Obesity and Its Metabolic Complications: The Role of Adipokines and the Relationship between Obesity, Inflammation, Insulin Resistance, Dyslipidemia and Nonalcoholic Fatty Liver Disease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
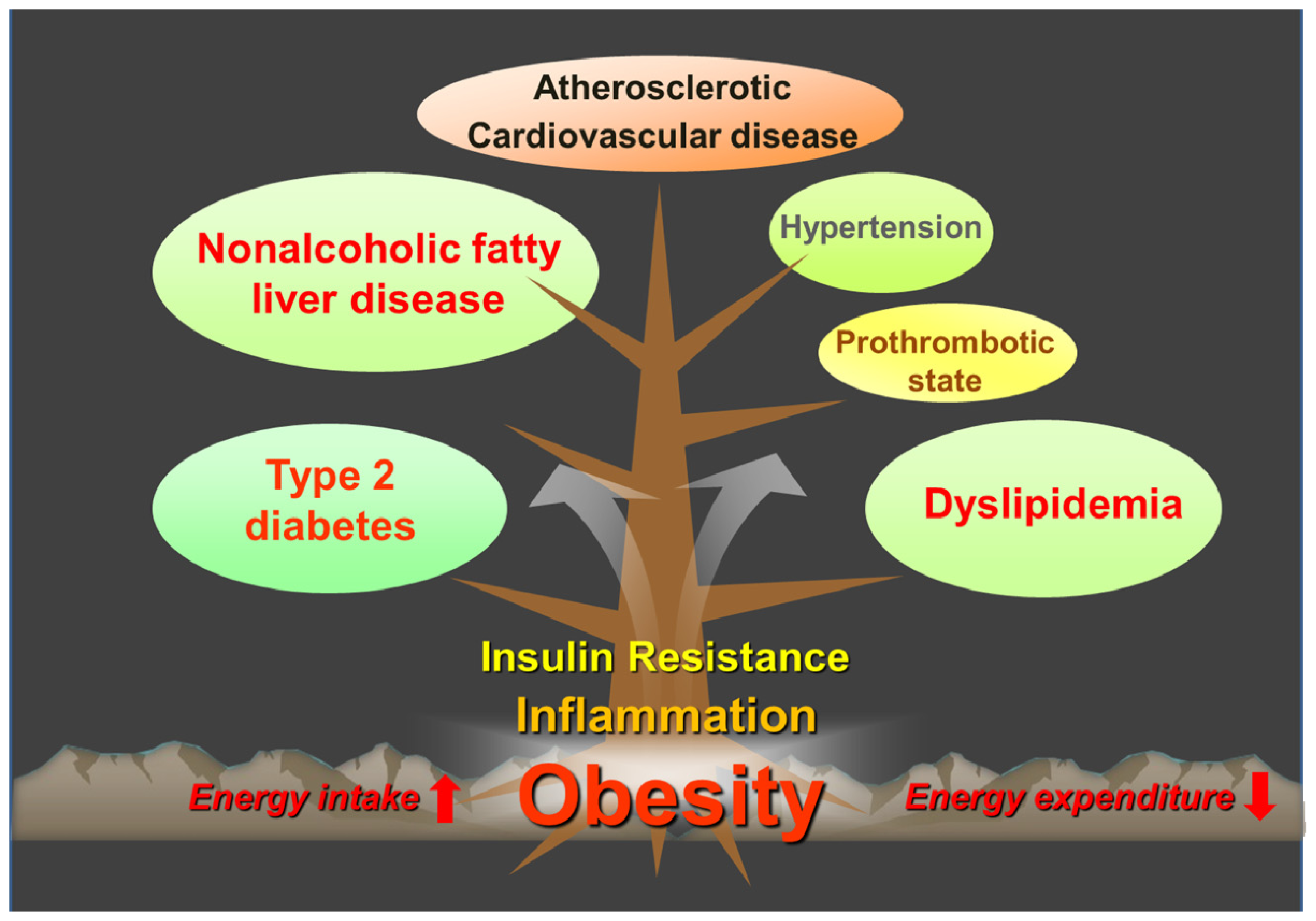
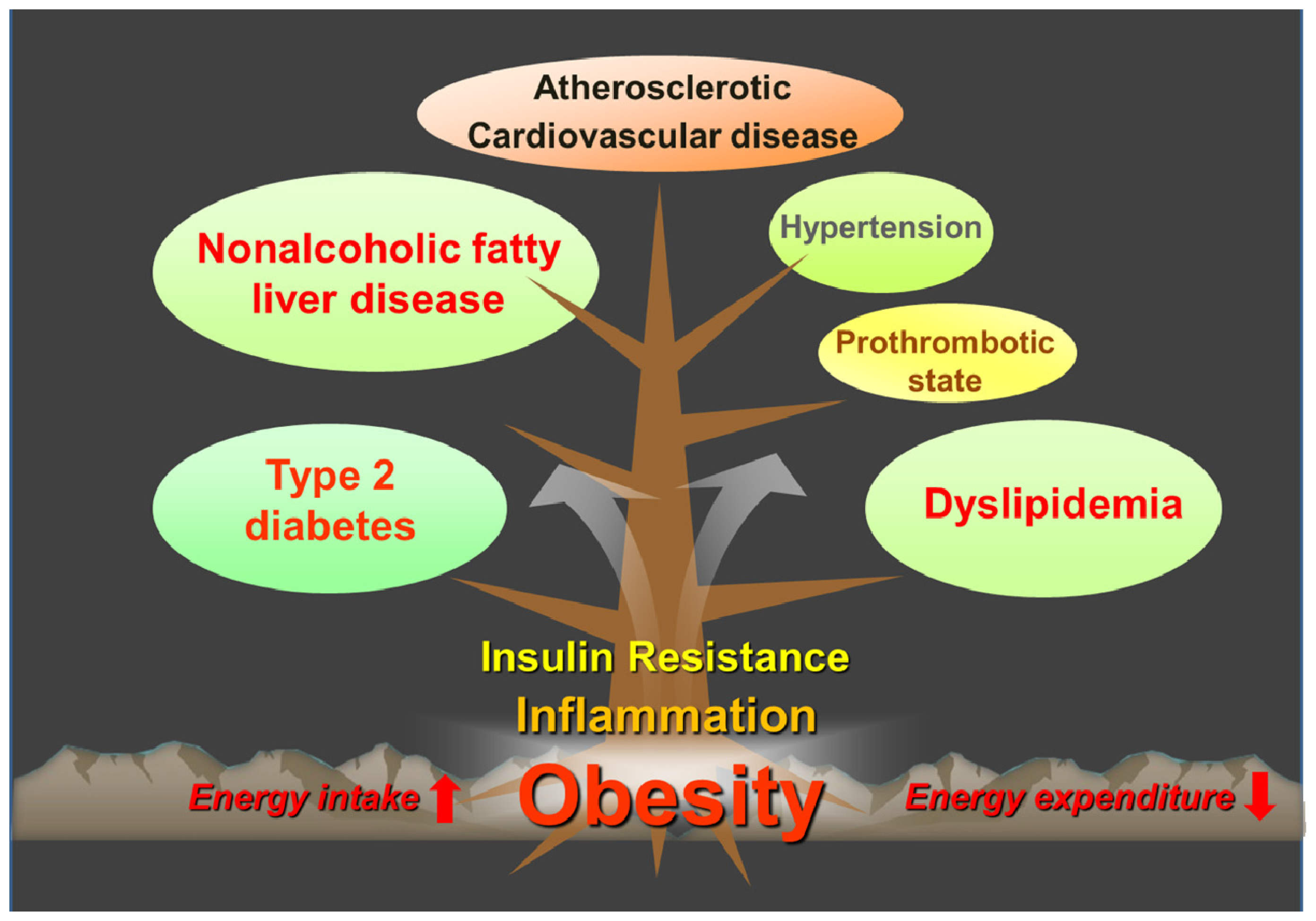
:1. Introduction
