Prediction of Protein S-Nitrosylation Sites Based on Adapted Normal Distribution Bi-Profile Bayes and Chou’s Pseudo Amino Acid Composition

Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
2.1.1. Sequence Analysis of S-Nitrosylation Sites
2.1.2. Performance of the BPB, BRABSB, ANBPB and RANS Prediction Models

{kind=link}
| Sequence Encoding Scheme | W1 | Sn (%) | Sp (%) | Acc (%) | MCC |
|---|---|---|---|---|---|
| BPB + Ecomposition a + Scomposition b | 2 | 65.31 | 65.63 | 65.52 | 0.2933 |
| BRABSB + Ecomposition + Scomposition | 2.5 | 73.09 | 58.16 | 63.14 | 0.2949 |
| ANBPB + Ecomposition + Scomposition | 2 | 67.60 | 64.29 | 65.39 | 0.3014 |
| RANS + Ecomposition + Scomposition | 2.5 | 63.90 | 61.42 | 62.24 | 0.2391 |
2.1.3. Comparison of the Performance of iSNO-ANBPB with Current Computational Approaches
| Dataset | Methods | Sn (%) | Sp (%) | Acc (%) | MCC |
|---|---|---|---|---|---|
| Li training dataset | Li et al. [15] | 42.86 | 70.98 | 61.61 | 0.1381 |
| iSNO-ANBPB | 67.60 | 64.29 | 65.39 | 0.3014 | |
| Xu dataset | GPS-SNO a | 18.88 | 89.63 | 56.07 | 0.1210 |
| GPS-SNO b | 28.04 | 81.98 | 56.39 | 0.1193 | |
| GPS-SNO c | 45.01 | 73.33 | 59.90 | 0.1915 | |
| iSNO-PseAAC | 67.01 | 68.15 | 67.62 | 0.3515 | |
| iSNO-ANBPB | 67.33 | 73.78 | 70.77 | 0.4146 | |
| Li test dataset | SNOSite | 74.42 | 28.10 | 40.24 | 0.0248 |
| iSNO-AAPair | 27.91 | 80.17 | 66.46 | 0.0858 | |
| Li et al. [15] | 51.16 | 69.42 | 64.63 | 0.1886 | |
| iSNO-PseAAC | 58.14 | 63.64 | 62.20 | 0.1940 | |
| iSNO-ANBPB | 74.12 | 59.50 | 63.41 | 0.2984 |
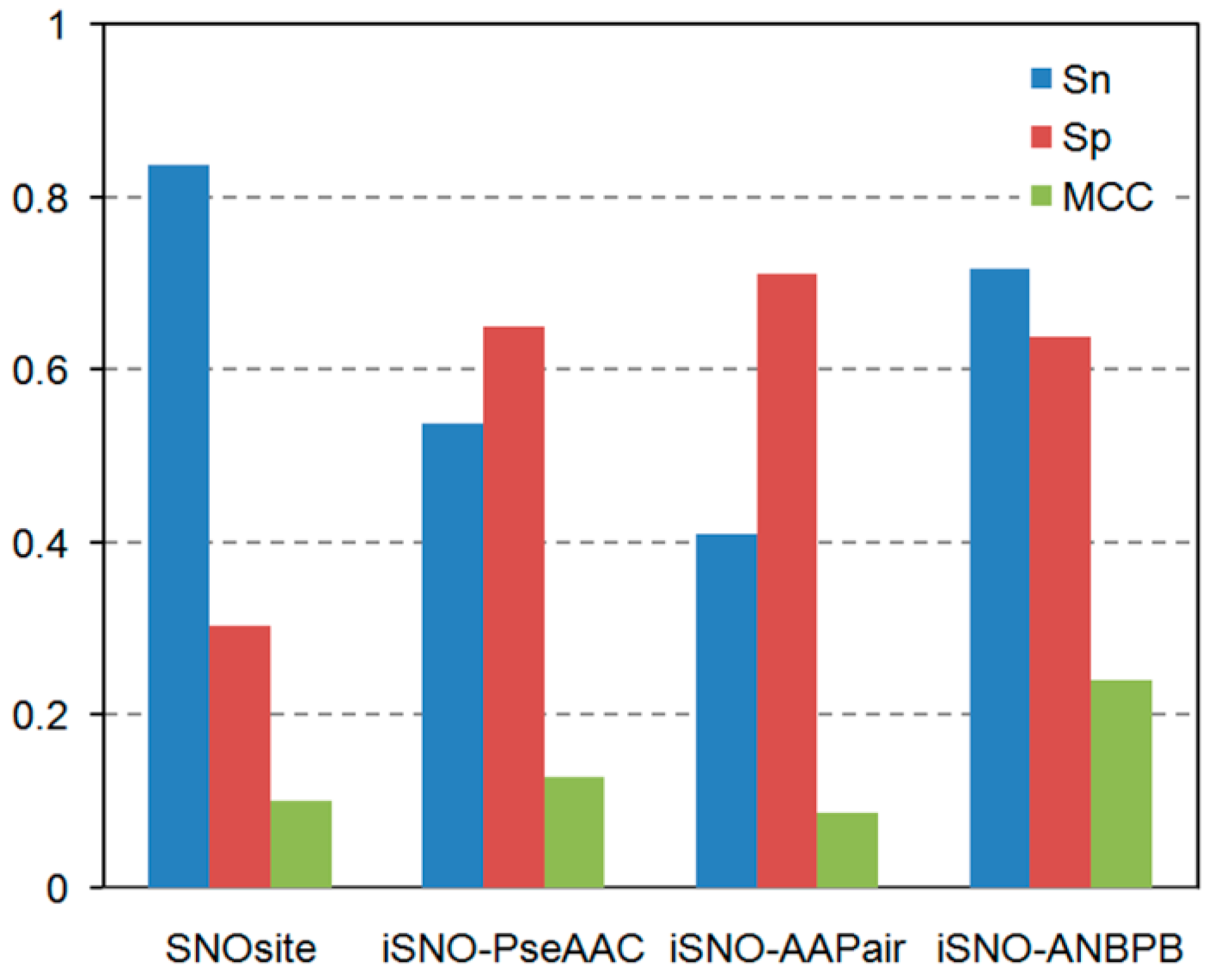
2.2. Discussion
3. Experimental Section
3.1. Datasets
3.2. Adapted Normal Distribution Bi-Profile Bayes Features Extraction (ANBPB)
 has a limiting cumulative distribution function which approximates a normal distribution N(0,1). Here, we modified the way of standard variable normalization to highlight and emphasize the distinction of each amino acid at the same position. We let Vj denote the standard variance of Xij (i = 1, 2, …, 20), i.e., the deviation of frequencies of each at the same jth position. And then we
has a limiting cumulative distribution function which approximates a normal distribution N(0,1). Here, we modified the way of standard variable normalization to highlight and emphasize the distinction of each amino acid at the same position. We let Vj denote the standard variance of Xij (i = 1, 2, …, 20), i.e., the deviation of frequencies of each at the same jth position. And then we  as the new normalization of Xij and deemed it obeys the standard normal distribution. The posterior probability pj (j = 1, 2, …, 2n) was coded by the adapted normal distribution as follows:
as the new normalization of Xij and deemed it obeys the standard normal distribution. The posterior probability pj (j = 1, 2, …, 2n) was coded by the adapted normal distribution as follows:
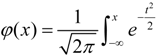 . For more details about this method, please refer to the original paper [24].
