1. Introduction
Coat color has long been a subject of interest to breeders and scientists [
1]. Not only is coat color a model phenotype for studying gene action and gene interactions, but also is important for goatskin, which is a valuable animal product. Mammalian coat color is almost totally dependent on either the presence or absence of melanin in skin and follicles [
2]. Therefore, it is necessary to understand the process of melanocyte formation. Melanocytes emerge from the neural crest, which is an early embryonic structure [
3]. In the late embryonic stage, neural crest cells differentiate into melanoblasts, which migrate to the skin basal layer, where they settle and are involved in the development of the hair follicles [
4]. The pigment cells reside in the bulb of the hair follicle and affect the coat color. Various factors affect mammalian coat color, including the composition, numbers, and arrangements of the melanin granules [
5]. Moreover, a number of genes such as
TYR,
MITF,
ASIP and
MC1R regulate the progress of hair follicle pigmentation. However, few studies have studied the regulatory mechanisms at the post-transcriptional level. MicroRNAs (miRNAs) are small non-protein-coding transcripts that regulate gene expression post-transcriptionally by binding to the 3'-untranslated region (3'-UTR) of the target messenger RNAs (mRNAs) thereby causing suppression of protein synthesis or mRNA cleavage [
6]. Increasing evidence shows that miRNAs play an important regulatory role in a variety of biological processes. The development of next-generation massively sequencing (NGMS) technologies, providing high throughput with low cost, have revolutionized genomic research, allowing many animal miRNAs to be identified and deposited in MiRBase (
http://www.mirbase.org/). To date, 24,521 entries representing hairpin precursor miRNAs, expressing 30,424 mature miRNAs products in 206 species have been identified and deposited in the public miRNA database miRBase (Release 20.0, June 2013). Among them, 2578 miRNAs were from human, 1908 from mouse, and 153 from sheep. Only a few studies identified miRNAs in goats (
Capra hircus) [
7,
8,
9], indicating that goat miRNAs still need to be sequenced.
In the last two years, significant progress has been made on the goat genome. The 2.66 Gb genome sequence data were obtained by combing short-read sequencing data and optical mapping data from a female Yunnan black goat. Meanwhile, 51 differentially expressed genes between the two types of hair follicles, the primary and secondary follicle, of a cashmere goat, were identified by comparative transcriptome analysis [
10]. Conserved miRNAs (346) were identified between dry period and peak lactation mammary gland tissues in the dairy goat [
11]. Five differentially expressed miRNAs were verified by quantitative PCR in the ovaries of pregnant and non-pregnant goats [
12]. Hair color is an important trait in the goat. Recently, several studies have tried to identify genes and miRNAs in goatskin and hair follicles. MiRNA data produced by Solexa sequencing among three follicular cycling stages in goatskin and hair follicles were reported [
13]. Similar studies focused on the identification of miRNAs in hair follicle and skin development [
14,
15,
16]. These studies enriched the goat hair follicle and skin miRNA database and enhanced our understanding of the process of miRNA regulation on development of skin and hair follicle. However, very few references are related to the mechanism of how miRNAs regulate coat color. Five differentially expressed miRNAs between the white and brown skin of alpaca were identified by quantitative PCR, including miR-211 and miR-202, which were significantly expressed in brown and white skins, respectively [
17]. MiR-137, which can downregulate the microphthalmia-associated transcription factor (MITF) was verified to influence the phenotype of coat color in transgenic mice overexpressing miR-137 [
18]. No studies have been attempted to identify miRNAs affecting coat color in the goat.
In this study, we sequenced miRNAs from black and white hair follicles collected from 1-year old crossbreed white and black coat color goats. These results provide new information on miRNA expression profiles in the goat and identify possible miRNA regulated pathways related to pigmentation in hair follicles.
