The Dipeptidyl Peptidase-4 Inhibitor Sitagliptin Protects against Dyslipidemia-Related Kidney Injury in Apolipoprotein E Knockout Mice

Abstract
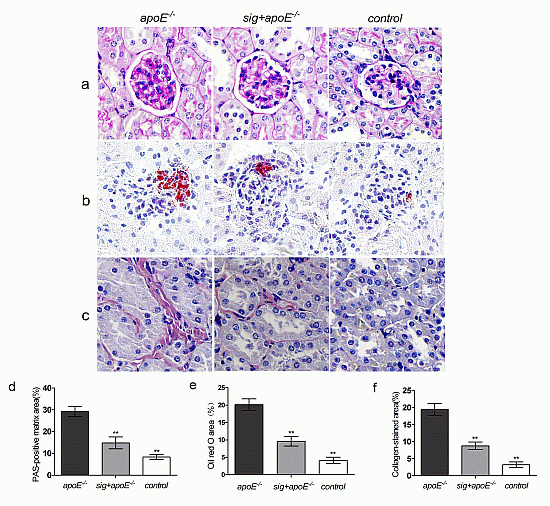
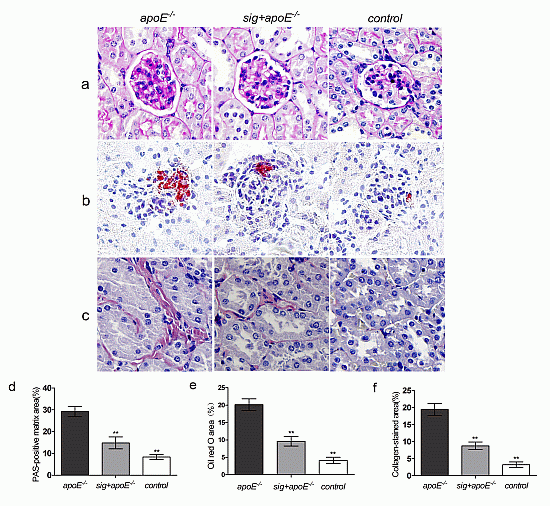
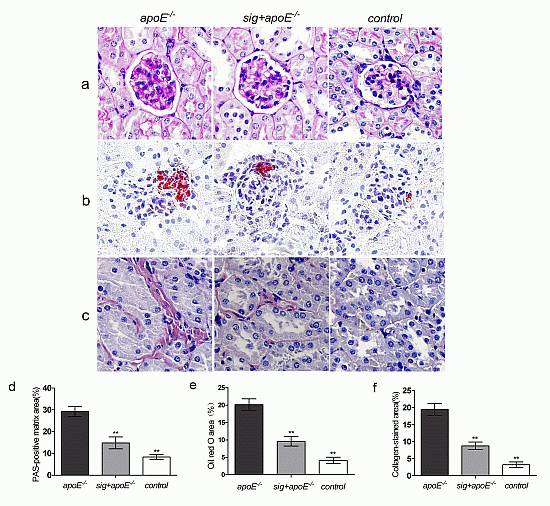
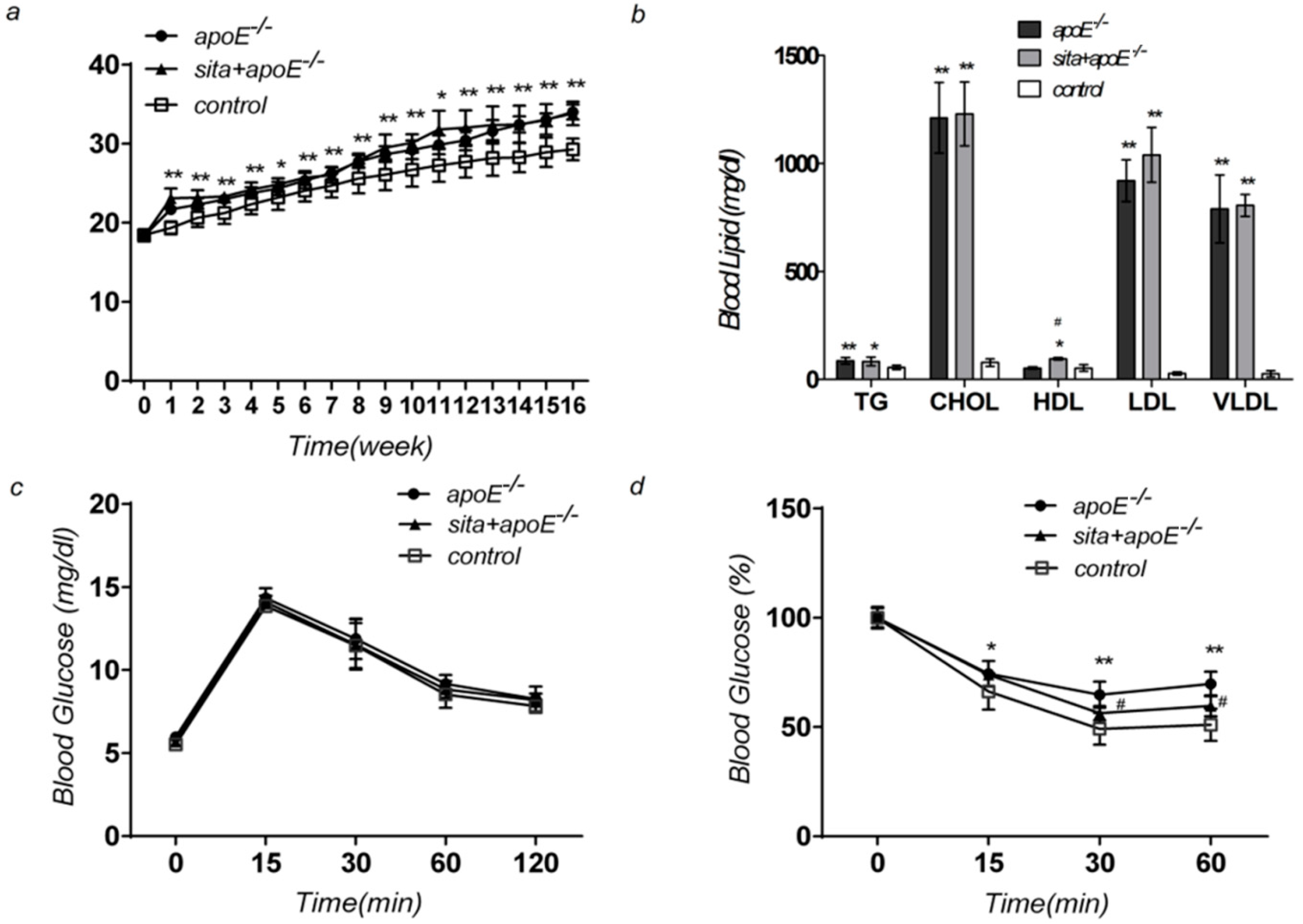
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Effect of Sitagliptin on Body Weight, Blood Glucose Level, Serum Lipid Level, and Insulin Sensitivity

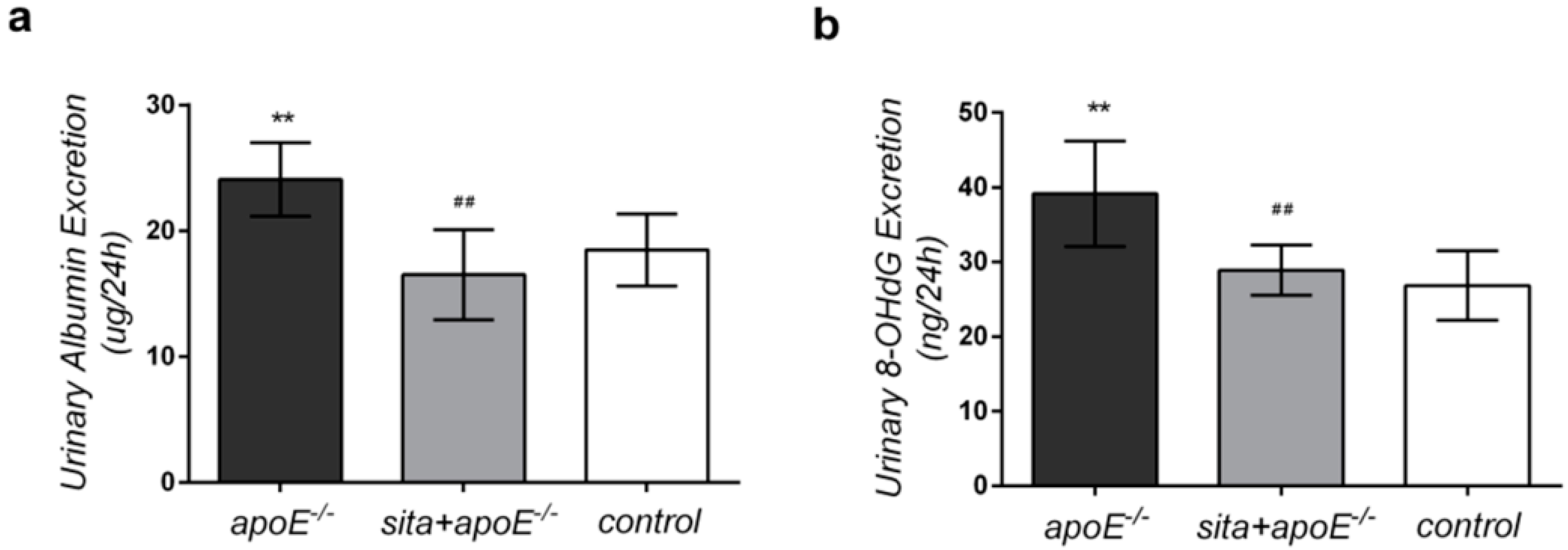
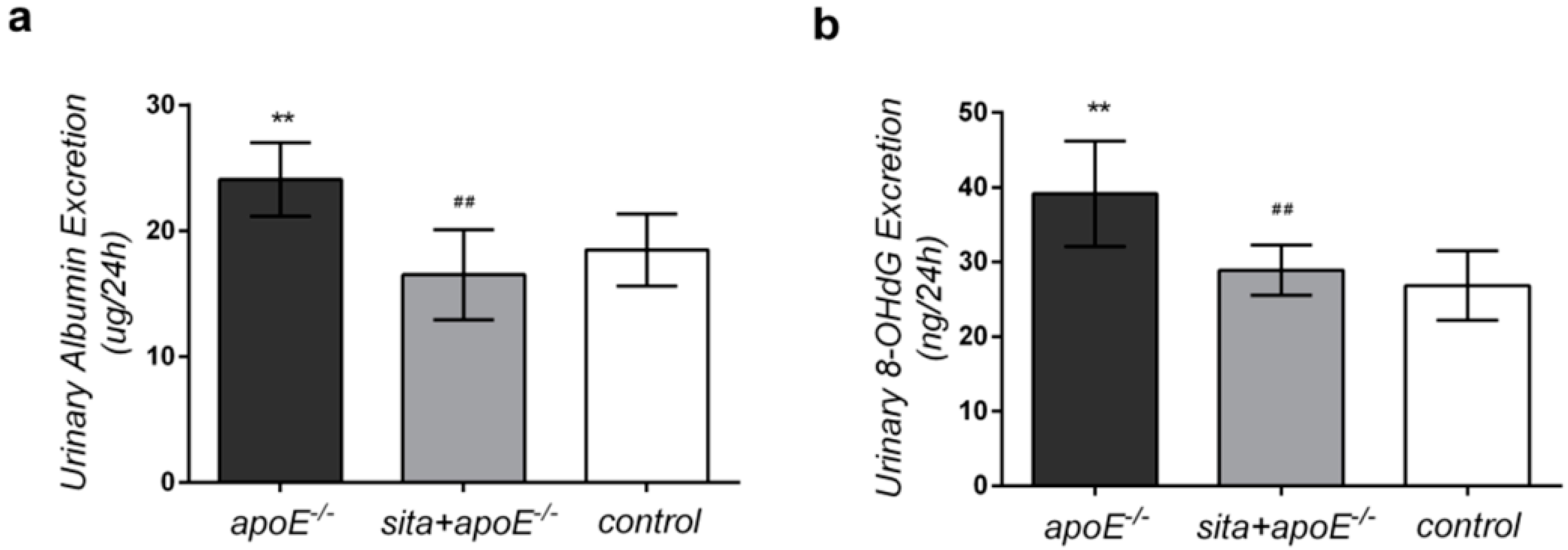
2.2. Effect of Sitagliptin on 24-h Urinary Albumin and 8-Hydroxy-2-deoxyguanosine (8-OHdG) Excretion
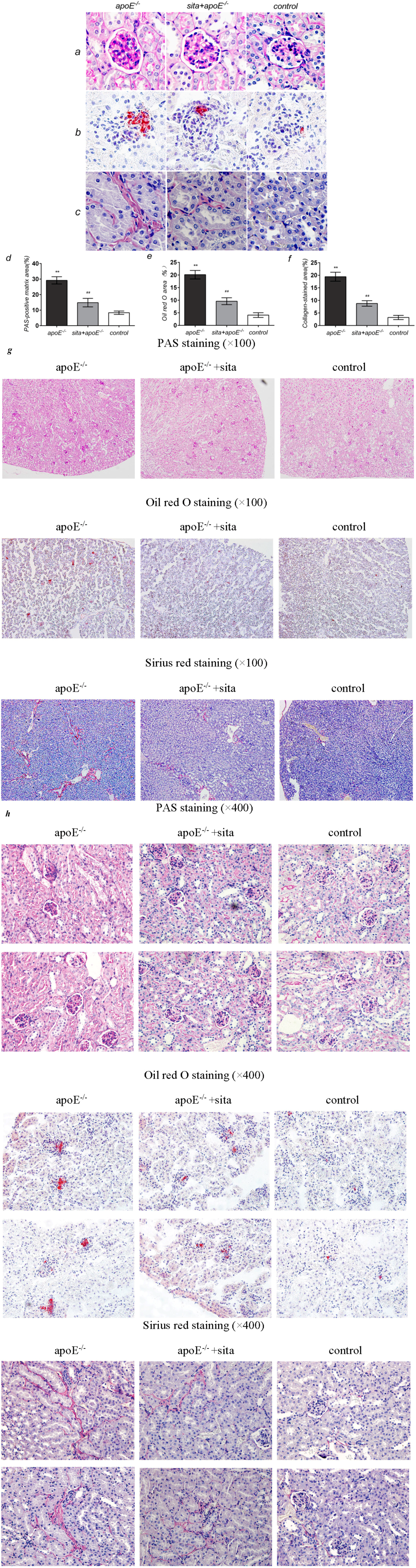
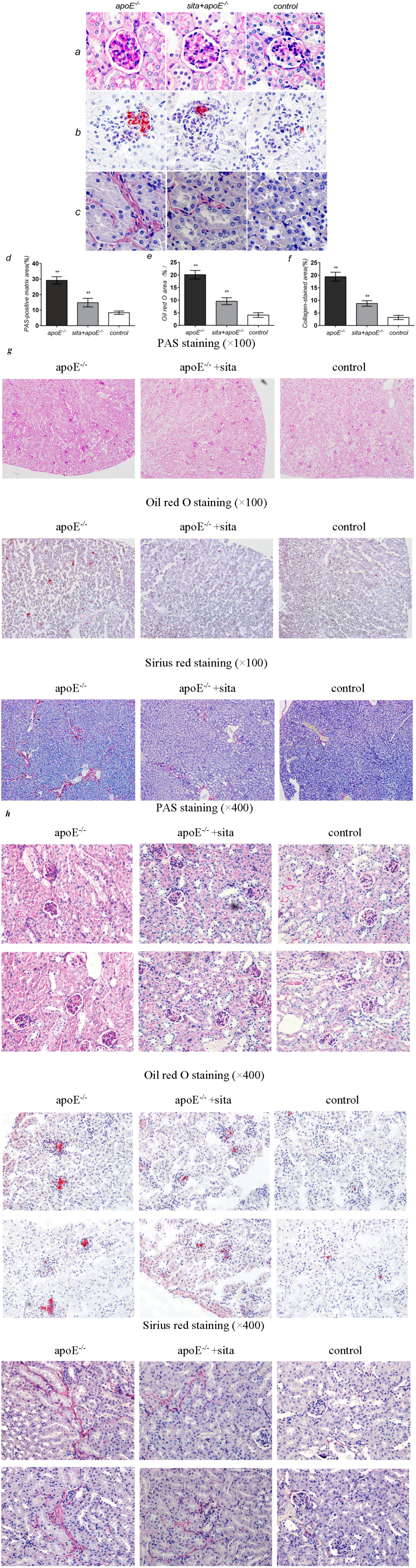
2.3. Effect of Sitagliptin on Dyslipidemia-Induced Pathological Changes of the Kidney
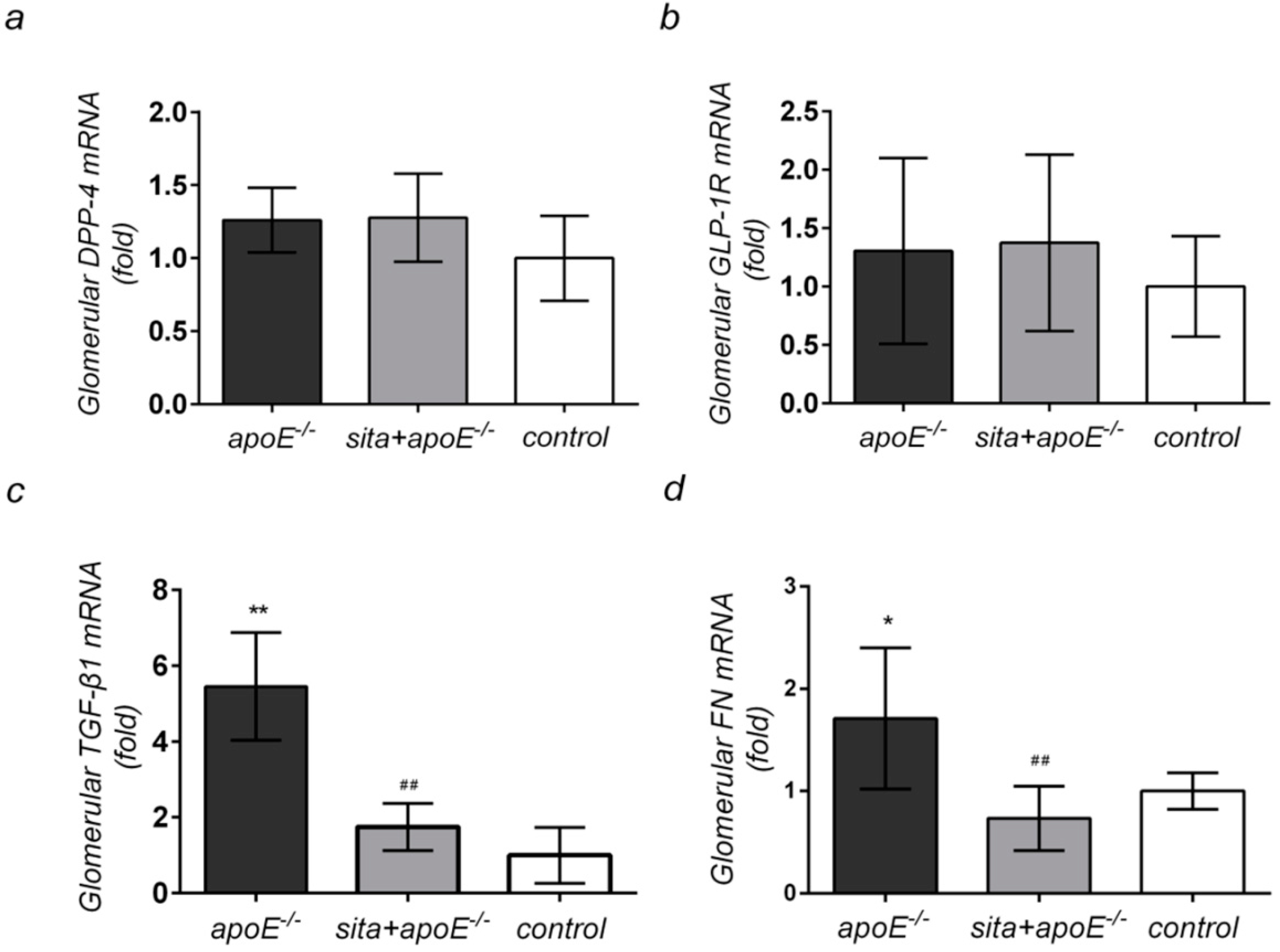
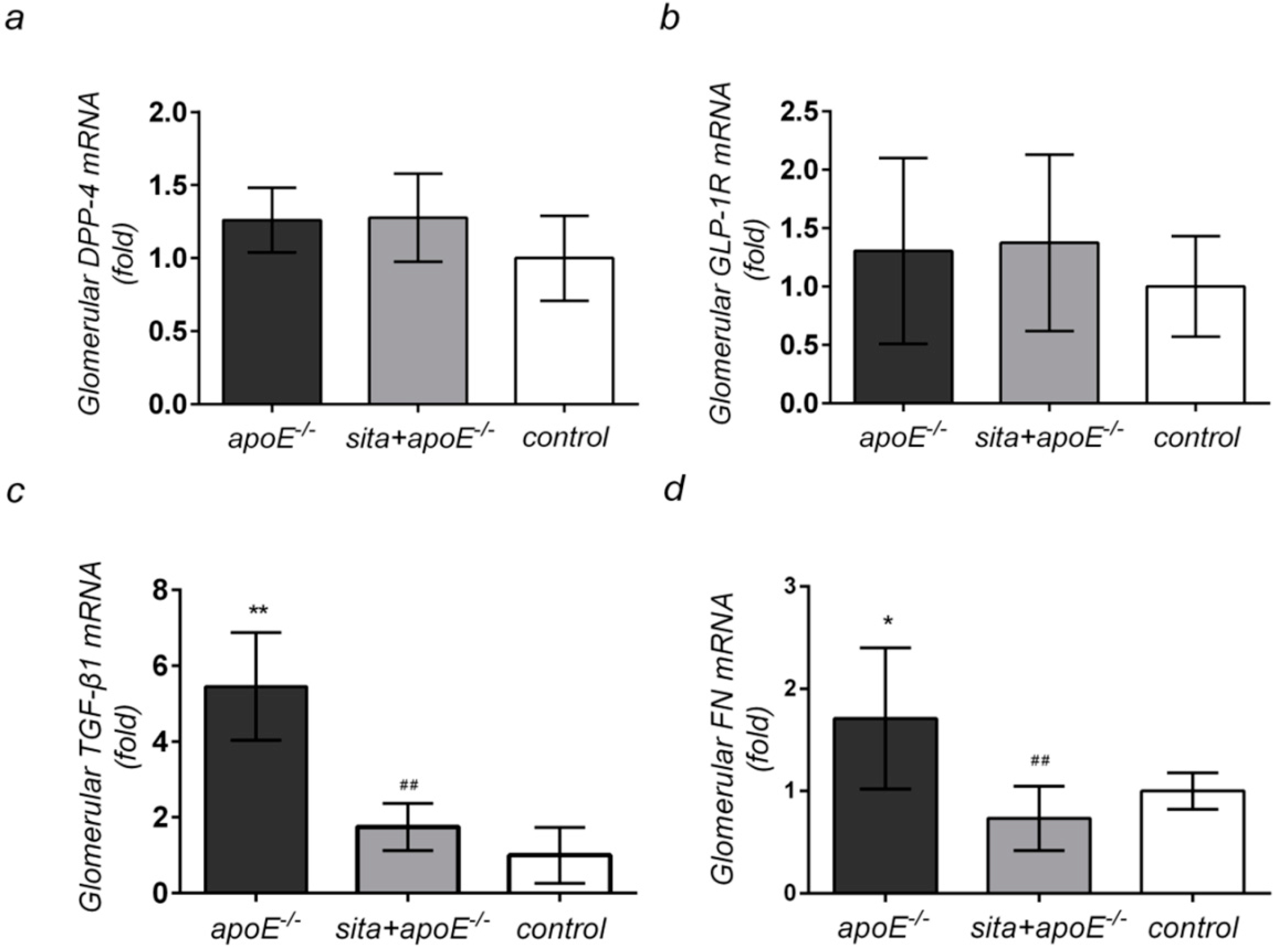
2.4. Effect of Sitagliptin Treatment on Renal Cortical mRNA Expression of DPP-4, GLP-1R, TGF-β1, and FN
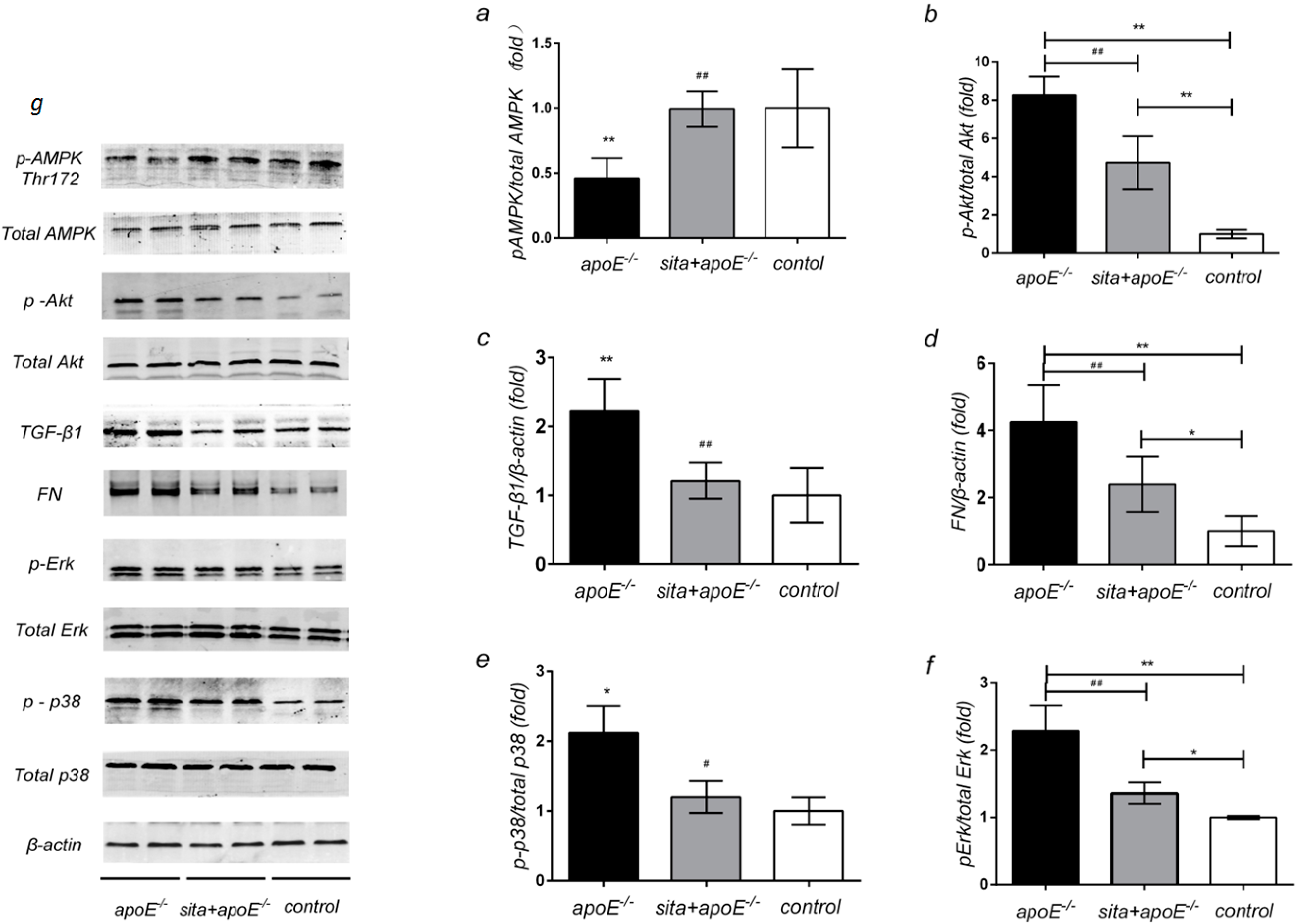
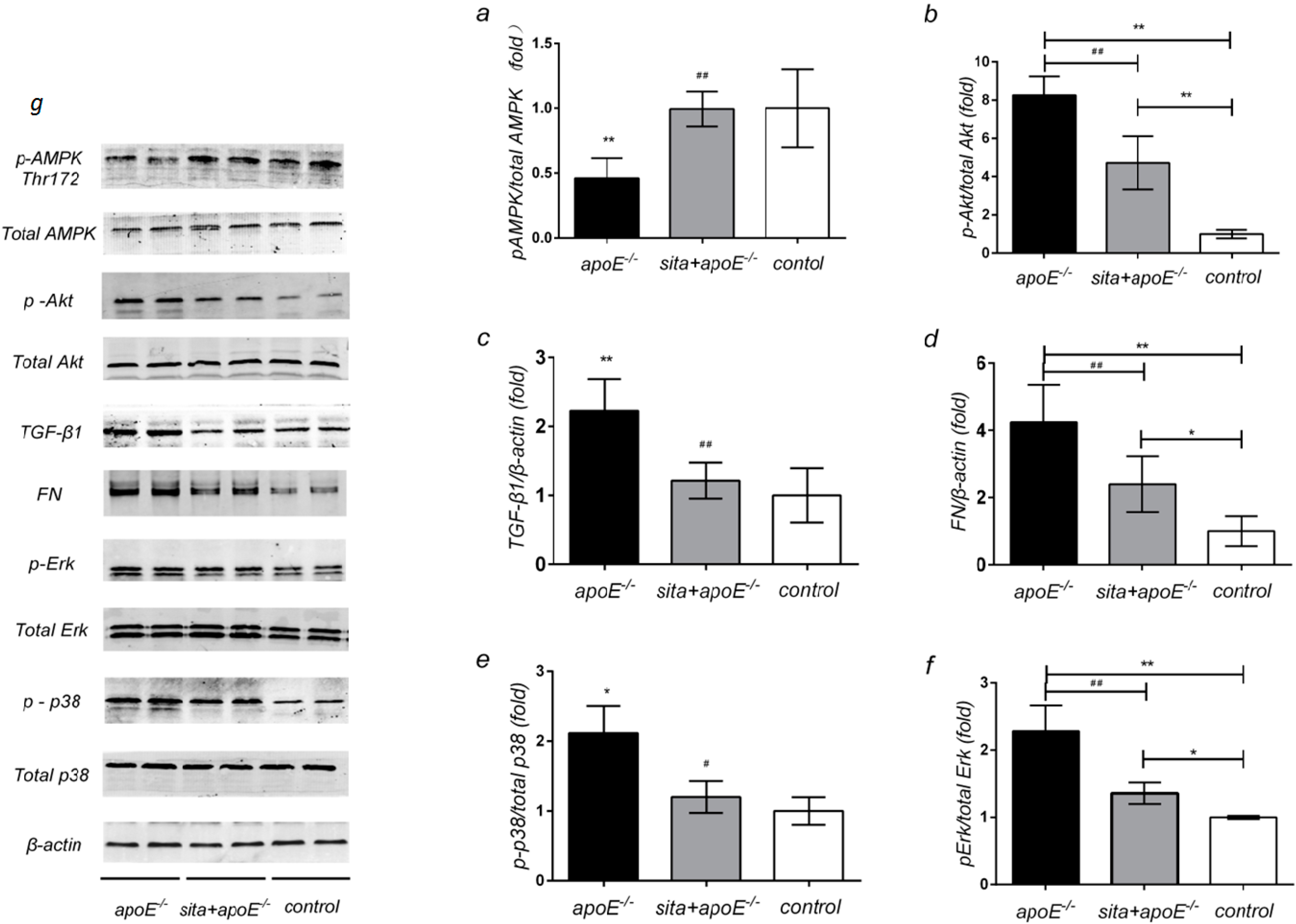
2.5. Effect of Sitagliptin on Renal Protein Expression of AMPK, Akt, TGF-β1, FN and MAPK Signaling Pathway
3. Discussion
4. Experimental Section
4.1. Animals
4.2. Analysis of Body Weight and Metabolic Profile
4.3. Assessment of Albuminuria and Urinary 8-OHdG
4.4. Renal Pathological Changes
4.5. cDNA Synthesis and Real-Time PCR
4.6. Western Blotting for Kidney Tissue
4.7. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Anderson, S.; King, A.J.; Brenner, B.M. Hyperlipidemia and glomerular sclerosis: An alternative viewpoint. Am. J. Med. 1989, 87, 34N–38N. [Google Scholar]
- Attman, P.O.; Alaupovic, P.; Samuelsson, O. Lipoprotein abnormalities as a risk factor for progressive nondiabetic renal disease. Kidney Int. Suppl. 1999, 71, S14–S17. [Google Scholar]
- Trevisan, R.; Dodesini, A.R.; Lepore, G. Lipids and renal disease. J. Am. Soc. Nephrol. 2006, 17, S145–S147. [Google Scholar] [CrossRef]
- Appel, G. Lipid abnormalities in renal disease. Kidney Int. 1991, 39, 169–183. [Google Scholar] [CrossRef]