. For more details about this method, please refer to the original paper [24].3.3. Pseudo Amino Acid Composition Based on Electrostatic Charge and Secondary Structure
3.4. Feature Space
3.5. Support Vector Machine Implementation and Parameter Selection
3.6. Performance Assessments
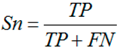
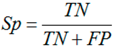
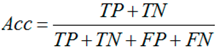
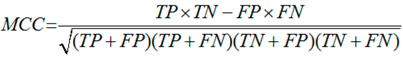
Supplementary Files
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Liu, M.; Talmadge, J.E.; Ding, S.J. Development and application of site-specific proteomic approach for study protein S-nitrosylation. Amino Acids 2012, 42, 1541–1551. [Google Scholar] [CrossRef]
- Tuteja, N.; Tuteja, M.R.; Misra, M.K. Nitric oxide as a unique bioactive signaling messenger in physiology and pathophysiology. J. Biomed. Biotechnol. 2004, 4, 227–237. [Google Scholar] [CrossRef]
- Lane, P.; Hao, G.; Gross, S.S. S-nitrosylation is emerging as a specific and fundamental posttranslational protein modification: Head-to-head comparison with O-phosphorylation. Sci. STKE 2001, 86. [Google Scholar] [CrossRef]
- Forrester, M.T.; Foster, M.W.; Benhar, M.; Stamler, J.S. Detection of protein S-nitrosylation with the biotin-switch technique. Free Radic. Biol. Med. 2009, 46, 119–126. [Google Scholar] [CrossRef]
- Forrester, M.T.; Thompson, J.W.; Foster, M.W.; Nogueira, L.; Moseley, M.A.; Stamler, J.S. Proteomic analysis of S-nitrosylation and denitrosylation by resin-assisted capture. Nat. Biotechnol. 2009, 27, 557–559. [Google Scholar] [CrossRef]
- Foster, M.W.; McMahon, T.J.; Stamler, J.S. S-nitrosylation in health and disease. Trends Mol. Med. 2003, 9, 160–168. [Google Scholar] [CrossRef]
- Lim, K.H.; Ancrile, B.B.; Kashatus, D.F.; Counter, C.M. Tumour maintenance is mediated by eNOS. Nature 2008, 452, 646–649. [Google Scholar] [CrossRef]
- Mannick, J.B.; Schonhoff, C.M. Measurement of protein S-nitrosylation during cell signaling. Methods Enzymol. 2008, 440, 231–242. [Google Scholar] [CrossRef]
- Jaffrey, S.R.; Snyder, S.H. The biotin switch method for the detection of S-nitrosylated proteins. Sci. STKE 2001, 86. [Google Scholar] [CrossRef]
- Huang, B.; Chen, C. An ascorbate-dependent artifact that interferes with the interpretation of the biotin switch assay. Free Radic. Biol. Med 2006, 41, 562–567. [Google Scholar] [CrossRef]
- Hao, G.; Derakhshan, B.; Shi, L.; Campagne, F.; Gross, S.S. SNOSID, a proteomic method for identification of cysteine S-nitrosylation sites in complex protein mixtures. Proc. Natl. Acad. Sci. USA 2006, 103, 1012–1017. [Google Scholar] [CrossRef]
- Xue, Y.; Liu, Z.; Gao, X.; Jin, C.; Wen, L.; Yao, X.B.; Ren, J. GPS-SNO: Computational prediction of protein S-nitrosylation sites with a modified GPS algorithm. PLoS One 2010, 5, e11290. [Google Scholar]
- Lee, T.Y.; Chen, Y.J.; Lu, T.C.; Huang, H.D.; Chen, Y.J. SNOSite: Exploiting maximal dependence decomposition to identify cysteine S-Nitrosylation with substrate site specificity. PLoS One 2011, 6, e21849. [Google Scholar]