3. Discussion
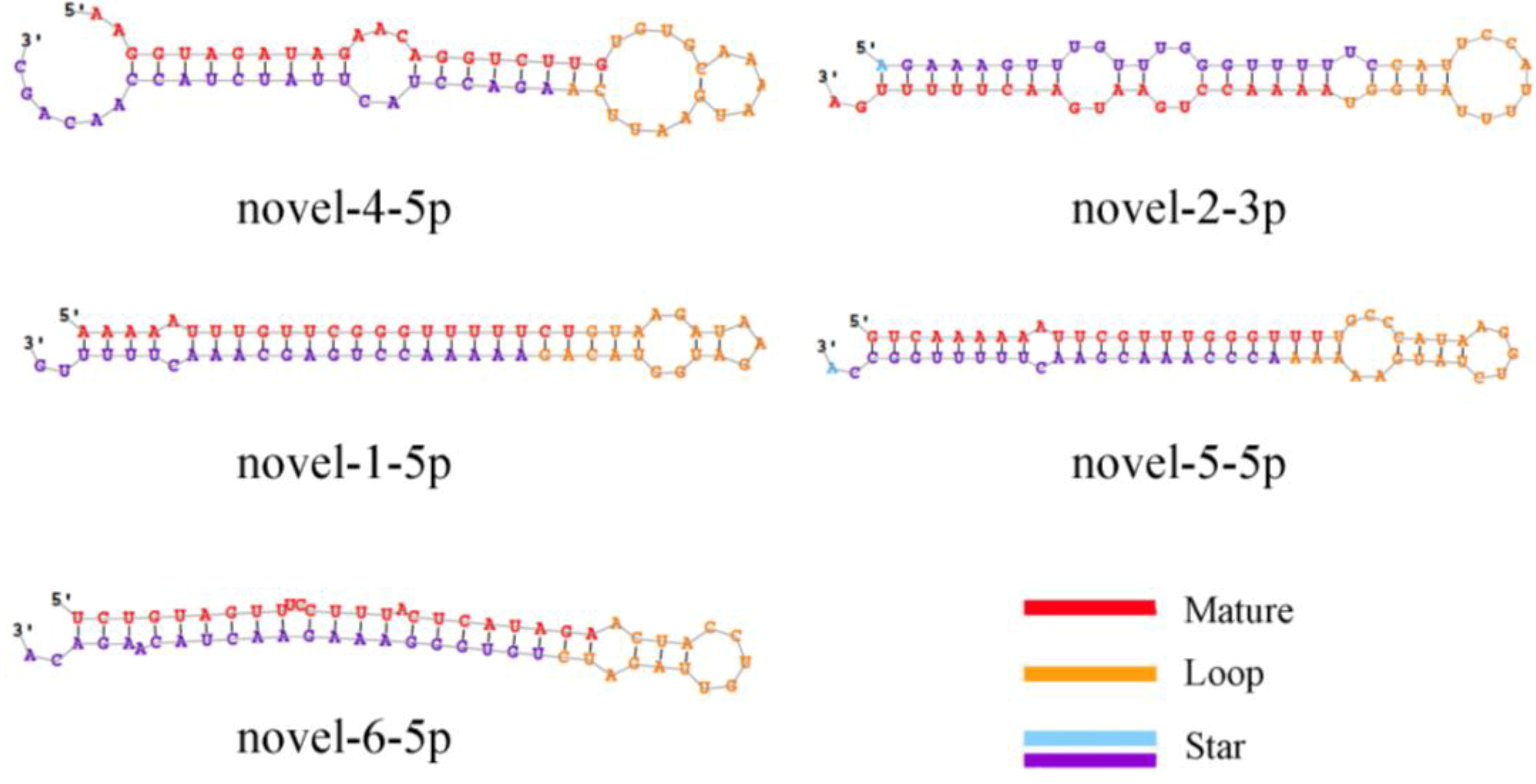
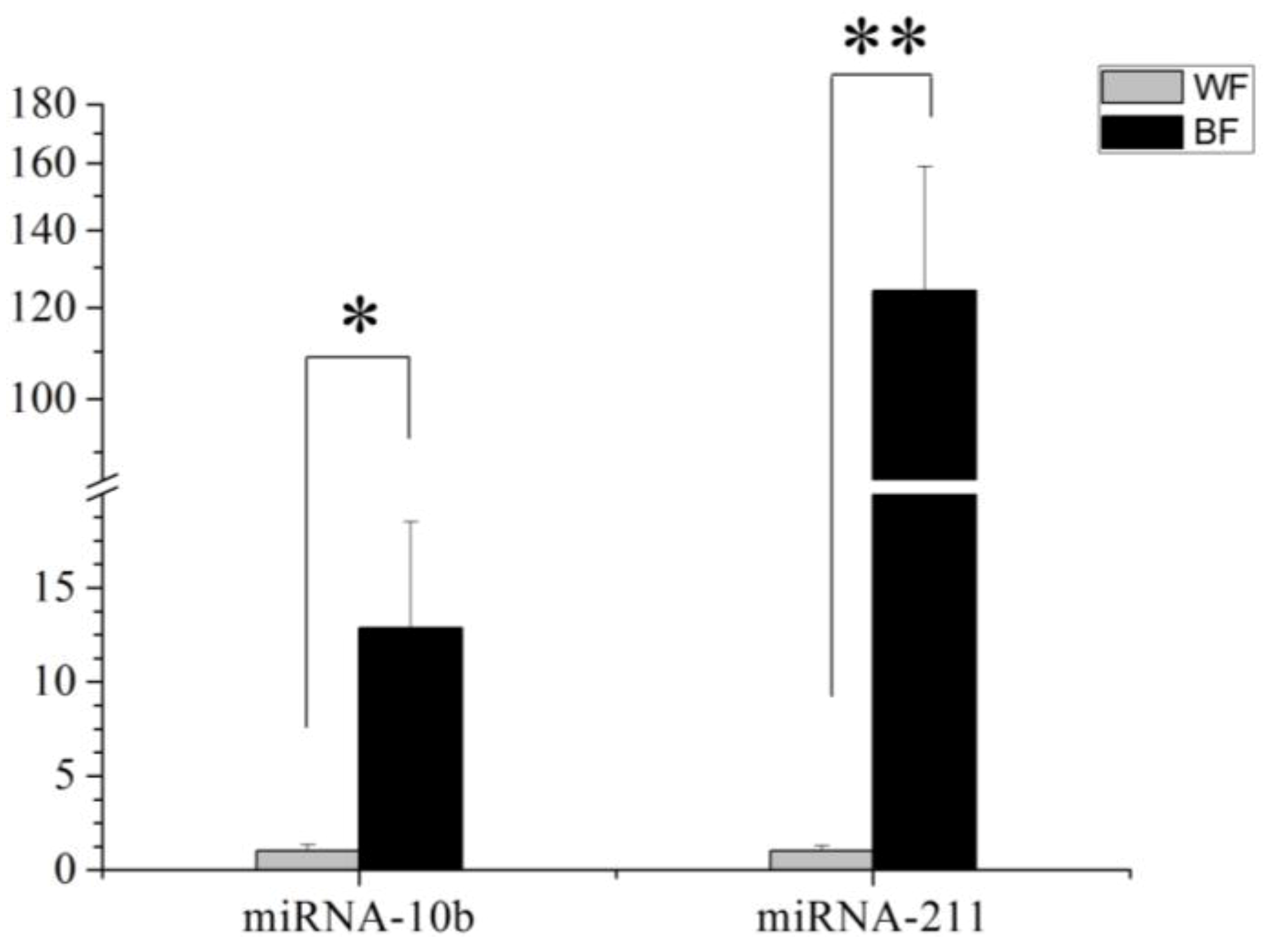
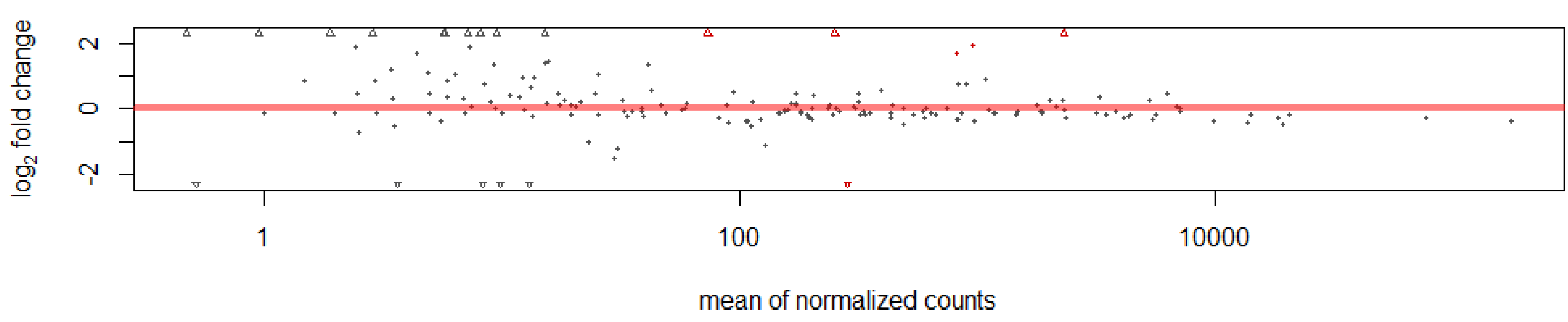
This study identified 205 conserved miRNAs and nine novel miRNAs by RNA-Seq in goat hair follicles. Six differentially expressed miRNAs were predicted in two types of hair follicles tissues. Most miRNAs were upregulated in black follicles, only miR-1307 was downregulated in white hair follicle. However, the expression of the novel miRNA was too low to perform further detection. Two differentially expressed miRNAs, miR-10b and miR-211, were verified by qPCR. Our results thus offer new information on goat hair follicle expressed miRNAs.