- Samuelsson, O.; Mulec, H.; Knight-Gibson, C.; Attman, P.O.; Kron, B.; Larsson, R.; Weiss, L.; Wedel, H.; Alaupovic, P. Lipoprotein abnormalities are associated with increased rate of progression of human chronic renal insufficiency. Nephrol. Dial. Transplant. 1997, 12, 1908–1915. [Google Scholar] [CrossRef]
- Hunsicker, L.G.; Adler, S.; Caggiula, A.; England, B.K.; Greene, T.; Kusek, J.W.; Rogers, N.L.; Teschan, P.E. Predictors of the progression of renal disease in the modification of diet in renal disease study. Kidney Int. 1997, 51, 1908–1919. [Google Scholar]
- Maschio, G.; Oldrizzi, L.; Rugiu, C.; Loschiavo, C. Serum lipids in patients with chronic renal failure on long-term, protein-restricted diets. Am. J. Med. 1989, 87, 51N–54N. [Google Scholar]
- Wen, M.; Segerer, S.; Dantas, M.; Brown, P.A.; Hudkins, K.L.; Goodpaster, T.; Kirk, E.; LeBoeuf, R.C.; Alpers, C.E. Renal injury in apolipoprotein E-deficient mice. Lab. Investig. 2002, 82, 999–1006. [Google Scholar] [CrossRef]
- Decleves, A.E.; Mathew, A.V.; Cunard, R.; Sharma, K. AMPK mediates the initiation of kidney disease induced by a high-fat diet. J. Am. Soc. Nephrol. 2011, 22, 1846–1855. [Google Scholar] [CrossRef]
- Sharma, K.; Ramachandrarao, S.; Qiu, G.; Usui, H.K.; Zhu, Y.; Dunn, S.R.; Ouedraogo, R.; Hough, K.; McCue, P.; Chan, L.; et al. Adiponectin regulates albuminuria and podocyte function in mice. J. Clin. Investig. 2008, 118, 1645–1656. [Google Scholar]
- Nangaku, M. Energy policy of the kidney: Launch of AMPK as a novel therapeutic target. Am. J. Physiol. Renal. Physiol. 2013, 305, F977–F978. [Google Scholar] [CrossRef]
- Satriano, J.; Sharma, K.; Blantz, R.C.; Deng, A. Induction of AMPK activity corrects early pathophysiological alterations in the subtotal nephrectomy model of chronic kidney disease. Am. J. Physiol. Renal. Physiol. 2013, 305, F727–F733. [Google Scholar] [CrossRef]
- Lee, M.J.; Feliers, D.; Mariappan, M.M.; Sataranatarajan, K.; Mahimainathan, L.; Musi, N.; Foretz, M.; Viollet, B.; Weinberg, J.M.; Choudhury, G.G.; et al. A role for AMP-activated protein kinase in diabetes-induced renal hypertrophy. Am. J. Physiol. Renal. Physiol. 2007, 292, F617–F627. [Google Scholar]