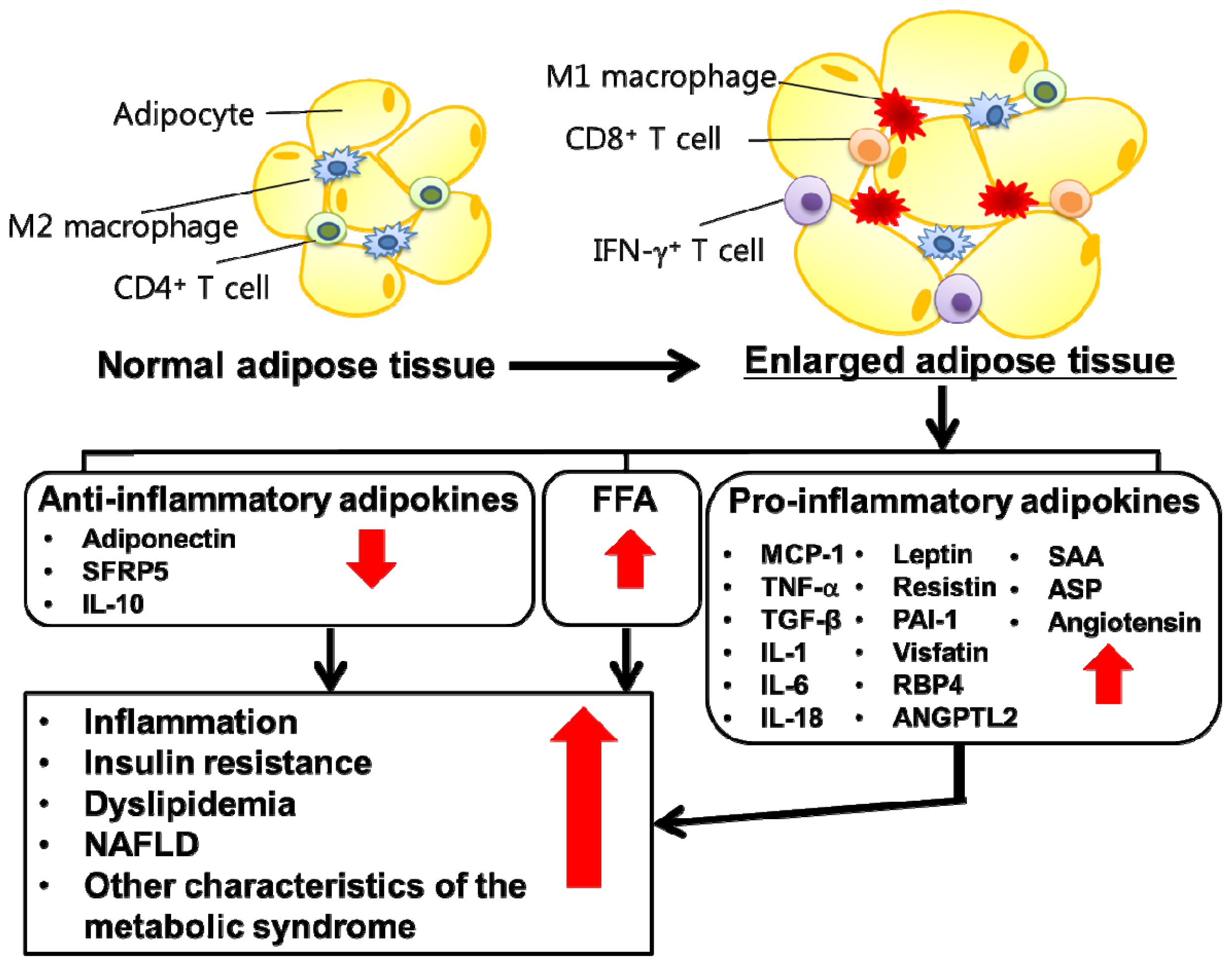
2. Function of Adipose Tissue
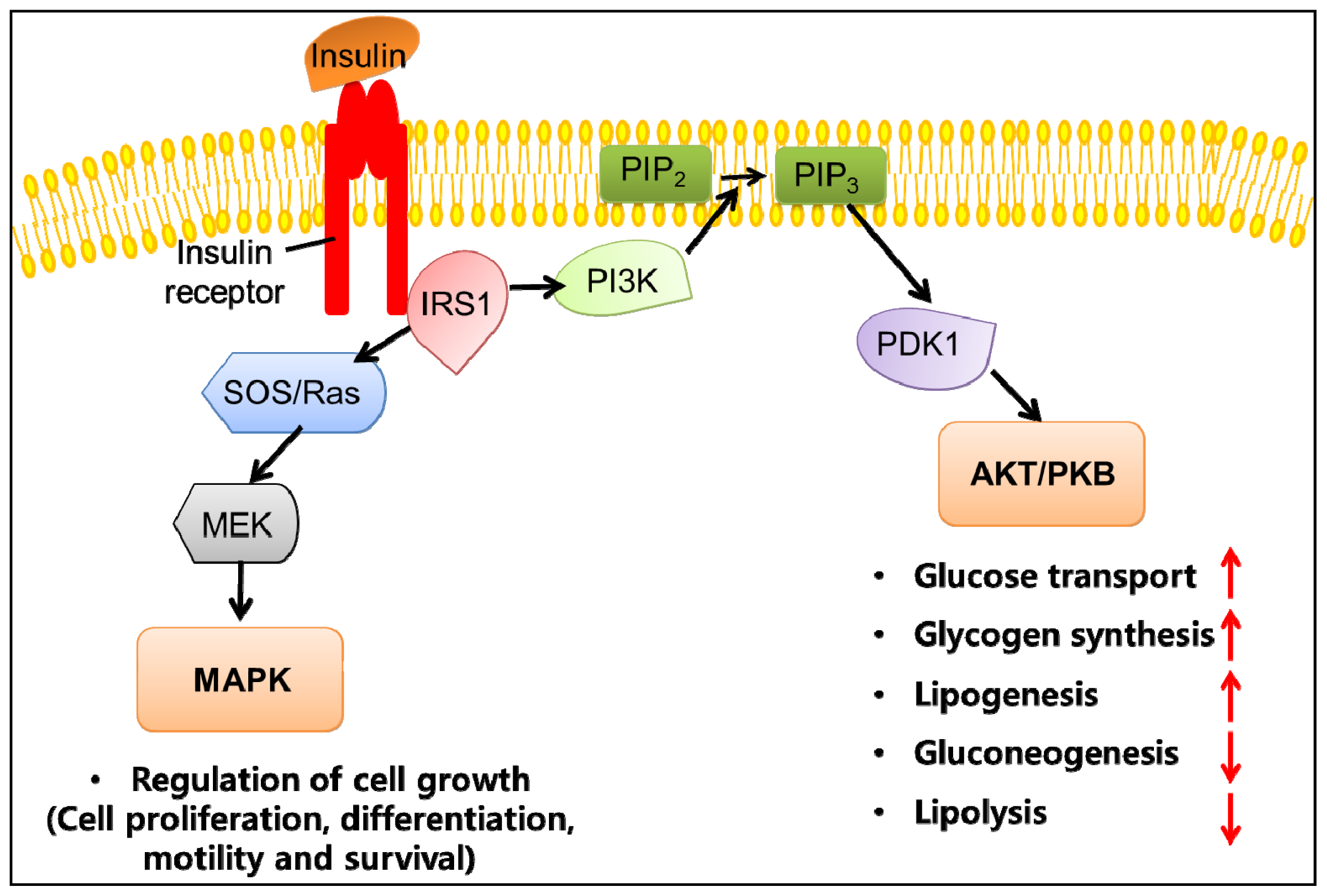
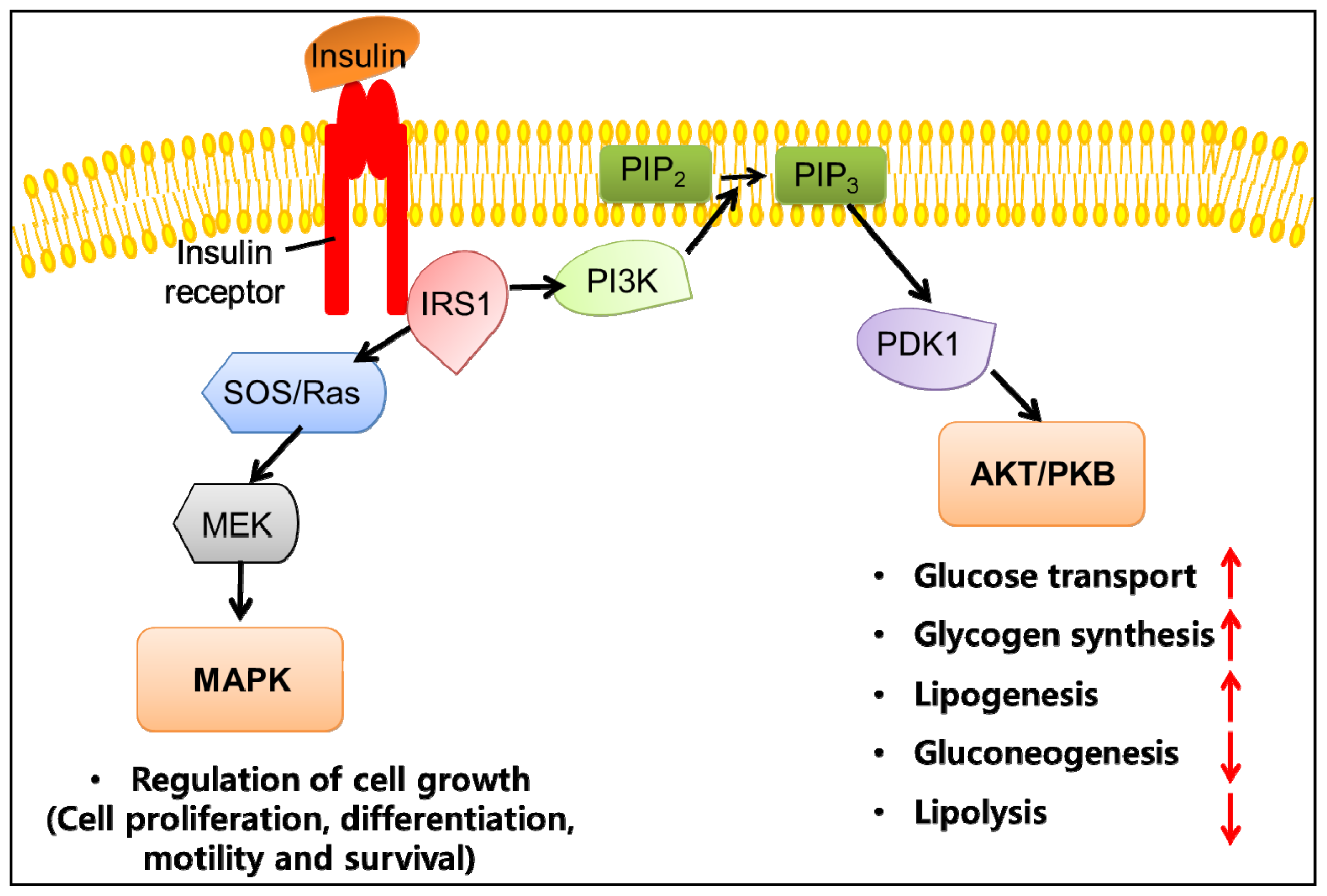
3. Obesity and Insulin Resistance