We obtained 214 miRNAs in this study. However, only a few miRNAs were differentially expressed between white and black hair follicles. One reason may be that the samples we used were from adult goats. Most of the relevant biological processes such as melanoblast differentiation, migration and maturation are completed during embryonic development. Thus, more differentially expressed miRNAs might be identified during early development than in the adult hair follicles where miRNAs may be involved in fewer biological events. In adult hair follicles, genes and miRNAs are probably involved in more functions related to melanocyte stem cell differentiation into melanocytes or the proliferation of melanocytes. Nevertheless, the identification of miRNAs in skin add a new dimension in the regulatory networks and identified novel players in hair follicle color formation [
23]. Our results may prompt further studies on how miRNAs affect hair follicle development, differentiation and pigmentation.
In our study, the MAPK signaling pathway was the major pathway involving 27 genes, and targeted by five differentially expressed miRNAs including miR10b. The MAPK family proteins, such as p38, ERK and JNK, play critical roles in melanogenesis [
24]. Most studies reported that the p38 MAPK signaling pathway activates MITF, which can up-regulate the expression of melanogenic enzymes [
25]. However, the ERK and/or JNK/SAPK pathways cause down-regulation of melanin synthesis by downregulating MITF [
26]. The detailed mechanism involving p38 MAPK in melanin synthesis is not completely understood.