- Kovacic, S.; Soltys, C.L.; Barr, A.J.; Shiojima, I.; Walsh, K.; Dyck, J.R. Akt activity negatively regulates phosphorylation of AMP-activated protein kinase in the heart. J. Biol. Chem. 2003, 278, 39422–39427. [Google Scholar]
- Dhillon, S. Sitagliptin: A review of its use in the management of type 2 diabetes mellitus. Drugs 2010, 70, 489–512. [Google Scholar] [CrossRef]
- Baggio, L.L.; Drucker, D.J. Harnessing the therapeutic potential of glucagon-like peptide-1: A critical review. Treat. Endocrinol. 2002, 1, 117–125. [Google Scholar]
- Usdin, T.B.; Mezey, E.; Button, D.C.; Brownstein, M.J.; Bonner, T.I. Gastric inhibitory polypeptide receptor, a member of the secretin-vasoactive intestinal peptide receptor family, is widely distributed in peripheral organs and the brain. Endocrinology 1993, 133, 2861–2870. [Google Scholar]
- Baggio, L.L.; Drucker, D.J. Biology of incretins: GLP-1 and GIP. Gastroenterology 2007, 132, 2131–2157. [Google Scholar] [CrossRef]
- Monami, M.; Lamanna, C.; Desideri, C.M.; Mannucci, E. DPP-4 inhibitors and lipids: Systematic review and meta-analysis. Adv. Ther. 2012, 29, 14–25. [Google Scholar] [CrossRef]
- Parlevliet, E.T.; Wang, Y.; Geerling, J.J.; Schroder-Van, D.E.J.; Picha, K.; O’Neil, K.; Stojanovic-Susulic, V.; Ort, T.; Havekes, L.M.; Romijn, J.A.; et al. GLP-1 receptor activation inhibits VLDL production and reverses hepatic steatosis by decreasing hepatic lipogenesis in high-fat-fed APOE*3-Leiden mice. PLoS One 2012, 7, e49152. [Google Scholar]
- Tremblay, A.J.; Lamarche, B.; Deacon, C.F.; Weisnagel, S.J.; Couture, P. Effect of sitagliptin therapy on postprandial lipoprotein levels in patients with type 2 diabetes. Diabetes Obes. Metab. 2011, 13, 366–373. [Google Scholar] [CrossRef]
- Hsieh, J.; Longuet, C.; Baker, C.L.; Qin, B.; Federico, L.M.; Drucker, D.J.; Adeli, K. The glucagon-like peptide 1 receptor is essential for postprandial lipoprotein synthesis and secretion in hamsters and mice. Diabetologia 2012, 53, 552–561. [Google Scholar]
