Nrf2-Mediated HO-1 Induction Coupled with the ERK Signaling Pathway Contributes to Indirect Antioxidant Capacity of Caffeic Acid Phenethyl Ester in HepG2 Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. CAPE (Caffeic Acid Phenethyl Ester) Increases ARE (Antioxidant Response Element)-Luciferase Activity and HO-1 (Heme Oxygenase-1) Expression
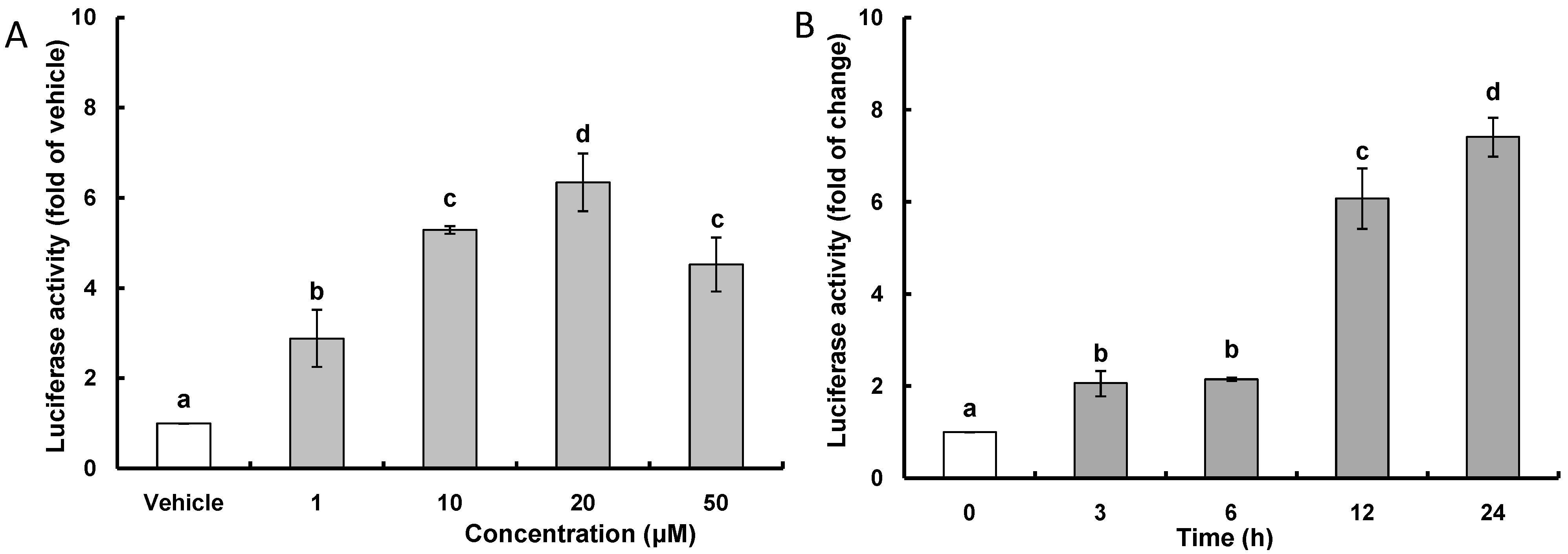
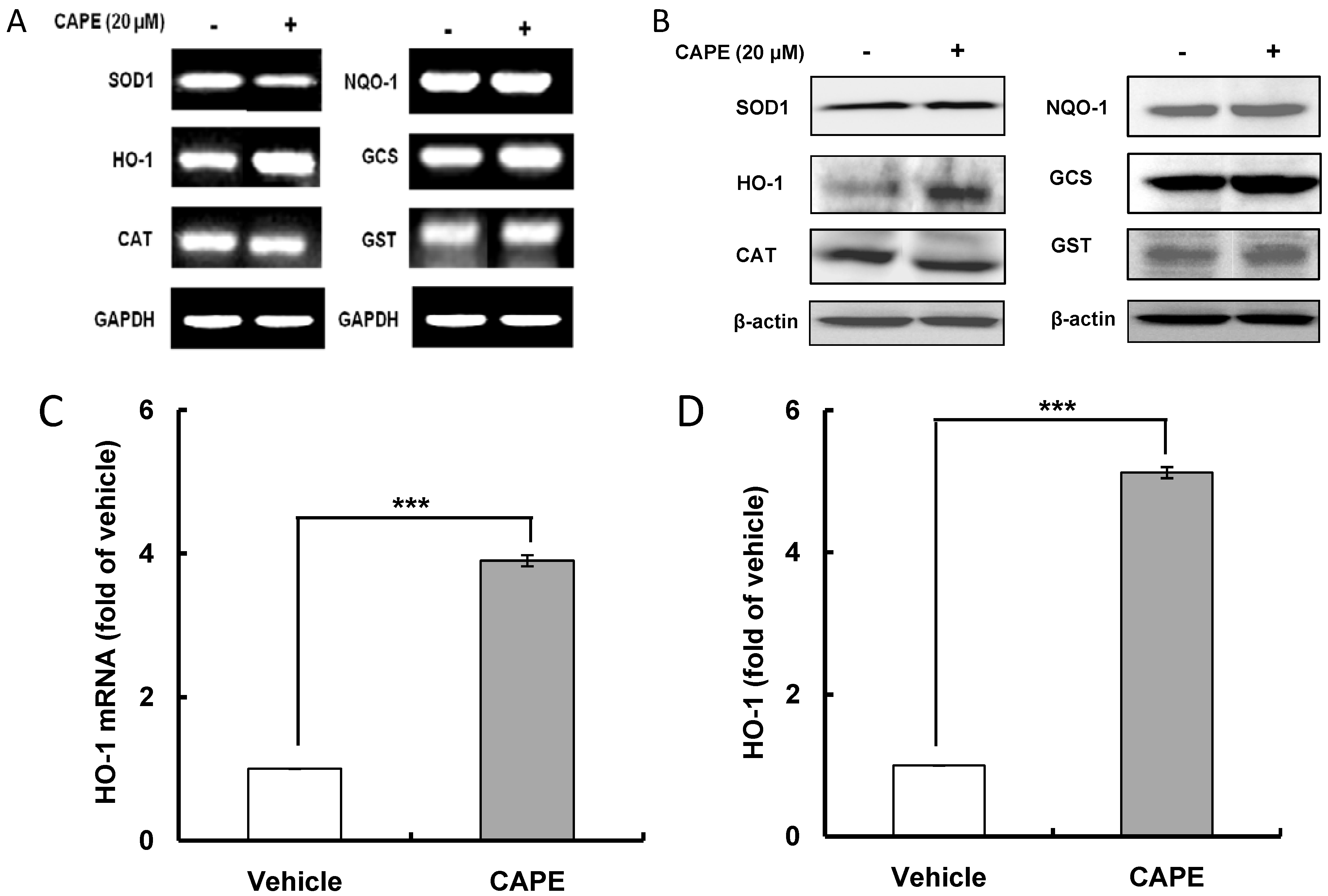
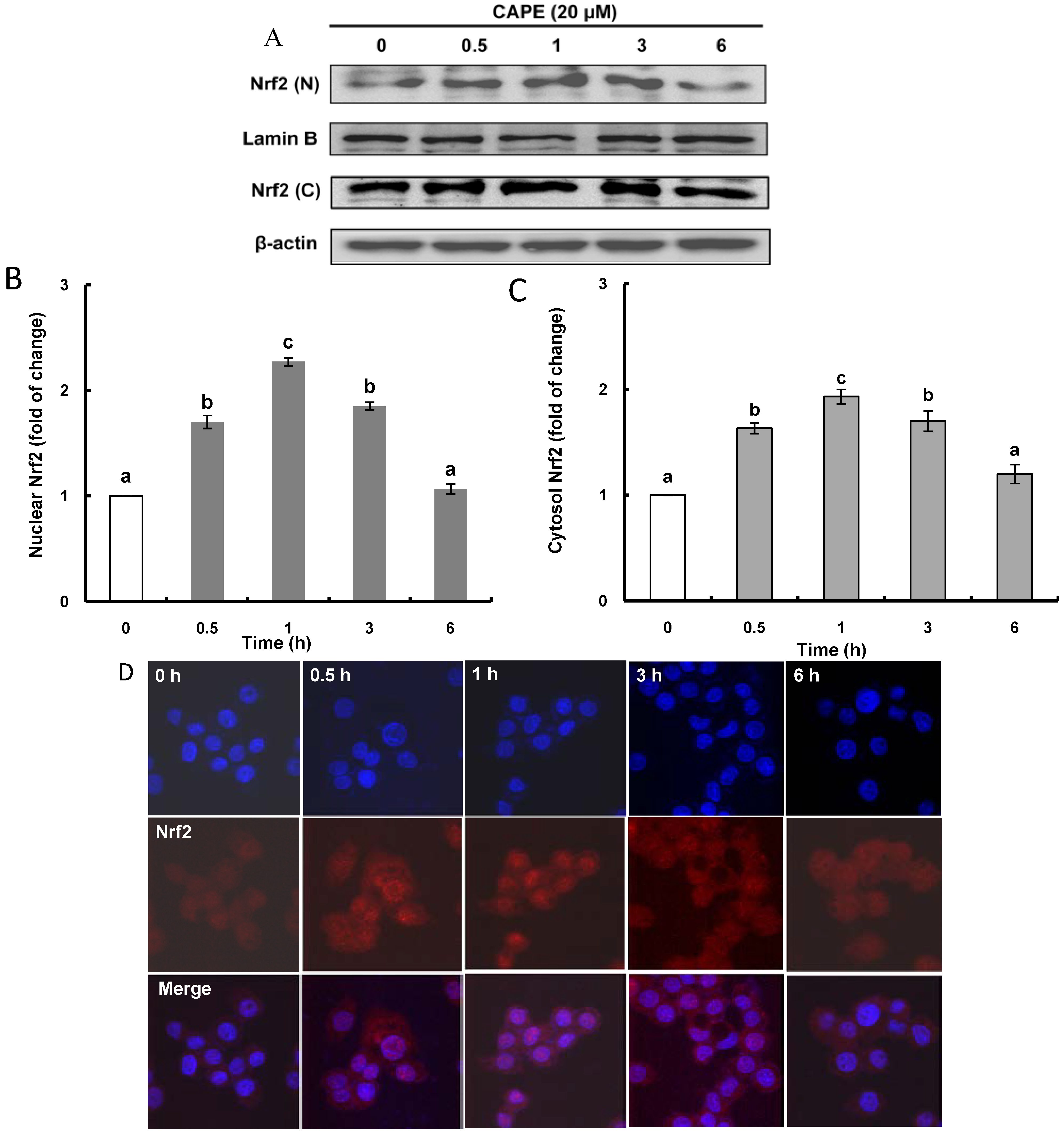
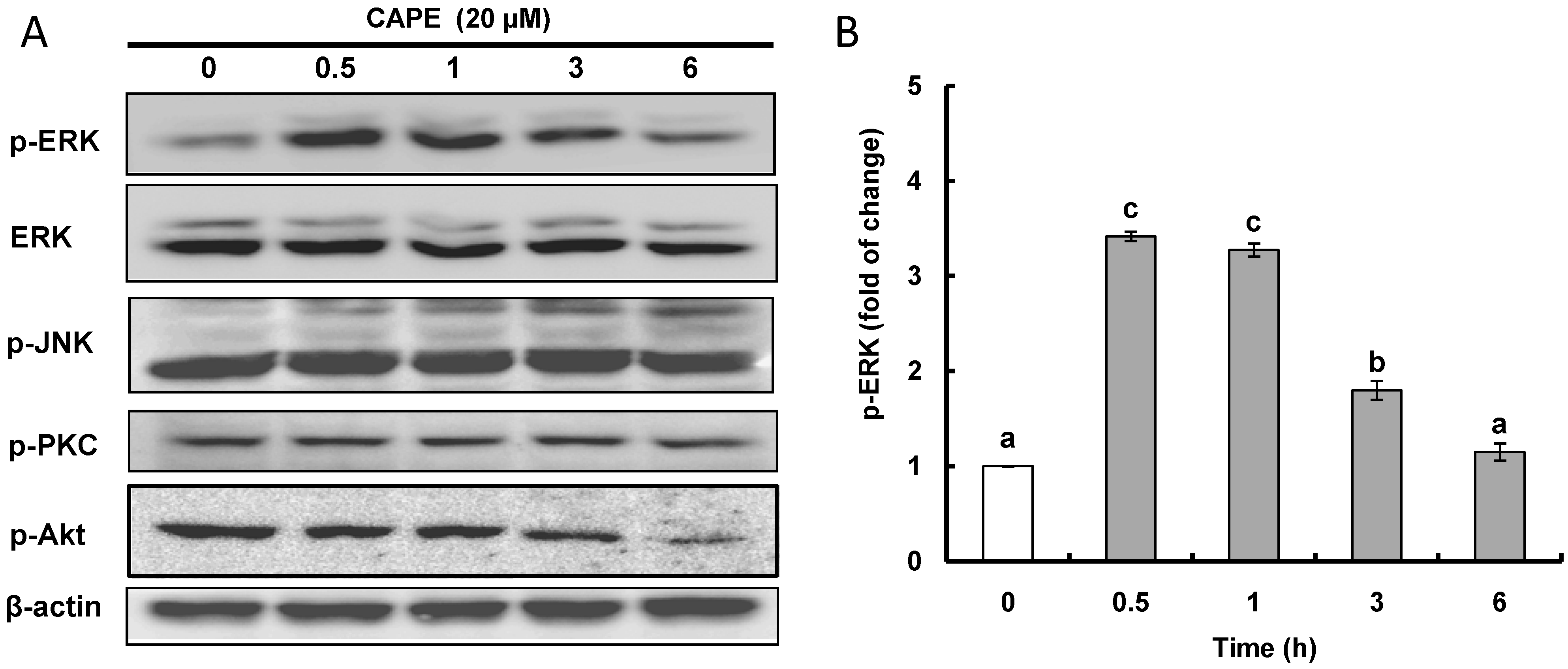
2.2. CAPE Activates ERK (Extracellular Signal-Regulated Kinase) Leading to Nrf2 (Nuclear Transcription Factor-Erythroid 2-Related Factor 2) Accumulation in the Nucleus

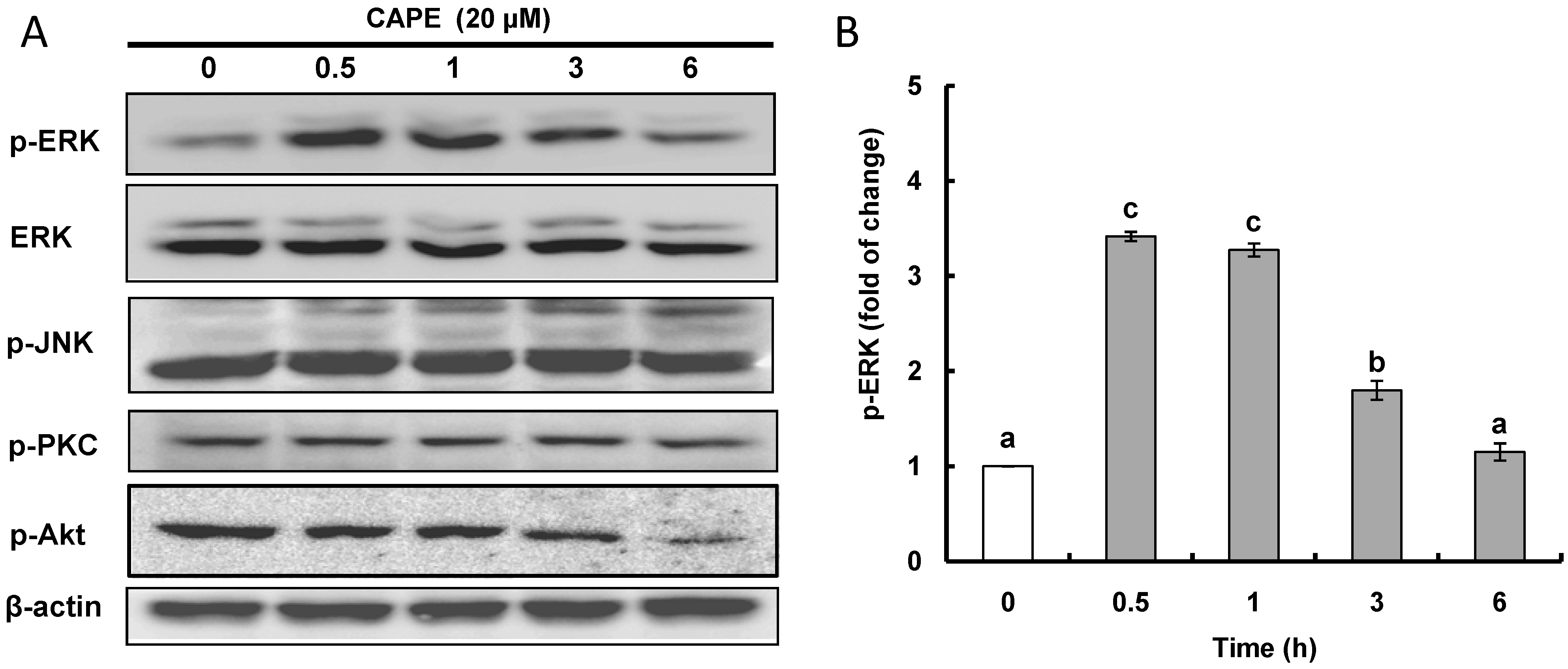
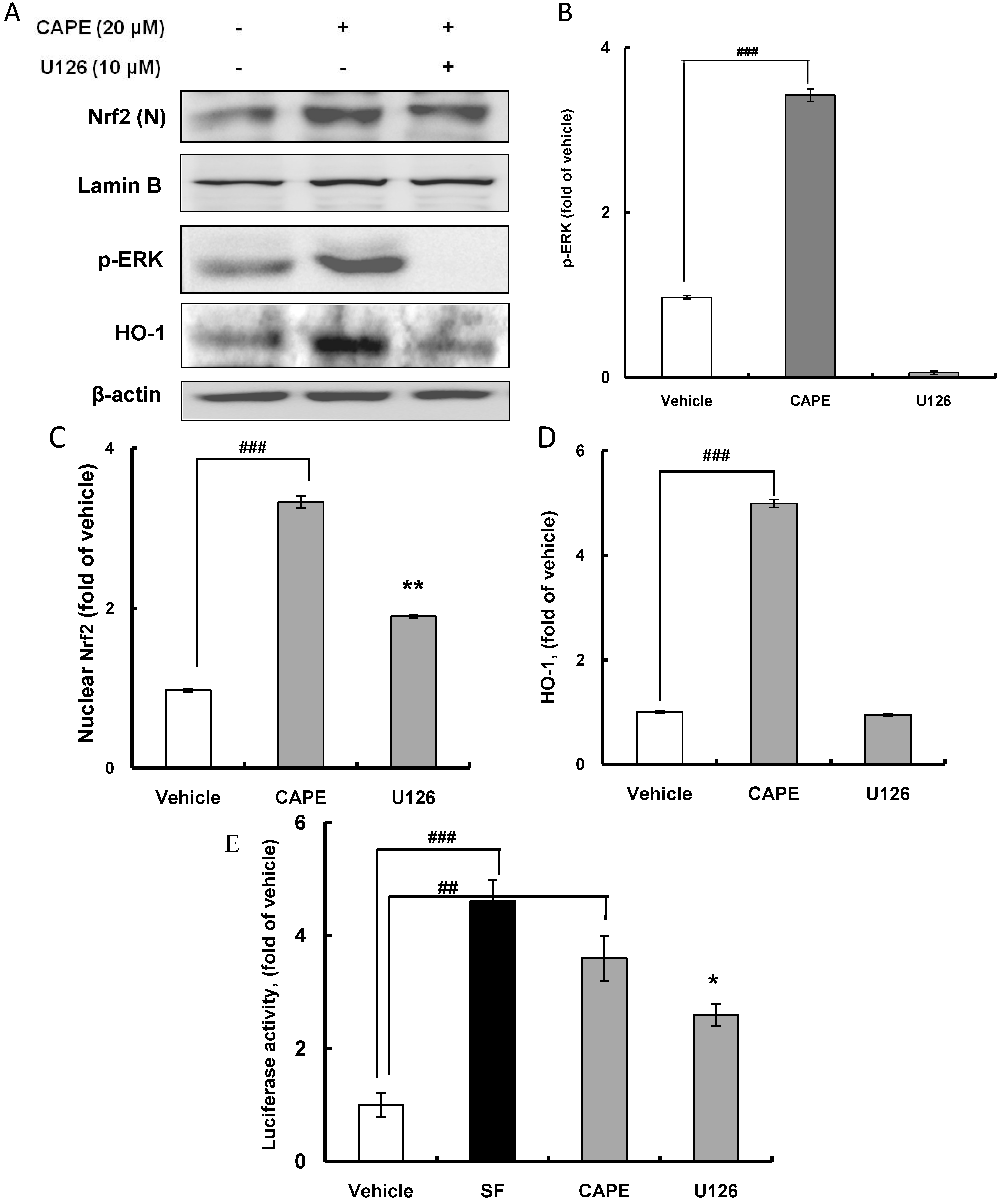
2.3. CAPE Induces HO-1 Expression through the ERK-Nrf2 Signaling Pathway
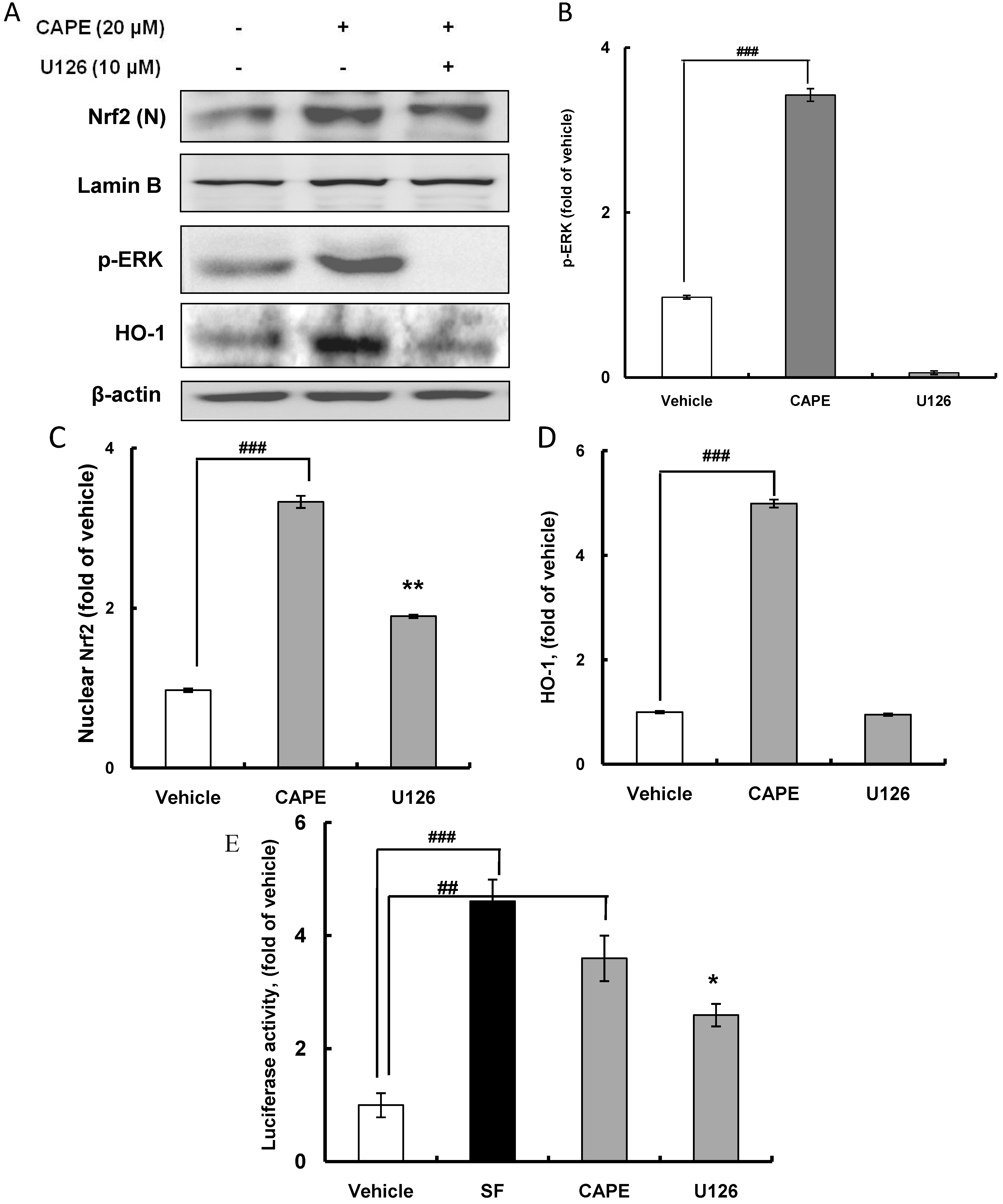
2.4. CAPE Partially Attenuates Cellular Oxidative Stress Induced by AAPH (2,2'-Azobis (2-amidinopropane) dihydrochloride) or H2O2 through Inducing HO-1 Expression
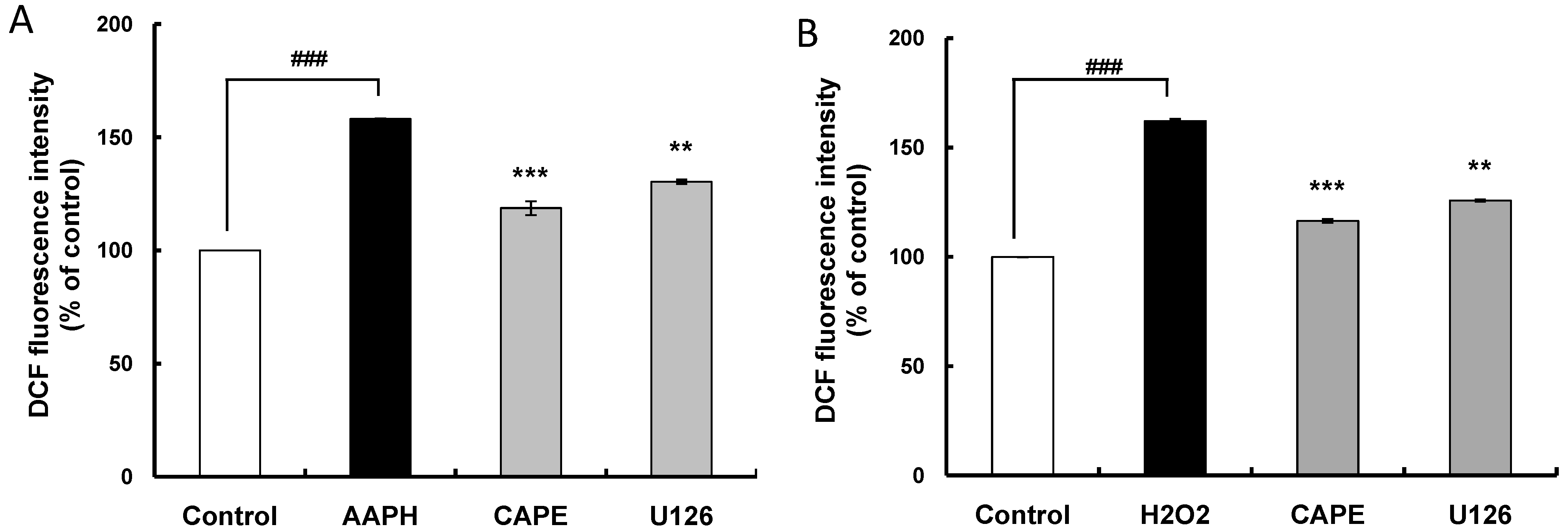
3. Experimental Section
3.1. Reagents
3.2. Transient Transfection and Antioxidant Response Element (ARE)-Luciferase Assay
3.3. Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
3.4. Western Blot Analysis
3.5. Observation of Fluorescence Imaging for Nrf2 and Nucleus
3.6. Cell Viability Assay