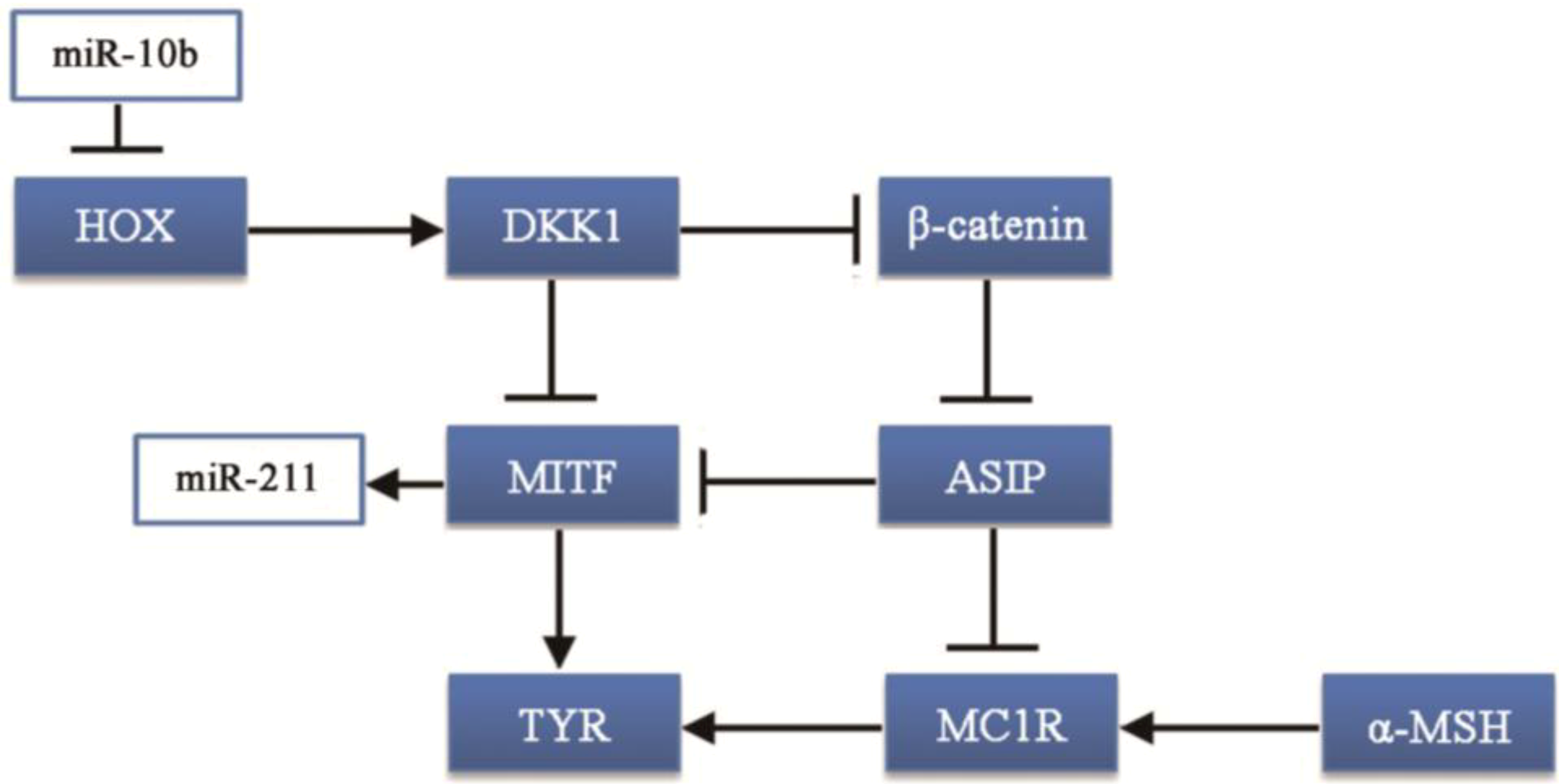
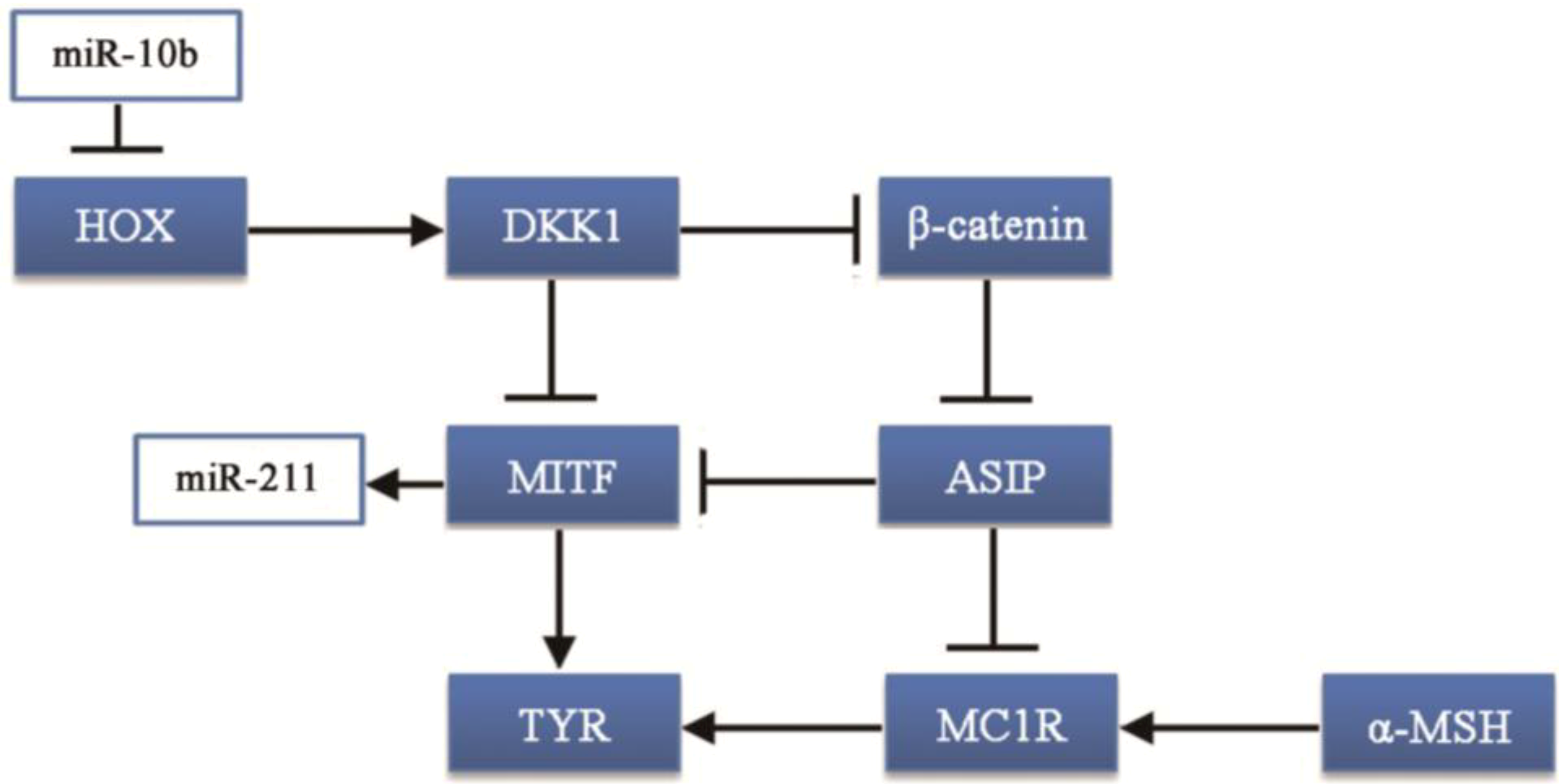
MiR-10b was one of the most abundant and differentially expressed miRNA in black hair follicles, with approximately 64,534.56 reads. MiR-10b takes part in carcinogenesis: miR-10b can suppress the translation of the HOXD10 gene leading to increased RHOC expression and AKT phosphorylation [
27,
28]. Although currently there are no studies on the impact of the HOXD10 gene on melanocytes or the production of melanin, many studies show that genes in the HOX gene family are related to the development of hair follicles, especially HOXC13 [
29,
30]. Thus, the HOX gene family may be associated with coat color formation. Moreover, the HOXA10 gene can upregulate the Dickkopf 1 (DKK1) gene [
31], which regulates skin pigmentation. DKK1 can inhibit the function and proliferation of melanocytes by suppressing β-catenin and microphthalmia-associated transcription factor (MITF) [
32,
33], which can promote the synthesis of melanin.
Our results showed that miR-10b regulates the
DVL3 gene in the Notch pathway. Notch is an evolutionarily conserved local cell-signaling pathway that participates in a variety of cellular processes such as cell fate specification, differentiation, proliferation, apoptosis, adhesion, epithelial-mesenchymal transition, migration and angiogenesis [
34], and the development of hair follicles [
35]. Melanocytes produce melanin and are tightly linked with hair regeneration cycles [