- Fujita, H.; Taniai, H.; Murayama, H.; Ohshiro, H.; Hayashi, H.; Sato, S.; Kikuchi, N.; Komatsu, T.; Komatsu, K.; Komatsu, K.; et al. DPP-4 inhibition with alogliptin on top of angiotensin II type 1 receptor blockade ameliorates albuminuria via up-regulation of SDF-1α in type 2 diabetic patients with incipient nephropathy. Endocr. J. 2014, 61, 159–166. [Google Scholar] [CrossRef]
- Mega, C.; de Lemos, E.T.; Vala, H.; Fernandes, R.; Oliveira, J.; Mascarenhas-Melo, F.; Teixeira, F.; Reis, F. Diabetic nephropathy amelioration by a low-dose sitagliptin in an animal model of type 2 diabetes (Zucker diabetic fatty rat). Exp. Diabetes Res. 2011, 2011, 162092. [Google Scholar]
- Liu, W.J.; Xie, S.H.; Liu, Y.N.; Kim, W.; Jin, H.Y.; Park, S.K.; Shao, Y.M.; Park, T.S. Dipeptidyl peptidase IV inhibitor attenuates kidney injury in streptozotocin-induced diabetic rats. J. Pharmacol.Exp. Ther. 2012, 340, 248–255. [Google Scholar] [CrossRef]
- Joo, K.W.; Kim, S.; Ahn, S.Y.; Chin, H.J.; Chae, D.W.; Lee, J.; Han, J.S.; Na, K.Y. Dipeptidyl peptidase IV inhibitor attenuates kidney injury in rat remnant kidney. BMC Nephrol. 2013, 14, 98. [Google Scholar] [CrossRef]
- Chen, Y.T.; Tsai, T.H.; Yang, C.C.; Sun, C.K.; Chang, L.T.; Chen, H.H.; Chang, C.L.; Sung, P.H.; Zhen, Y.Y.; Leu, S.; et al. Exendin-4 and sitagliptin protect kidney from ischemia-reperfusion injury through suppressing oxidative stress and inflammatory reaction. J. Transl. Med. 2013, 11, 270. [Google Scholar] [CrossRef]
- Moorhead, J.F.; Chan, M.K.; El-Nahas, M.; Varghese, Z. Lipid nephrotoxicity in chronic progressive glomerular and tubulo-interstitial disease. Lancet 1982, 2, 1309–1311. [Google Scholar]
- Kees-Folts, D.; Diamond, J.R. Relationship between hyperlipidemia, lipid mediators, and progressive glomerulosclerosis in the nephrotic syndrome. Am. J. Nephrol. 1993, 13, 365–375. [Google Scholar] [CrossRef]
- Schaeffner, E.S.; Kurth, T.; Curhan, G.C.; Glynn, R.J.; Rexrode, K.M.; Baigent, C.; Buring, J.E.; Gaziano, J.M. Cholesterol and the risk of renal dysfunction in apparently healthy men. J. Am. Soc. Nephrol. 2013, 14, 2084–2091. [Google Scholar]