3.7. Cellular Antioxidant Capacity
3.8. Statistical Analysis
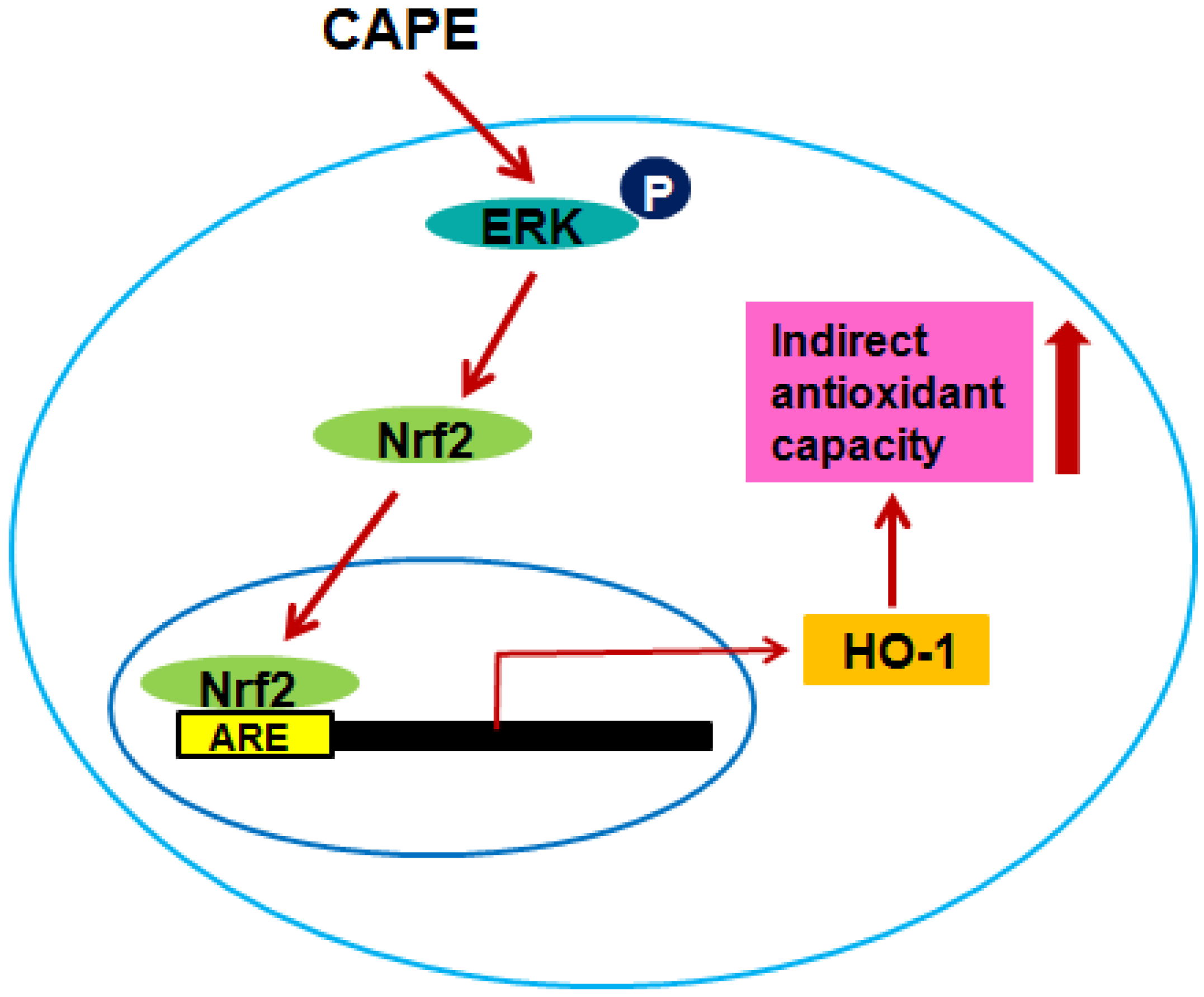
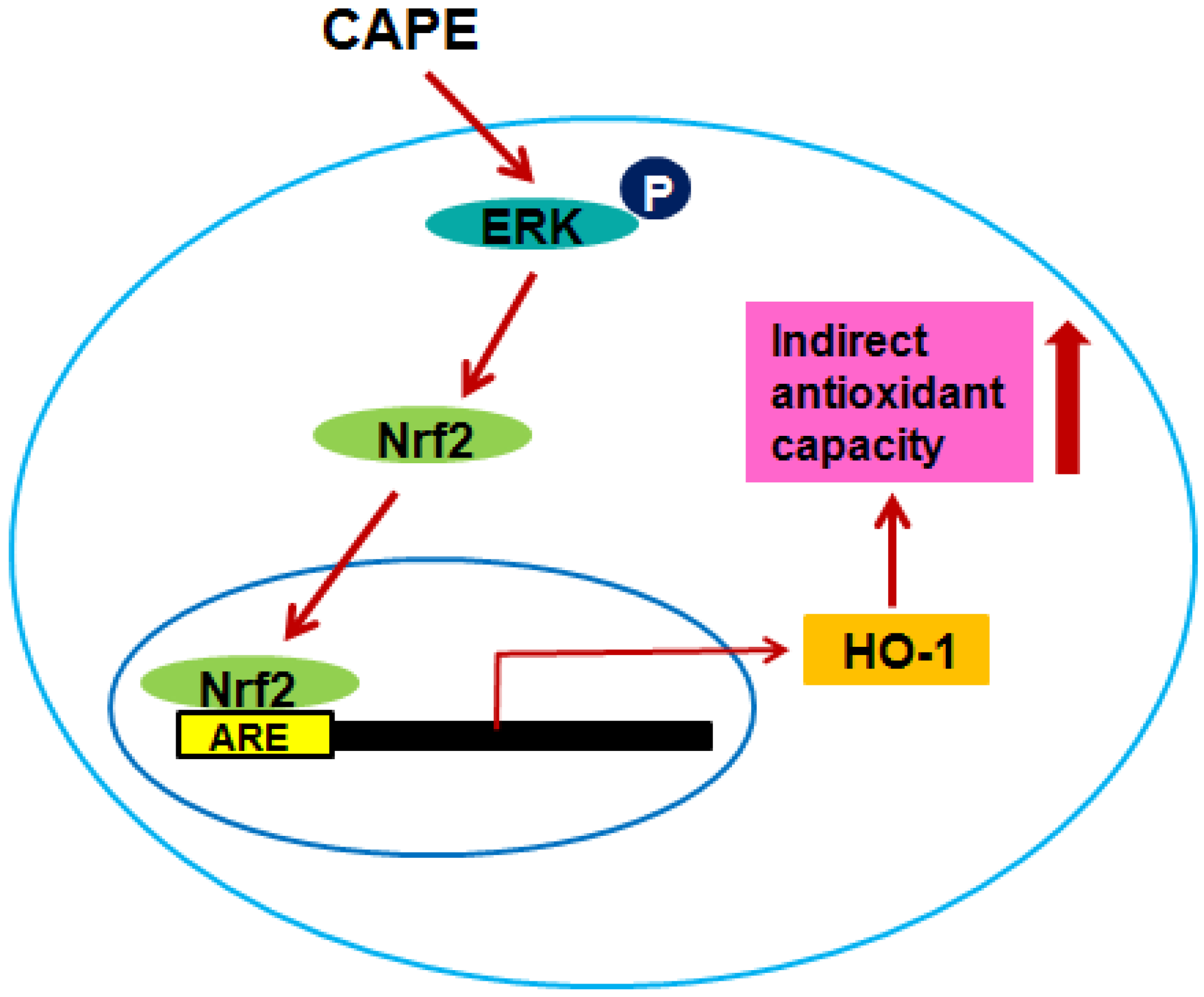
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dinkova-Kostova, A.T.; Talalay, P. Direct and indirect antioxidant properties of inducers of cytoprotective proteins. Mol. Nutr. Food Res. 2008, 52, S128–S138. [Google Scholar]
- Dinkova-Kostova, A.T.; Cheah, J.; Samouilov, A.; Zweiser, J.L.; Bozak, R.E.; Hicks, R.J.; Talalay, P. Phenolic Michael reaction acceptors: combined direct and indirect antioxidant defense against electrophiles and oxidants. Med. Chem. 2007, 3, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Wang, X.J. Induction of the Keap1/Nrf2/ARE pathway by oxidizable diphenol. Chem. Biol. Interact. 2011, 192, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Bryan, H.K.; Olayanju, A.; Goldring, C.E.; Park, B.K. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem. Pharmacol. 2013, 85, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Paonessa, J.D.; Zhang, Y. Mechanism of chemical activation of Nrf2. PLoS One 2012, 7, 1–7. [Google Scholar]
- Park, J.H.; Lee, J.K.; Kim, H.S.; Chung, S.T.; Eom, J.H.; Kim, K.A.; Chung, S.J.; Paik, S.Y.; Oh, H.Y. Immunomodulatory effect of caffeic acid phenethyl ester in Balb/c mice. Int. Immunopharmacol. 