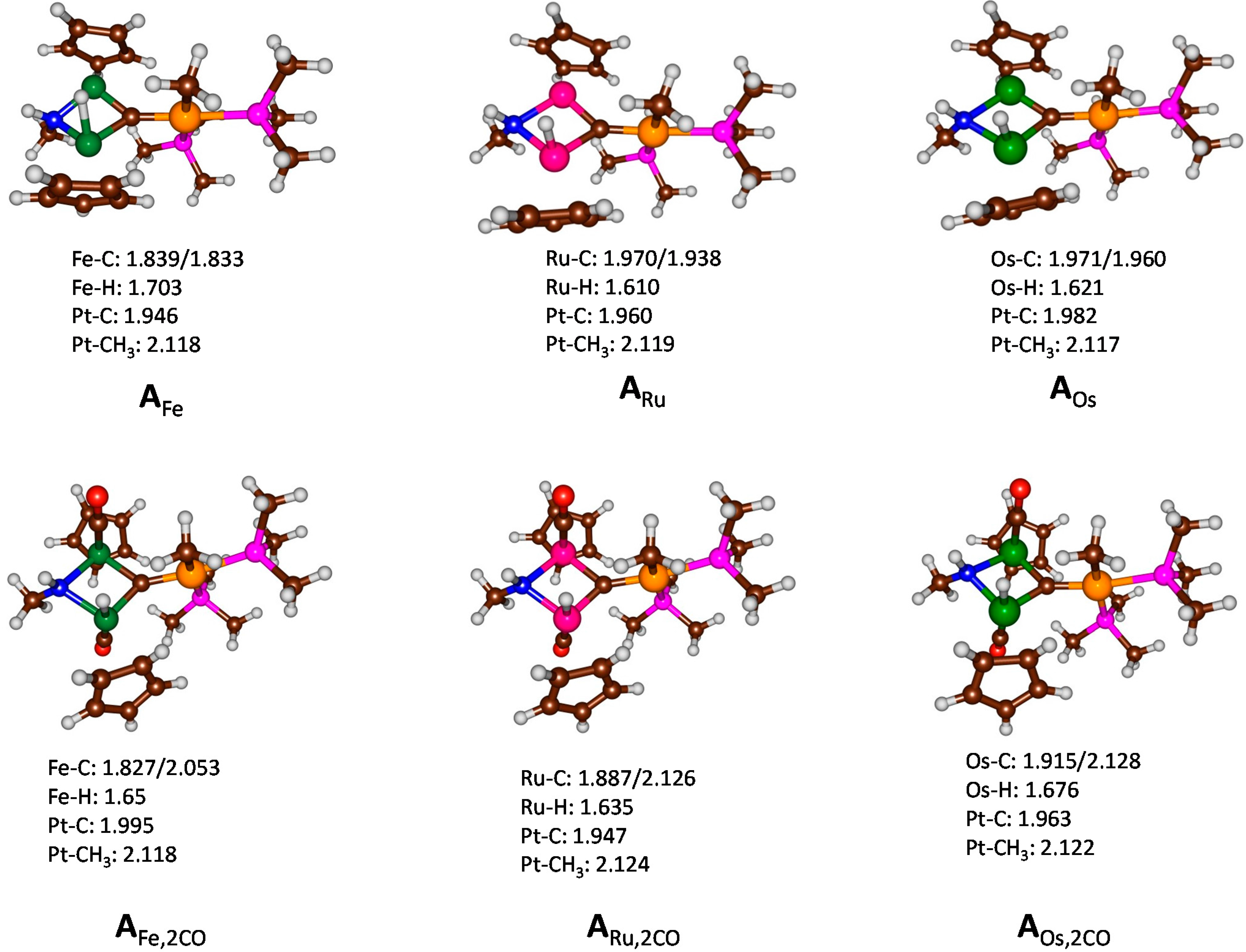

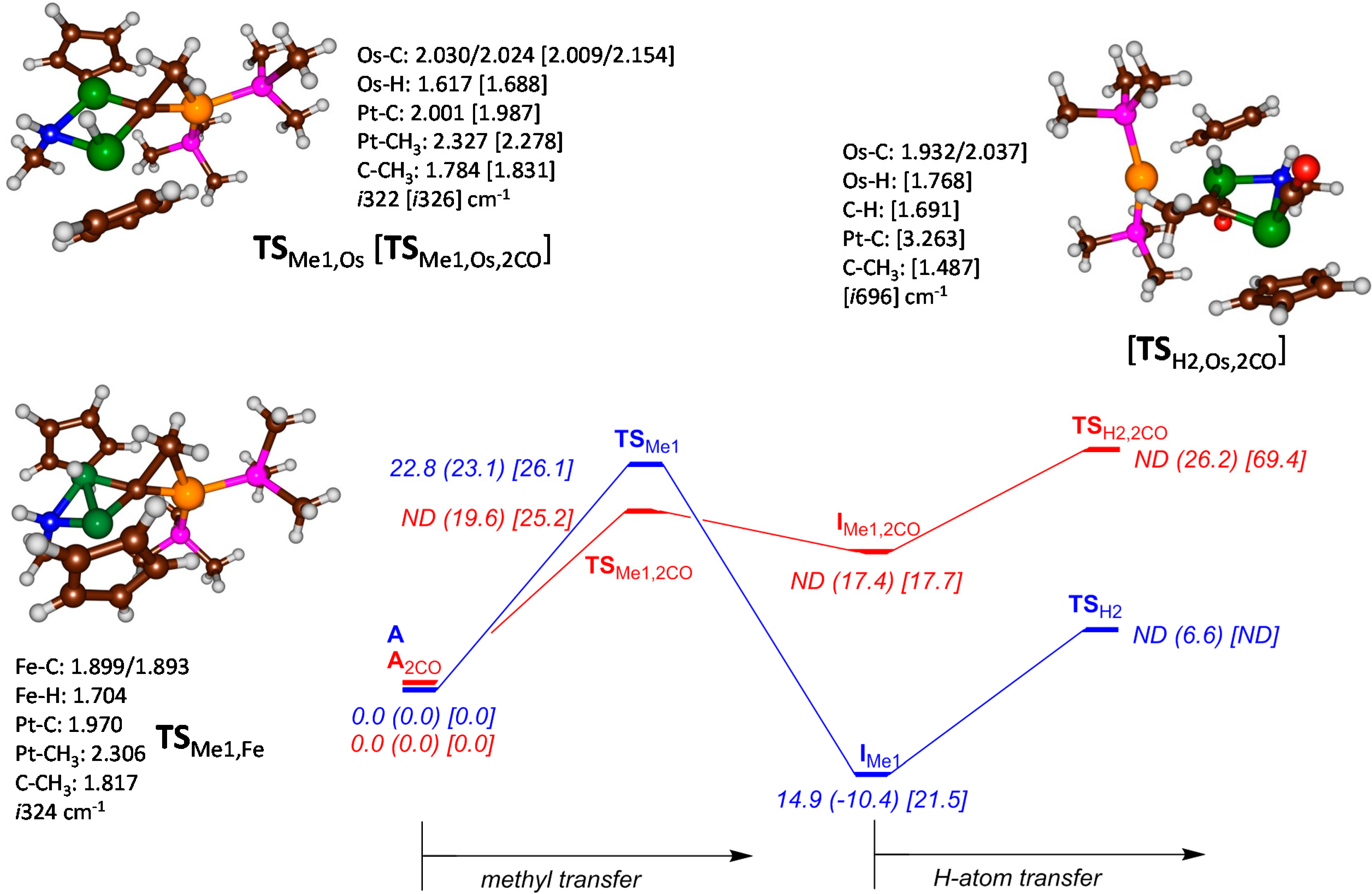

{kind=link}
{kind=link}
{kind=link}
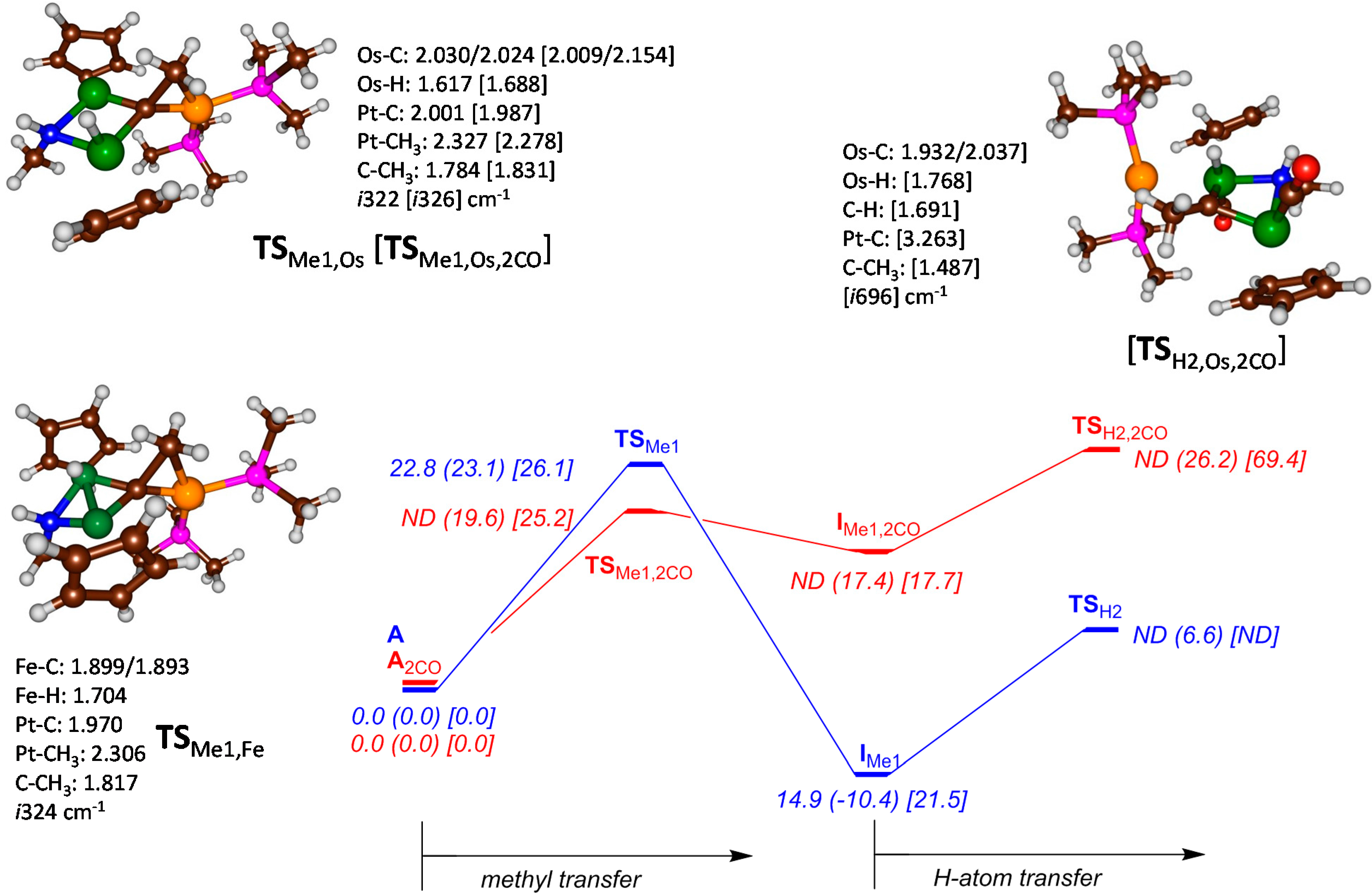{kind=link}
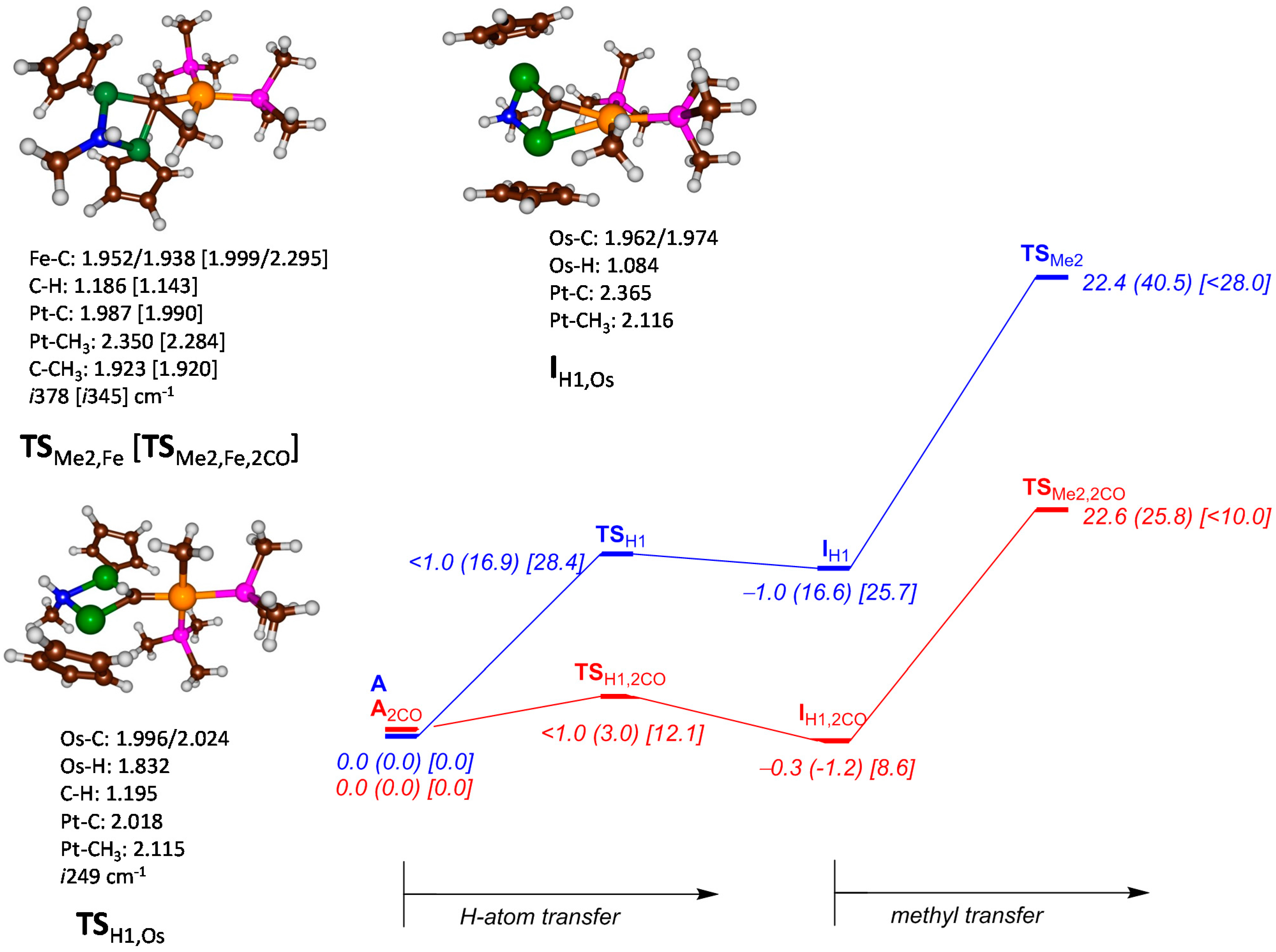
{kind=link}
{kind=link}
| Structure | B3LYP | B3LYP-D3 | M06-L |
|---|---|---|---|
| AFe | 0.00 | 0.00 | 0.00 |
| TSMe1,Fe | 24.31 | 24.69 | 23.89 |
| IMe1,Fe | 17.92 | 18.44 | 17.86 |
| IH1,Fe | 5.21 | 5.91 | 9.07 |
| TSMe2,Fe | 24.88 | 25.87 | 34.57 |
| AOs | 0.00 | 0.00 | 0.00 |
| TSMe1,Os | 26.63 | 25.63 | 23.88 |
| IMe1,Os | 22.80 | 20.97 | 19.30 |
| TSH1,Os | 28.33 | 30.05 | 25.16 |
| IH1,Os | 26.89 | 26.36 | 20.01 |
| AOs,2CO | 0.00 | 0.00 | 0.00 |
| TSMe1,Os,2CO | 25.79 | 24.70 | 23.30 |
| IMe1,Os,2CO | 18.92 | 18.08 | 15.70 |
| TSH1,Os,2CO | 11.44 | 11.69 | 10.70 |
| IH1,Os,2CO | 8.35 | 9.47 | 7.06 |
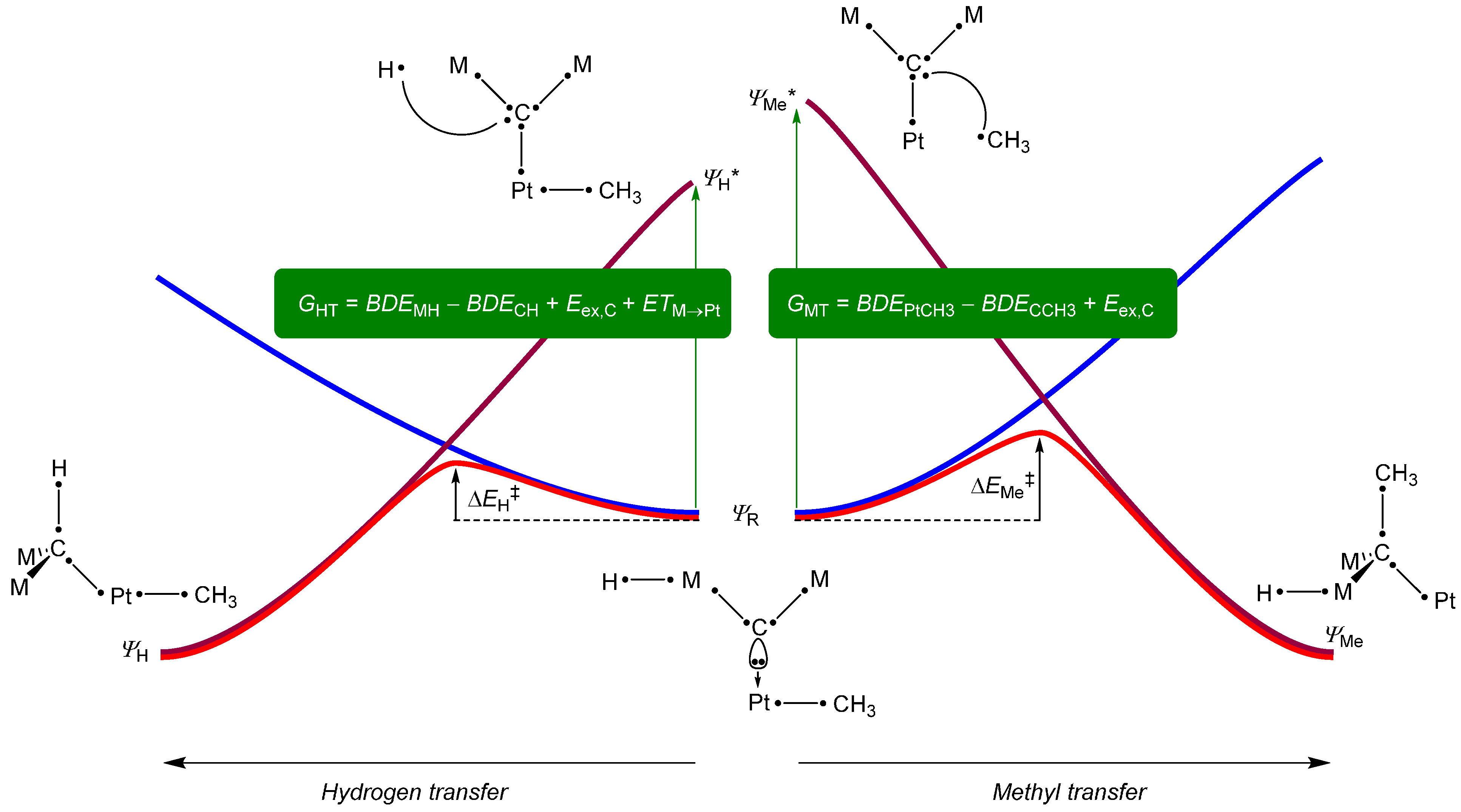
3. Experimental Section
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fischer, F.; Tropsch, H. Über die direketer synthese von Erdöl-Kohlenwasserstoffen bei gewöhnlichem. Berichte 1926, 59, 830–831. [Google Scholar]
- Rofer-de Poorter, C.K. A comprehensive mechanism for the Fischer-Tropsch synthesis. Chem. Rev. 1981, 81, 447–474. [Google Scholar] [CrossRef]