- Li, Y.X.; Shao, Y.H.; Jing, L.; Deng, N.Y. An efficient support vector machine approach for identifying protein S-nitrosylation sites. Protein Pept. Lett. 2011, 18, 573–587. [Google Scholar] [CrossRef]
- Li, B.Q.; Hu, L.L.; Niu, S.; Cai, Y.D.; Chou, K.C. Predict and analyze S-nitrosylation modification sites with the mRMR and IFS approaches. J.Proteome Res. 2012, 75, 1654–1665. [Google Scholar]
- Xu, Y.; Ding, J.; Wu, L.Y.; Chou, K.C. iSNO-PseAAC: Predict cysteine S-nitrosylation sites in proteins by incorporating position specific amino acid propensity into pseudo amino acid composition. PLoS One 2013, 8, e55844. [Google Scholar]
- Xu, Y.; Shao, X.J.; Wu, L.Y.; Deng, N.Y.; Chou, K.C. iSNO-AAPair: Incorporating amino acid pairwise coupling into PseAAC for predicting cysteine S-nitrosylation sites in proteins. Peer J. 2013, 1, e171. [Google Scholar] [CrossRef]
- Shao, J.; Xu, D.; Tsai, S.N.; Wang, Y.; Ngai, S.M. Computational identification of protein methylation sites through bi-profile Bayes feature extraction. PLoS One 2009, 4, e4920. [Google Scholar] [CrossRef]
- Song, J.; Tan, H.; Shen, H.; Mahmood, K.; Boyd, S.E.; Webb, G.I.; Akutsu, T.; Whisstock, J.C. Cascleave: Towards more accurate prediction of caspase substrate cleavage sites. Bioinformatics 2010, 26, 752–760. [Google Scholar] [CrossRef]
- Wee, J.K.; Simarmata, D.; Kam, Y.W. SVM-based prediction of linear B-cell epitopes using Bayes feature extraction. BMC Genomics 2010, 11, S21. [Google Scholar] [CrossRef]
- Jia, C.Z.; Liu, T.; Chang, A.K.; Zhai, Y. Prediction of mitochondrial proteins of malaria parasite using bi-profile Bayes feature extraction. Biochimie 2011, 93, 778–782. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Q.; Sun, M.; Guo, D. High accuracy prediction of bacterial type III secreted effectors based on position-specific amino acid composition profiles. Bioinformatics 2011, 27, 777–784. [Google Scholar] [CrossRef]
- Shao, J.; Xu, D.; Hu, L.; Kwan, Y.W.; Wang, Y.; Kong, X.; Ngai, S.M. Systematic analysis of human lysine acetylation proteins and accurate prediction of human lysine acetylation through bi-relative adapted binomial score Bayes feature representation. Mol. Biosyst. 2012, 8, 2964–2973. [Google Scholar] [CrossRef]
- Jia, C.Z.; Liu, T.; Wang, Z.P. O-GlcNAcPRED: A sensitive predictor to capture protein O-GlcNAcylation sites. Mol. BioSyst. 2013, 9, 2909–2913. [Google Scholar] [CrossRef]
- Jia, C.Z.; Zhang, Y.S.; Wang, Z.P. SulfoTyrP: A high accuracy predictor of protein sulfotyrosine sites. Match Commun. Math. Comput. Chem. 2014, 71, 227–240. [Google Scholar]
- Chou, K.C. Some remarks on protein attribute prediction and pseudo amino acid composition (50th Anniversary Year Review). J. Theor. Biol. 2011, 273, 236–247. [Google Scholar] [CrossRef]
- Chen, W.; Feng, P.M.; Lin, H.; Chou, K.C. iRSpot-PseDNC: Identify recombination spots with pseudo dinucleotide composition. Nucleic Acids Res 2013, 41, e68. [Google Scholar] [CrossRef]