2004, 4, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Baird, L.; Dinkova-Kostova, A.T. The cytoprotective role of the Keap1-Nrf2 pathway. Arch. Toxicol. 2011, 85, 241–272. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Tong, K.I.; Yamamoto, M. Molecular mechanism activation of Nrf2-Keap1 pathway in regulation of adaptive response to electrophiles. Free Radic. Biol. Med. 2004, 36, 1208–1213. [Google Scholar] [CrossRef] [PubMed]
- Son, S.; Lewis, B.A. Free radical scavenging and antioxidant activity of caffeic acid amide and ester analogues: Structure-activity relationship. J. Agric. Food Chem. 2002, 50, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Longo, R.; Vanella, A. Antioxidant activity: Role of caffeic acid phenethyl ester and galangian. Fitoterapia 2002, 73 (Suppl. 1), S21–S29. [Google Scholar] [CrossRef]
- Ahn, M.R.; Kunimass, K.; Kumazawa, S.; Nakayama, T.; Kaji, K.; Uto, Y.; Hori, H.; Nagasawa, H.; Ohta, T. Correlation between antiangiogenic activity and antioxidant activity of various components from propolis. Mol. Nutr. Food Res. 2009, 53, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Stavchansky, S.; Bowman, P.D.; Kerwin, S.M. Cytoprotective effect of caffeic acid phenethyl ester (CAPE) and catechol ring-fluorinated CAPE derivatives against menadione-induced oxidative stress in human endothelial cells. Bioorg. Med. Chem. 2006, 14, 4879–4887. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, H.R.; Uz, E.; Yucel, N.; Altuntas, I.; Ozcelik, N. Protective effect of caffeic acid phenethyl ester (CAPE) on lipid peroxidation and antioxidant enzymes in diabetic rat liver. J. Biochem. Mol. Toxicol. 2004, 18, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Okutan, H.; Ozcelik, N.; Yilmaz, H.R.; Uz, E. Effects of caffeic acid phenethyl ester on lipid peroxidation and antioxidant enzymes in diabetic rat heart. Clin. Biochem. 2005, 38, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Mapesa, J.O.; Waldschmit, N.; Schmoeller, I.; Blume, C.; Hofmann, T.; Mahungu, S.; Clavel, T.; Haller, D. Catechols in caffeic acid phenethyl ester are essential for inhibition on TNF-mediated IP-10 expression through NF-κB-dependent but Ho-1- and p38-independent mechanisms in mouse intestinal epithelial cells. Mol. Nutr. Food Res. 2011, 55, 1850–1861. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, F.; Maffia, P.; Pinto, L.; Ianaro, A.; Russo, A.; Capasso, F.; Lalenti, A. Phytochemical compounds involved in the anti-inflammatory effects propolis extract. Fitoterapia 2002, 73 (Suppl. 1), S53–S63. [Google Scholar]
- Ho, C.C.; Chou, M.Y.; Chen, F.L.; Hu, C.C.; Chen, C.S.; Lu, G.Y.; Yang, C.C. Effects of CAPE-like compounds on HIV replication in vitro and modulation of cytokines in vivo. J. Antimicrob. Chemother. 