36]. In the hair follicle, melanocyte and melanocytes stem cells numbers are maintained in a dynamic balance. In the cell cycle of the hair follicle, melanocytes proliferate during the hair growth phase and are depleted during the regression phase; the new melanocyte is produced by the differentiation and proliferation of melanocyte stem cells [
37,
38,
39]. The Notch signaling pathway plays a key role in melanoblasts, melanocyte stem cells, keratinocytes and melanocytes [
40]. Many studies reported that lack of Notch signaling can lead to the reduction of the number of melanocytes which can cause the coat color [
41,
42,
43]. Interestingly, HOX, the Notch signaling pathway and the Wnt/β-catenin signaling pathway interact via cross-talk [
44,
45]. Taken together, miR-10b could be an important regulator in goat coat color formation.
MiR-211 had a similar expression pattern to miR-10b. Most studies of this miRNA are on cancer, with few reports on hair follicle development or coat color. One study reported that miR-211 is highly expressed in brown alpaca skin via white alpaca skin expression [
17]. That result is consistent with our study, however, the mechanism has not been investigated. MITF promotes the expression of many genes in pigment cell and regulates melanocyte development by increasing the expression of enzymes, involved in melanin synthesis and melanosome biogenesis [
46]. Studies have shown that miR-211 is induced by the expression of MITF [
47,
48]. This could explain why the expression of miR-211 is higher in black follicles than in white follicles. However, how miR-211 regulates melanocytes or melanin synthesis is not clear.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}