- Schulz, H. Short history and present trends of Fischer-Tropsch synthesis. Appl. Catal. A Gen. 1999, 186, 3–12. [Google Scholar] [CrossRef]
- Khodakov, A.Y.; Chu, W.; Fongarland, P. Advances in the development of novel cobalt Fischer-Tropsch catalysts for synthesis of long-chain hydrocarbons and clean fuels. Chem. Rev. 2007, 107, 1692–1744. [Google Scholar] [CrossRef] [PubMed]
- Maitlis, P.M.; Zanotti, V. The role of electrophilic species in the Fischer-Tropsch reaction. Chem. Commun. 2009, 13, 1619–1634. [Google Scholar] [CrossRef] [PubMed]
- Tsakoumis, N.E.; Rønning, M.; Borg, Ø.; Rytter, E.; Holmen, A. Deactivation of cobalt based Fischer-Tropsch catalysts: A review. Catal. Today 2010, 154, 162–182. [Google Scholar] [CrossRef]
- Shetty, S.; van Santen, R.A. CO dissociation on Ru and Co surfaces: The initial step in the Fischer-Tropsch synthesis. Catal. Today 2011, 171, 168–173. [Google Scholar] [CrossRef]
- Inderwildi, O.R.; Jenkins, S.J. In-silico investigations in heterogeneous catalysis—Combustion and synthesis of small alkanes. Chem. Soc. Rev. 2008, 37, 2274–2309. [Google Scholar] [CrossRef] [PubMed]
- Perkins, P.; Vollhardt, K.P.C. Plymer-supported .eta.5-cyclopentadienylcobalt. An immobilized “homogenous” Fischer-Tropsch catalyst. J. Am. Chem. Soc. 1979, 101, 3985–3987. [Google Scholar] [CrossRef]