2005, 56, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Chung, T.W.; Moon, S.K.; Chang, Y.C.; Ko, J.H.; Lee, Y.C.; Cho, G.; Kim, S.H.; Kim, J.G.; Kim, C.H. Novel and therapeutic effect of caffeic acid phenethyl ester on hepatocarcinoma cells: Complete regression of hepatoma growth and metastasis by dual mechanism. FASEB J. 2004, 18, 1670–1681. [Google Scholar] [CrossRef] [PubMed]
- Balogun, E.; Hoque, M.; Gong, P.; Killeen, E.; Green, C.J.; Foresti, R.; Alam, J.; Motterlini, R. Curcumin activates the haem oxygenase-1 gene via regulation of Nrf2 and the antioxidant-responsive element. Biochem. J. 2003, 371, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Alía, M.; Ramos, S.; Mateos, R.; Bravo, L.; Goya, L. Response of the antioxidant defense system to ter-buty hydroperoxide and hydrogen peroxide in a human hepatoma cell line (HepG2). J. Biochem. Mol. Toxicol. 2005, 19, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Goya, L.; Mateos, T.; Bravo, L. Effect of the olice oil phenol hydroxytyrosol on human hepatoma HepG2 cells. Eur. J. Nutr. 2007, 46, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Stavchansky, S.; Zhao, B.; Byum, J.A.; Kerwin, S.; Bowman, P.D. Cytoprotection of human endothelial cells from menadione cytotoxicity by caffeci acid phenethyl ester: The role of heme oxygenase-1. Eur. J. Pharmacol. 