- Feng, P.M.; Chen, W.; Lin, H.; Chou, K.C. iHSP-PseRAAAC: Identifying the heat shock protein families using pseudo reduced amino acid alphabet composition. Anal. Biochem. 2013, 442, 118–125. [Google Scholar] [CrossRef]
- Xiao, X.; Min, J.L.; Wang, P.; Chou, K.C. iCDI-PseFpt: Identify the channel-drug interaction in cellular networking with PseAAC and molecular fingerprints. J. Theor. Biol. 2013, 337, 71–79. [Google Scholar] [CrossRef]
- Lin, S.X.; Lapointe, J. Theoretical and experimental biology in one. J. Biomed. Sci. Eng. 2013, 6, 435–442. [Google Scholar] [CrossRef]
- Chou, K.C.; Shen, H.B. Recent progress in protein subcellular location prediction. Anal. Biochem. 2007, 370, 1–16. [Google Scholar] [CrossRef]
- BLASTclust. Available online: http://toolkit.tuebingen.mpg.de/blastclust (accessed on 1 December 2010).
- Chou, K.C. Prediction of protein cellular attributes using pseudo amino acid composition. Proteins: Struct. Funct. Genet 2001, 43, 246–255. [Google Scholar] [CrossRef]
- Chou, K.C. Using amphiphilic pseudo amino acid composition to predict enzyme subfamily classes. Bioinformatics 2005, 21, 10–19. [Google Scholar] [CrossRef]
- Nanni, L.; Lumini, A. Genetic programming for creating Chou’s pseudo amino acid based features for submitochondria localization. Amino Acids 2008, 34, 653–660. [Google Scholar] [CrossRef]
- Sahu, S.S.; Panda, G. A novel feature representation method based on Chou’s pseudo amino acid composition for protein structural class prediction. Comput. Biol. Chem. 2010, 34, 320–327. [Google Scholar] [CrossRef]
- Nanni, L.; Lumini, A; Gupta, D.; Garg, A. Identifying bacterial virulent proteins by fusing a set of classifiers based on variants of Chou’s pseudo amino acid composition and on evolutionary information. IEEE/ACM Trans. Comput. Biol. Bioinform 2012, 9, 467–475. [Google Scholar] [CrossRef]
- Mohammad Beigi, M.; Behjati, M.; Mohabatkar, H. Prediction of metalloproteinase family based on the concept of Chou’s pseudo amino acid composition using a machine learning approach. J. Struct. Funct. Genomics 2011, 12, 191–197. [Google Scholar] [CrossRef]
- Mohabatkar, H.; Mohammad Beigi, M.; Esmaeili, A. Prediction of GABAA receptor proteins using the concept of Chou’s pseudo-amino acid composition and support vector machine. J. Theor. Biol. 2011, 281, 18–23. [Google Scholar]
- Zou, D.; He, Z.; He, J.; Xia, Y. Supersecondary structure prediction using Chou’s pseudo amino acid composition. J. Comput. Chem. 2011, 32, 271–278. [Google Scholar] [CrossRef]
- Mohabatkar, H. Prediction of cyclin proteins using Chou’s pseudo amino acid composition. Protein Peptide Lett. 2010, 17, 1207–1214. [Google Scholar] [CrossRef]
- Georgiou, D.N.; Karakasidis, T.E.; Nieto, J.J.; Torres, A. Use of fuzzy clustering technique and matrices to classify amino acids and its impact to Chou’s pseudo amino acid composition. J. Theor. Biol. 2009, 257, 17–26. [Google Scholar] [CrossRef]
- Esmaeili, M.; Mohabatkar, H.; Mohsenzadeh, S. Using the concept of Chou’s pseudo amino acid composition for risk type prediction of human papillomaviruses. J. Theor. Biol. 2010, 263, 203–209. [Google Scholar] [CrossRef]