- Inderwildi, O.R.; King, D.A.; Jenkins, S.J. Fischer-Tropsch synthesis of liquid fuels: Lessons from homogenous catalysis. Phys. Chem. Chem. Phys. 2009, 11, 11110–11112. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, S.; Morita, H.; Karitani, K.; Fujiwara, H.; Matsuzaka, H. A bimetallic Ru2Pt complex containing a trigonal-planar μ3-carbido ligand: Formation, structure, and reactivity relevant to the Fischer-Tropsch process. J. Am. Chem. Soc. 2009, 131, 18026–18027. [Google Scholar] [CrossRef] [PubMed]
- Sainna, M.A.; Singh, D.; Kumar, D.; de Visser, S.P. A trimetal carbene with reactivity reminiscent of Fischer-Tropsch catalysis. Organometallics 2015, 34, 1651–1660. [Google Scholar] [CrossRef]
- Sharma, P.K.; de Visser, S.P.; Ogliaro, F.; Shaik, S. Is the ruthenium analogue of Compound I of cytochrome P450 an efficient oxidant? A theoretical investigation of the methane hydroxylation reaction. J. Am. Chem. Soc. 2003, 125, 2291–2300. [Google Scholar] [CrossRef] [PubMed]
- Sallmann, M.; Kumar, S.; Chernev, P.; Nehrkorn, J.; Schnegg, A.; Kumar, D.; Dau, H.; Limberg, C.; de Visser, S.P. Structure and mechanism leading to formation of the cysteine sulfinate product complex of a biomimetic cysteine dioxygenase model. Chem. Eur. J. 2015, 21, 7470–7479. [Google Scholar] [CrossRef] [PubMed]