- Taneda, S.; Hudkins, K.L.; Cui, Y.; Farr, A.G.; Alpers, C.E.; Segerer, S. Growth factor expression in a murine model of cryoglobulinemia. Kidney Int. 2003, 63, 576–590. [Google Scholar] [CrossRef]
- Viollet, B.; Foretz, M.; Guigas, B.; Horman, S.; Dentin, R.; Bertrand, L.; Hue, L.; Andreelli, F. Activation of AMP-activated protein kinase in the liver: A new strategy for the management of metabolic hepatic disorders. J. Physiol. 2006, 574, 41–53. [Google Scholar] [CrossRef]
- Zeng, Y.; Li, C.; Guan, M.; Zheng, Z.; Li, J.; Xu, W.; Wang, L.; He, F.; Xue, Y. The DPP-4 inhibitor sitagliptin attenuates the progress of atherosclerosis in apolipoprotein-E-knockout mice via AMPK- and MAPK-dependent mechanisms. Cardiovasc. Diabetol. 2014, 13, 32. [Google Scholar] [CrossRef]
- Rask-Madsen, C.; Li, Q.; Freund, B.; Feather, D.; Abramov, R.; Wu, I.H.; Chen, K.; Yamamoto-Hiraoka, J.; Goldenbogen, J.; Sotiropoulos, K.B.; et al. Loss of insulin signaling in vascular endothelial cells accelerates atherosclerosis in apolipoprotein E null mice. Cell Metab. 2012, 11, 379–89. [Google Scholar]
- Matsubara, J.; Sugiyama, S.; Sugamura, K.; Nakamura, T.; Fujiwara, Y.; Akiyama, E.; Kurokawa, H.; Nozaki, T.; Ohba, K.; Konishi, M.; et al. A dipeptidyl peptidase-4 inhibitor, des-fluoro-sitagliptin, improves endothelial function and reduces atherosclerotic lesion formation in apolipoprotein E-deficient mice. J. Am. Coll. Cardiol. 2012, 59, 265–276. [Google Scholar] [CrossRef]
- Deacon, C.F.; Holst, J.J. Dipeptidyl peptidase IV inhibition as an approach to the treatment and prevention of type 2 diabetes: A historical perspective. Biochem. Biophys. Res. Commun. 2002, 294, 1–4. [Google Scholar] [CrossRef]
- Conarello, S.L.; Li, Z.; Ronan, J.; Roy, R.S.; Zhu, L.; Jiang, G.; Liu, F.; Woods, J.; Zycband, E.; Moller, D.E.; et al. Mice lacking dipeptidyl peptidase IV are protected against obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2003, 100, 6825–6830. [Google Scholar] [CrossRef]
- Fujita, H.; Morii, T.; Fujishima, H.; Sato, T.; Shimizu, T.; Hosoba, M.; Tsukiyama, K.; Narita, T.; Takahashi, T.; Drucker, D.J.; et al. The protective roles of GLP-1R signaling in diabetic nephropathy: Possible mechanism and therapeutic potential. Kidney Int. 2014, 85, 579–589. [Google Scholar] [CrossRef]