4. Role of Adipose Tissue-Produced Adipokines in Insulin Resistance
4.1. CCL2/MCP-1 and Other Chemokines
4.2. TNF-α
4.3. IL-6 and IL-18
4.4. Leptin
4.5. Resistin
4.6. PAI-1
4.7. Visfatin
4.8. RBP4
4.9. ANGPTL2
4.10. Adiponectin
4.11. SFRP5
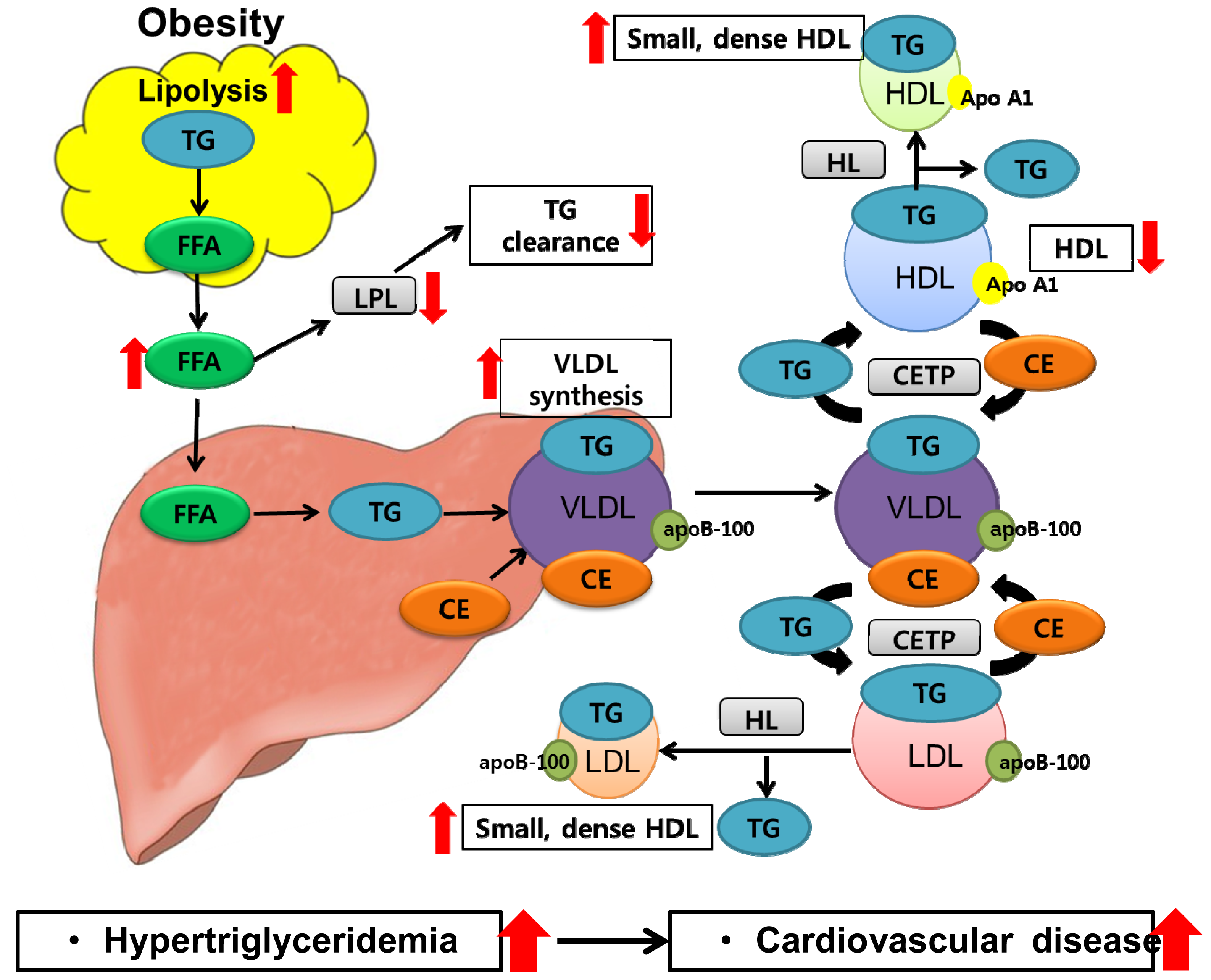
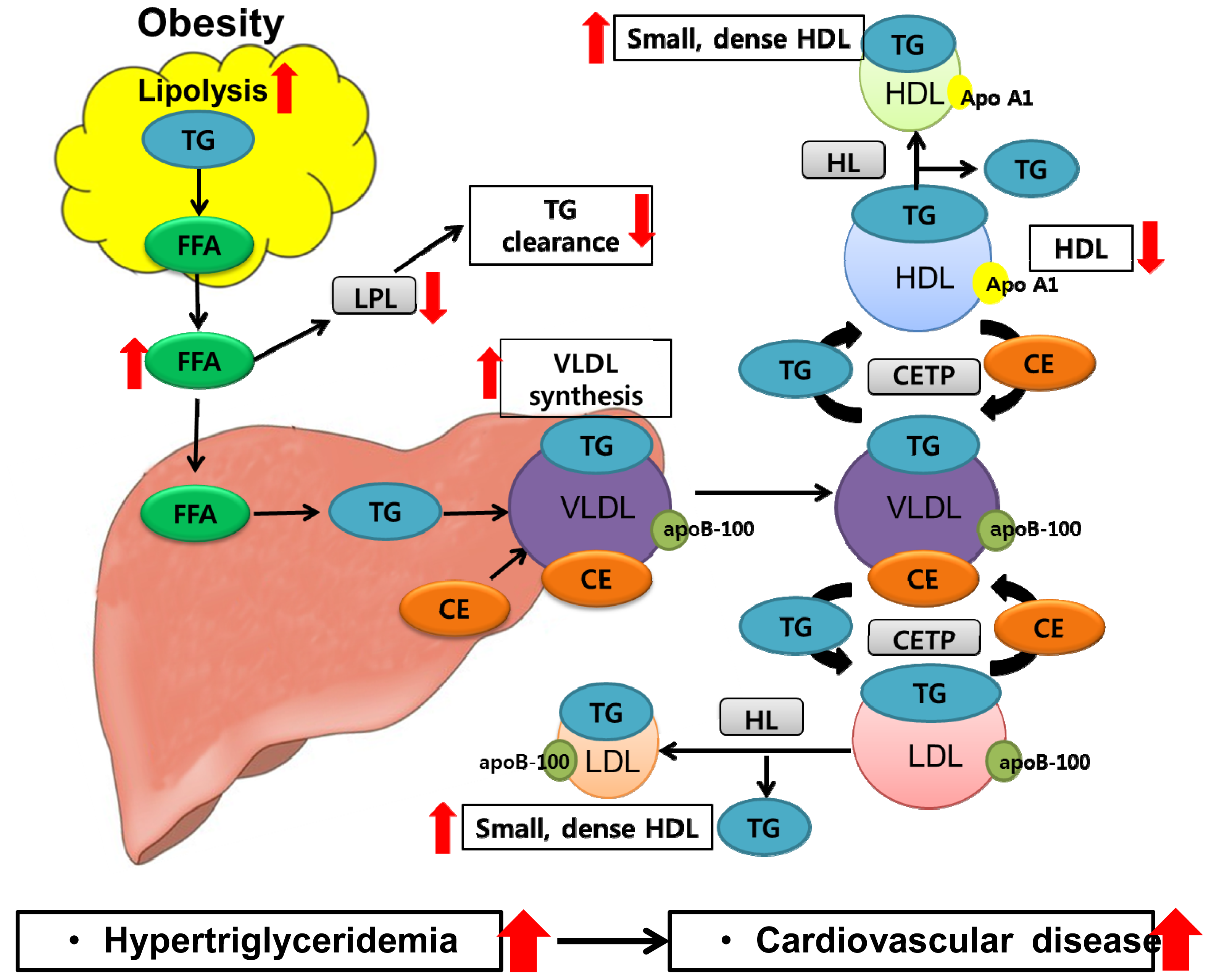
5. Obesity and Dyslipidemia