- İşci, Ü.; Faponle, A.S.; Afanasiev, P.; Albrieux, F.; Briois, V.; Ahsen, V.; Dumoulin, F.; Sorokin, A.B.; de Visser, S.P. Site-selective formation of an iron(IV)-oxo species at the more electron-rich iron atom of heteroleptic μ-nitrido diiron phthalocyanines. Chem. Sci. 2015, 6, 5063–5075. [Google Scholar]
- De Visser, S.P.; Ogliaro, F.; Sharma, P.K.; Shaik, S. What factors affect the regioselectivity of oxidation by cytochrome P450? A DFT study of allylic hydroxylation and double bond epoxidation in a model reaction. J. Am. Chem. Soc. 2002, 124, 11809–11826. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P. Substitution of hydrogen by deuterium changes the regioselectivity of ethylbenzene hydroxylation by an oxo-iron-porphyrin catalyst. Chem. Eur. J. 2006, 12, 8168–8177. [Google Scholar] [CrossRef] [PubMed]
- Pratter, S.M.; Konstantinovics, C.; di Giuro, C.L.M.; Leitner, E.; Kumar, D.; de Visser, S.P.; Grogan, G.; Straganz, G.D. Inversion of enantio-selectivity of a mononuclear non-heme iron(II)-dependent hydroxylase by tuning the interplay of metal-center geometry and protein structure. Angew. Chem. Int. Ed. 2013, 52, 9677–9681. [Google Scholar] [CrossRef] [PubMed]
- Shaik, S.; Kumar, D.; de Visser, S.P. A valence bond modeling of trends in hydrogen abstraction barriers and transition states of hydroxylation reactions catalyzed by cytochrome P450 enzymes. J. Am. Chem. Soc. 2008, 130, 10128–10140. [Google Scholar] [CrossRef] [PubMed]