- Ishibashi, Y.; Nishino, Y.; Matsui, T.; Takeuchi, M.; Yamagishi, S. Glucagon-like peptide-1 suppressesadvanced glycation end product-induced monocyte chemoattractant protein-1 expression in mesangial cells by reducing advanced glycation end product receptor level. Metabolism 2011, 60, 1271–1277. [Google Scholar] [CrossRef]
- Walker, P.D.; Kaushal, G.P.; Shah, S.V. Meprin A, the major matrix degrading enzyme in renal tubules, produces a novel nidogen fragment in vitro and in vivo. Kidney Int. 1998, 53, 1673–1680. [Google Scholar] [CrossRef]
- Nogueira-Machado, J.A.; Volpe, C.M.; Veloso, C.A.; Chaves, M.M. HMGB1, TLR and RAGE: A functional tripod that leads to diabetic inflammation. Expert Opin. Ther. Targets 2011, 15, 1023–1035. [Google Scholar] [CrossRef]
- Hocher, B.; Reichetzeder, C.; Alter, M.L. Renal and cardiac effects of DPP4 inhibitors—From preclinical development to clinical research. Kidney Blood Press. Res. 2012, 36, 65–84. [Google Scholar] [CrossRef]
- Jin, H.Y.; Liu, W.J.; Park, J.H.; Baek, H.S.; Park, T.S. Effect of dipeptidyl peptidase-IV (DPP-IV) inhibitor (Vildagliptin) on peripheral nerves in streptozotocin-induced diabetic rats. Arch. Med. Res. 2009, 40, 536–544. [Google Scholar] [CrossRef]
- Abd, E.M.D.; Elshazly, S.M. Renoprotective effect of sitagliptin against hypertensive nephropathy induced by chronic administration of L-NAME in rats: Role of GLP-1 and GLP-1 receptor. Eur. J. Pharmacol. 2013, 720, 158–165. [Google Scholar] [CrossRef]
- Munoz-Garcia, B.; Moreno, J.A.; Lopez-Franco, O.; Sanz, A.B.; Martin-Ventura, J.L.; Blanco, J.; Jakubowski, A.; Burkly, L.C.; Ortiz, A.; Egido, J.; et al. Tumor necrosis factor-like weak inducer of apoptosis (TWEAK) enhances vascular and renal damage induced by hyperlipidemic diet in ApoE-knockout mice. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 2061–2068. [Google Scholar] [CrossRef]
- Manson, S.R.; Song, J.B.; Hruska, K.A.; Austin, P.F. HDAC dependent transcriptional repression of BMP-7 potentiates TGF-β mediated renal fibrosis in obstructive uropathy. J. Urol. 2014, 191, 242–252. [Google Scholar] [CrossRef]