6. Role of Adipose Tissue-Produced Adipokines in Dyslipidemia
6.1. Cytokines
6.2. SAA
6.3. Adiponectin
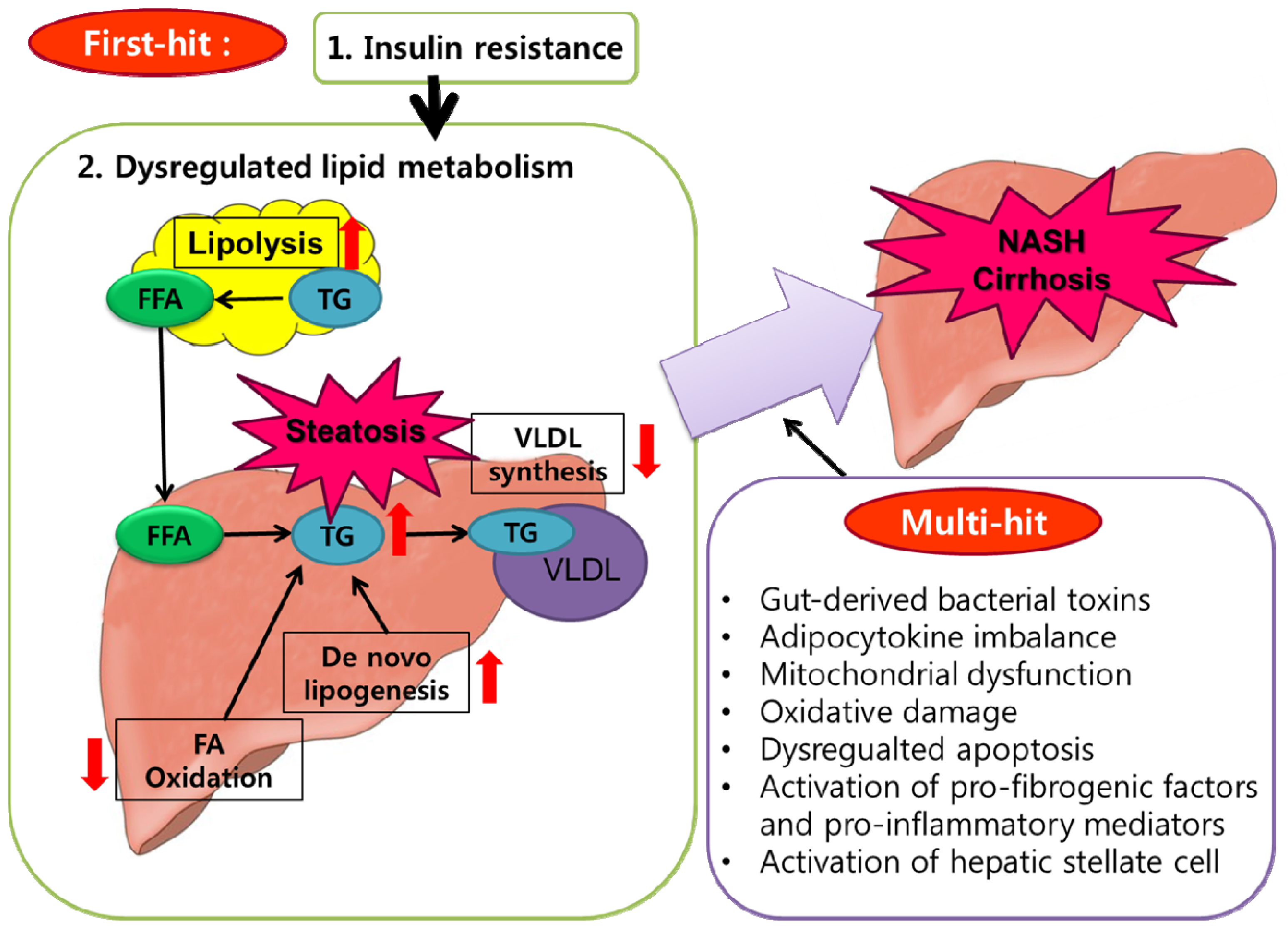
7. Obesity and NAFLD
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Obesity and Overweight. Available online: http://www.who.int/mediacentre/factsheets/fs311/en/ (accessed on 5 May 2013).