- Latifi, R.; Bagherzadeh, M.; de Visser, S.P. Origin of the correlation of the rate constant of substrate hydroxylation by nonheme iron(IV)-oxo complexes with the bond-dissociation energy of the C–H bond of the substrate. Chem. Eur. J. 2009, 15, 6651–6662. [Google Scholar] [CrossRef] [PubMed]
- De Visser, S.P. Trends in substrate hydroxylation reactions by heme and nonheme iron(IV)-oxo oxidants give correlations between intrinsic properties of the oxidant with barrier height. J. Am. Chem. Soc. 2010, 132, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Karamzadeh, B.; Sastry, G.N.; de Visser, S.P. What factors influence the rate constant of substrate epoxidation by Compound I of cytochrome P450 and analogous iron(IV)-oxo oxidants. J. Am. Chem. Soc. 2010, 132, 7656–7667. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Sastry, G.N.; de Visser, S.P. Effect of the axial ligand on substrate sulfoxidation mediated by iron(IV)-oxo porphyrin cation radical oxidants. Chem. Eur. J. 2011, 17, 6196–6205. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Latifi, R.; Kumar, S.; Rybak-Akimova, E.V.; Sainna, M.A.; de Visser, S.P. Rationalization of the barrier height for para-Z-styrene epoxidation by iron(IV)-oxo porphyrins with variable axial ligands. Inorg. Chem. 2013, 52, 7968–7979. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Faponle, A.S.; Quesne, M.G.; Sainna, M.A.; Zhang, J.; Franke, A.; Kumar, D.; van Eldik, R.; Liu, W.; de Visser, S.P. Drug metabolism by cytochrome P450 enzymes: What distinguishes the pathways leading to substrate hydroxylation over desaturation? Chem. Eur. J. 2015, 21, 9083–9092. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Becke, A.D. Density functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Sainna, M.A.; Kumar, S.; Kumar, D.; Fornarini, S.; Crestoni, M.E.; de Visser, S.P. A comprehensive test set of epoxidation rate constants by iron(IV)-oxo porphyrin complexes. Chem. Sci. 2015, 6, 1516–1529. [Google Scholar] [CrossRef]
- Vardhaman, A.K.; Sastri, C.V.; Kumar, D.; de Visser, S.P. Nonheme ferric hydroperoxo intermediates are efficient oxidants of bromide oxidation. Chem. Commun. 2011, 47, 11044–11046. [Google Scholar] [CrossRef] [PubMed]
- Vardhaman, A.K.; Barman, P.; Kumar, S.; Sastri, C.V.; Kumar, D.; de Visser, S.P. Comparison of the reactivity of nonheme iron(IV)-oxo versus iron(IV)-imido complexes: Which is the better oxidant? Angew. Chem. Int. Ed. 2013, 52, 12288–12292. [Google Scholar] [CrossRef] [PubMed]