- Huang, X.; Wen, D.; Zhang, M.; Xie, Q.; Ma, L.; Guan, Y.; Ren, Y.; Chen, J.; Hao, C.M. Sirt1 activation ameliorates renal fibrosis by inhibiting the TGF-β/Smad3 pathway. J. Cell. Biochem. 2014. [Google Scholar] [CrossRef]
- Lopez-Hernandez, F.J.; Lopez-Novoa, J.M. Role of TGF-β in chronic kidney disease: An integration of tubular, glomerular and vascular effects. Cell Tissue Res. 2012, 347, 141–154. [Google Scholar] [CrossRef]
- Chen, Z.X.; Xie, X.Y.; Yu, R.C.; Zhang, J.; Lei, M.X. Hyperlipidemia induced by high fat diet ingestion activates TGF-β/Smad signaling pathway in the kidney of diabetic rats. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2008, 33, 906–912. (In Chinese) [Google Scholar]
- Yanagita, M. Inhibitors/antagonists of TGF-β system in kidney fibrosis. Nephrol. Dial. Transplant. 2012, 27, 3686–3691. [Google Scholar] [CrossRef]
- Kato, M. TGF-β-induced signaling circuit loops mediated by microRNAs as new therapeutic targets for renal fibrosis? Kidney Int. 2013, 84, 1067–1069. [Google Scholar] [CrossRef]
- Liu, N.; He, S.; Ma, L.; Ponnusamy, M.; Tang, J.; Tolbert, E.; Bayliss, G.; Zhao, T.C.; Yan, H.; Zhuang, S. Blocking the class I histone deacetylase ameliorates renal fibrosis and inhibits renal fibroblast activation via modulating TGF-β and EGFR signaling. PLoS One 2013, 8, e54001. [Google Scholar]
- Meng, X.M.; Chung, A.C.; Lan, H.Y. Role of the TGF-β/BMP-7/Smad pathways in renal diseases. Clin. Sci. 2013, 124, 243–254. [Google Scholar] [CrossRef]
- Lan, H.Y. Diverse roles of TGF-β/Smads in renal fibrosis and inflammation. Int. J. Biol. Sci. 2011, 7, 1056–1067. [Google Scholar] [CrossRef]
- Dai, Y.; Palade, P.; Wang, X.; Mercanti, F.; Ding, Z.; Dai, D.; Mehta, J.L. High fat diet causes renal fibrosis in LDLr-null mice through MAPK-NF-κB pathway mediated by Ox-LDL. J. Cardiovasc. Pharmacol. 2014, 63, 158–166. [Google Scholar] [CrossRef]
- Gao, X.; Wu, G.; Gu, X.; Fu, L.; Mei, C. Kruppel-like factor 15 modulates renal interstitial fibrosis by ERK/MAPK and JNK/MAPK pathways regulation. Kidney Blood Press. Res. 2013, 37, 631–640. [Google Scholar] [CrossRef]