2008, 591, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Stavchansky, S.; Kerwin, S.M.; Bowman, P.D. Structure-activity relationships in the cytoprotective effect of caffeic acid phenethyl ester (CAPE) and fluorinated derivatives: Effects on heme oxygenae-1 induction and antioxidant activities. Eur. J. Pharmacol. 2010, 635, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Scapagnini, G.; Foresti, R.; Calabrese, V.; Gluffrida Stella, A.M.; Green, C.J.; Motterlini, R. Caffeic acid phenethyl ester and curcumin: A novel class of heme oxygenase-1 inducers. Mol. Pharmacol. 2002, 61, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Scapagnini, G.; Vasto, S.; Abraham, N.G.; Caruso, C.; Zella, D.; Fabio, G. Modulation of Nrf2/ARE pathway by food polyphenols: a nutritional neuroprotective stragety for cognitive and neurodegenerative disorders. Mol. Neurobiol. 2011, 44, 192–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K.; Tanaka, I.; Nakahishi, I.; Kurematsu, A.; Yakumaru, H.; Ikota, N.; Ishihara, H. Drastic effect of several caffeic acid derivatives on the induction of heme oxygenase-1 expression revealed by quantitative real-time RT-PCR. Biofactors 2006, 28, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Yasui, N.; Nishiyama, E.; Juman, S.; Negishi, H.; Miki, T.; Yamori, Y.; Ikeda, K. Caffeic acid phenethyl ester suppresses oxidative stress in 3T3-L1 adipocytes. J. Asian Nat. Prod. Res. 2013, 15, 1189–1196. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Chen, H.; Bai, H.; Zhang, W.; Qin, X.; Zhang, X.; Li, W.; Liang, X.; Hai, C. Suppression of nuclear factor erythroid 2-related factor 2 via extracellular signal-regulated kinase contributes to bleomycin-induced oxidative stress and fibrogenesis. Toxicol. Lett. 2013, 220, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Sun, X.C.; Wang, G.N.; Li, M.M.; Chi, M. Icariside II enhances Nrf2 nuclear translocation to upregulate phase II detoxifying enzyme expression coupled with the ERK, Akt and JNk signaling pathways. Molecules 2011, 16, 9234–9244. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.L.; Xu, C.J.; Pan, Z.; Keum, Y.S.; Kim, J.W.; Shen, G.X.; Yu, S.W.; Oo, K.T.; Ma, J.J.; Kong, A.T. Butylated hydroxyanisole regulates ARE-mediated gene expession via Nrf2 coupled with ERK and JNK signaling pathway in HepG2 cells. Mol. Carcinog. 2006, 45, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, W.S.; Yum, S.W.; Hong, S.C.; Oh, J.E.; Lee, J.W.; Kwak, M.K.; Park, E.J.; Na, D.H.; Jung, Y.J. Caffeic acid phenethyl ester activation of Nrf2 pathway is enhanced under oxidative state: Structural analysis and potential as a pathologically targeted therapeutic agent in treatment of colonic inflammation. Free Radic. Biol. Med. 2013, 65, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Grindel, B.J.; Rohe, B.; Safford, S.E.; Bennett, J.J.; Farach-Carson, M.C. Tumor necrosis factor-α treatment of HepG2 cells mobilizes a cytoplasmic pool of Erp57/1,25D3-MARRS to the nucleus. J. Cell. Biochem. 