- Vardhaman, A.K.; Barman, P.; Kumar, S.; Sastri, C.V.; Kumar, D.; de Visser, S.P. Mechanistic insight into halide oxidation by non-heme iron complexes. Haloperoxidase versus halogenase activity. Chem. Commun. 2013, 49, 10926–10928. [Google Scholar] [CrossRef] [PubMed]
- Jastrzebski, R.; Quesne, M.G.; Weckhuysen, B.M.; de Visser, S.P.; Bruijnincx, P.C.A. Experimental and computational evidence for the mechanism of intradiol catechol dioxygenation by non-heme iron(III) complexes. Chem. Eur. J. 2014, 20, 15686–15691. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Faponle, A.S.; Barman, P.; Vardhaman, A.K.; Sastri, C.V.; Kumar, D.; de Visser, S.P. Long-range electron transfer triggers mechanistic differences between iron(IV)-oxo and iron(IV)-imido oxidants. J. Am. Chem. Soc. 2014, 136, 17102–17115. [Google Scholar] [CrossRef] [PubMed]
- Faponle, A.S.; Quesne, M.G.; Sastri, C.V.; Banse, F.; de Visser, S.P. Differences and comparisons of the properties and reactivities of iron(III)-hydroperoxo complexes with saturated coordination sphere. Chem. Eur. J. 2015, 21, 1221–1236. [Google Scholar] [CrossRef] [PubMed]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- De Visser, S.P.; Quesne, M.G.; Martin, B.; Comba, P.; Ryde, U. Computational modelling of oxygenation processes in enzymes and biomimetic model complexes. Chem. Commun. 