- Zia Ur, R.; Khan, A. Identifying GPCRs and their types with Chou’s pseudo amino acid composition: An approach from multi-scale energy representation and position specific scoring matrix. Protein Peptide Lett. 2012, 19, 890–903. [Google Scholar] [CrossRef]
- Zhang, S.W.; Zhang, Y.L.; Yang, H.F.; Zhao, C.H.; Pan, Q. Using the concept of Chou’s pseudo amino acid composition to predict protein subcellular localization: An approach by incorporating evolutionary information and von Neumann entropies. Amino Acids 2008, 34, 565–572. [Google Scholar] [CrossRef]
- Hayat, M.; Khan, A. Discriminating outer membrane proteins with fuzzy K-nearest neighbor algorithms based on the general form of Chou’s PseAAC. Protein Peptide Lett. 2012, 19, 411–421. [Google Scholar] [CrossRef]
- Du, P.; Wang, X.; Xu, C.; Gao, Y. PseAAC-Builder: A cross-platform stand-alone program for generating various special Chou’s pseudo-amino acid compositions. Anal. Biochem. 2012, 425, 117–119. [Google Scholar]
- Cao, D.S.; Xu, Q.S.; Liang, Y.Z. Propy: A tool to generate various modes of Chou’s PseAAC. Bioinformatics 2013, 29, 960–962. [Google Scholar]
- Shen, H.B.; Chou, K.C. PseAAC: a flexible web-server for generating various kinds of protein pseudo amino acid composition. Anal. Biochems. 2008, 373, 386–388. [Google Scholar] [CrossRef]
- Marino, S.M.; Gladyshev, V.N. Structural analysis of cysteine S-nitrosylation: A modified acid-based motif and the emerging role of trans-nitrosylation. J. Mol. Biol. 2009, 395, 844–859. [Google Scholar] [CrossRef]
- Song, X.; Wang, M.; Chen, Y.P.; Wang, H.; Han, P.; Sun, H. Prediction of pre-miRNA with multiple stem-loops using pruning algorithm. Comput. Biol. Med. 2013, 43, 409–416. [Google Scholar] [CrossRef]
- Kazemian, H.B.; White, K.; Brown, D.P. Applications of evolutionary SVM to prediction of membrane alpha-helices. Expert Syst. Appl. 2013, 40, 3412–3420. [Google Scholar] [CrossRef]
- Chang, C.C.; Lin, C.J. LIBSVM: A Library for Support Vector Machines. Available online: http://www.csie.ntu.edu.tw/~cjlin/libsvm (accessed on 1 April 2014).
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jia, C.; Lin, X.; Wang, Z. Prediction of Protein S-Nitrosylation Sites Based on Adapted Normal Distribution Bi-Profile Bayes and Chou’s Pseudo Amino Acid Composition. Int. J. Mol. Sci. 2014, 15, 10410-10423. https://doi.org/10.3390/ijms150610410
Jia C, Lin X, Wang Z. Prediction of Protein S-Nitrosylation Sites Based on Adapted Normal Distribution Bi-Profile Bayes and Chou’s Pseudo Amino Acid Composition. International Journal of Molecular Sciences. 2014; 15(6):10410-10423. https://doi.org/10.3390/ijms150610410
Chicago/Turabian StyleJia, Cangzhi, Xin Lin, and Zhiping Wang. 2014. "Prediction of Protein S-Nitrosylation Sites Based on Adapted Normal Distribution Bi-Profile Bayes and Chou’s Pseudo Amino Acid Composition" International Journal of Molecular Sciences 15, no. 6: 10410-10423. https://doi.org/10.3390/ijms150610410