- Ma, X.H.; Zou, Y.; Zhang, Y.; He, L.Q. MAPK p38 pathway may be involved in renal function improvement in chronic renal failure rats treated with Jianpi Qinghua decoction. Zhejiang Da Xue Xue Bao Yi Xue Ban 2013, 42, 567–572. (In Chinese) [Google Scholar]
- Zdychova, J.; Komers, R. Emerging role of Akt kinase/protein kinase B signaling in pathophysiology of diabetes and its complications. Physiol. Res. 2005, 54, 1–16. [Google Scholar]
- Chung, H.W.; Lim, J.H.; Kim, M.Y.; Shin, S.J.; Chung, S.; Choi, B.S.; Kim, H.W.; Kim, Y.S.; Park, C.W.; Chang, Y.S. High-fat diet-induced renal cell apoptosis and oxidative stress in spontaneously hypertensive rat are ameliorated by fenofibrate through the PPARα-FoxO3a-PGC-1α pathway. Nephrol. Dial. Transplant. 2012, 27, 2213–2225. [Google Scholar] [CrossRef]
- Decleves, A.E.; Zolkipli, Z.; Satriano, J.; Wang, L.; Nakayama, T.; Rogac, M.; Le, T.P.; Nortier, J.L.; Farquhar, M.G.; Naviaux, R.K.; et al. Regulation of lipid accumulation by AMK-activated kinase in high fat diet-induced kidney injury. Kidney Int. 2014, 85, 611–623. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, J.; Guan, M.; Li, C.; Lyv, F.; Zeng, Y.; Zheng, Z.; Wang, C.; Xue, Y. The Dipeptidyl Peptidase-4 Inhibitor Sitagliptin Protects against Dyslipidemia-Related Kidney Injury in Apolipoprotein E Knockout Mice. Int. J. Mol. Sci. 2014, 15, 11416-11434. https://doi.org/10.3390/ijms150711416
Li J, Guan M, Li C, Lyv F, Zeng Y, Zheng Z, Wang C, Xue Y. The Dipeptidyl Peptidase-4 Inhibitor Sitagliptin Protects against Dyslipidemia-Related Kidney Injury in Apolipoprotein E Knockout Mice. International Journal of Molecular Sciences. 2014; 15(7):11416-11434. https://doi.org/10.3390/ijms150711416
Chicago/Turabian StyleLi, Jingjing, Meiping Guan, Chenzhong Li, Fuping Lyv, Yanmei Zeng, Zongji Zheng, Chengzhi Wang, and Yaoming Xue. 2014. "The Dipeptidyl Peptidase-4 Inhibitor Sitagliptin Protects against Dyslipidemia-Related Kidney Injury in Apolipoprotein E Knockout Mice" International Journal of Molecular Sciences 15, no. 7: 11416-11434. https://doi.org/10.3390/ijms150711416