2014, 50, 262–282. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Ortega, A.; Quesne, M.G.; Bui, S.; Heyes, D.J.; Steiner, R.A.; Scrutton, N.S.; de Visser, S.P. Catalytic mechanism of cofactor-free dioxygenases and how they circumvent spin-forbidden oxygenation of their substrates. J. Am. Chem. Soc. 2015, 137, 7474–7487. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parameterization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. 2006, 125, 194101. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sainna, M.A.; De Visser, S.P. Alkyl Chain Growth on a Transition Metal Center: How Does Iron Compare to Ruthenium and Osmium? Int. J. Mol. Sci. 2015, 16, 23369-23381. https://doi.org/10.3390/ijms161023369
Sainna MA, De Visser SP. Alkyl Chain Growth on a Transition Metal Center: How Does Iron Compare to Ruthenium and Osmium? International Journal of Molecular Sciences. 2015; 16(10):23369-23381. https://doi.org/10.3390/ijms161023369
Chicago/Turabian StyleSainna, Mala A., and Sam P. De Visser. 2015. "Alkyl Chain Growth on a Transition Metal Center: How Does Iron Compare to Ruthenium and Osmium?" International Journal of Molecular Sciences 16, no. 10: 23369-23381. https://doi.org/10.3390/ijms161023369