2011, 112, 2606–2615. [Google Scholar] [CrossRef] [PubMed]
- Lautraite, S.; Bigot-Lasserre, D.; Bars, R.; Carmichael, N. Optimization of cell-based assays for medium through screening of oxidative stress. Toxicol. In Vitro 2003, 17, 207–220. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kim, J.-K.; Jang, H.-D. Nrf2-Mediated HO-1 Induction Coupled with the ERK Signaling Pathway Contributes to Indirect Antioxidant Capacity of Caffeic Acid Phenethyl Ester in HepG2 Cells. Int. J. Mol. Sci. 2014, 15, 12149-12165. https://doi.org/10.3390/ijms150712149
Kim J-K, Jang H-D. Nrf2-Mediated HO-1 Induction Coupled with the ERK Signaling Pathway Contributes to Indirect Antioxidant Capacity of Caffeic Acid Phenethyl Ester in HepG2 Cells. International Journal of Molecular Sciences. 2014; 15(7):12149-12165. https://doi.org/10.3390/ijms150712149
Chicago/Turabian StyleKim, Jin-Kyoung, and Hae-Dong Jang. 2014. "Nrf2-Mediated HO-1 Induction Coupled with the ERK Signaling Pathway Contributes to Indirect Antioxidant Capacity of Caffeic Acid Phenethyl Ester in HepG2 Cells" International Journal of Molecular Sciences 15, no. 7: 12149-12165. https://doi.org/10.3390/ijms150712149