- Wang, Y.C.; McPherson, K.; Marsh, T.; Gortmaker, S.L.; Brown, M. Health and economic burden of the projected obesity trends in the USA and the UK. Lancet 2011, 378, 815–825. [Google Scholar]
- López-Velázquez, J.A.; Silva-Vidal, K.V.; Ponciano-Rodríguez, G.; Chávez-Tapia, N.C.; Arrese, M.; Uribe, M.; Méndez-Sánchez, N. The prevalence of nonalcoholic fatty liver disease in the Americas. Ann. Hepatol 2014, 13, 166–178. [Google Scholar]
- Carr, D.B.; Utzschneider, K.M.; Hull, R.L.; Kodama, K.; Retzlaff, B.M.; Brunzell, J.D.; Shofer, J.B.; Fish, B.E.; Knopp, R.H.; Kahn, S.E. Intra-abdominal fat is a major determinant of the National Cholesterol Education Program Adult Treatment Panel III criteria for the metabolic syndrome. Diabetes 2004, 53, 2087–2094. [Google Scholar]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Investig 2003, 112, 1821–1830. [Google Scholar]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar]
- Lumeng, C.N.; Saltiel, A.R. Inflammatory links between obesity and metabolic disease. J. Clin. Investig 2011, 121, 2111–2117. [Google Scholar]
- Kershaw, E.E.; Flier, J.S. Adipose tissue as an endocrine organ. J. Clin. Endocrinol. Metab 2004, 89, 2548–2556. [Google Scholar]
- Hauner, H. Secretory factors from human adipose tissue and their functional role. Proc. Nutr. Soc 2005, 64, 163–169. [Google Scholar]
- Halberg, N.; Wernstedt-Asterholm, I.; Scherer, P.E. The adipocyte as an endocrine cell. Endocrinol. Metab. Clin. N. Am 2008, 37, 753–768. [Google Scholar]
- Perseghin, G.; Ghosh, S.; Gerow, K.; Shulman, G.I. Metabolic defects in lean nondiabetic offspring of NIDDM parents: A cross-sectional study. Diabetes 1997, 46, 1001–1009. [Google Scholar]
- Boden, G. Obesity and free fatty acids. Endocrinol. Metab. Clin. N. Am 2008, 37, 635–646. [Google Scholar]
- Horowitz, J.F.; Coppack, S.W.; Paramore, D.; Cryer, P.E.; Zhao, G.; Klein, S. Effect of short-term fasting on lipid kinetics in lean and obese women. Am. J. Physiol 1999, 276, E278–E284. [Google Scholar]
- Large, V.; Reynisdottir, S.; Langin, D.; Fredby, K.; Klannemark, M.; Holm, C.; Arner, P. Decreased expression and function of adipocyte hormone-sensitive lipase in subcutaneous fat cells of obese subjects. J. Lipid Res 1999, 40, 2059–2066. [Google Scholar]
- Hellström, L.; Reynisdottir, S. Influence of heredity for obesity on adipocyte lipolysis in lean and obese subjects. Int. J. Obes. Relat. Metab. Disord 2000, 24, 340–344. [Google Scholar]
- McQuaid, S.E.; Hodson, L.; Neville, M.J.; Dennis, A.L.; Cheeseman, J.; Humphreys, S.M.; Ruge, T.; Gilbert, M.; Fielding, B.A.; Frayn, K.N.; et al. Downregulation of adipose tissue fatty acid trafficking in obesity: A driver for ectopic fat deposition? Diabetes 2011, 60, 47–55. [Google Scholar]
- Langin, D.; Dicker, A.; Tavernier, G.; Hoffstedt, J.; Mairal, A.; Rydén, M.; Arner, E.; Sicard, A.; Jenkins, C.M.; Viguerie, N.; et al. Adipocyte lipases and defect of lipolysis in human obesity. Diabetes 2005, 54, 3190–3197. [Google Scholar]
- Jocken, J.W.; Langin, D.; Smit, E.; Saris, W.H.; Valle, C.; Hul, G.B.; Holm, C.; Arner, P.; Blaak, E.E. Adipose triglyceride lipase and hormone-sensitive lipase protein expression is decreased in the obese insulin-resistant state. J. Clin. Endocrinol. Metab 2007, 92, 2292–2299. [Google Scholar]
- Karpe, F.; Dickmann, J.R.; Frayn, K.N. Fatty acids, obesity, and insulin resistance: Time for a reevaluation. Diabetes 2011, 60, 2441–2449. [Google Scholar]
- Hausman, D.B.; DiGirolamo, M.; Bartness, T.J.; Hausman, G.J.; Martin, R.J. The biology of white adipocyte proliferation. Obes. Rev 2001, 2, 239–254. [Google Scholar]
- Spalding, K.L.; Arner, E.; Westermark, P.O.; Bernard, S.; Buchholz, B.A.; Bergmann, O.; Blomqvist, L.; Hoffstedt, J.; Naslund, E.; Britton, T.; et al. Dynamics of fat cell turnover in humans. Nature 2008, 453, 783–787. [Google Scholar]
- Tchoukalova, Y.D.; Votruba, S.B.; Tchkonia, T.; Giorgadze, N.; Kirkland, J.L.; Jensen, M.D. Regional differences in cellular mechanisms of adipose tissue gain with overfeeding. Proc. Natl. Acad. Sci. USA 2010, 107, 18226–18231. [Google Scholar]
- Wang, Y.; Rimm, E.B.; Stampfer, M.J.; Willett, W.C.; Hu, F.B. Comparison of abdominal adiposity and overall obesity in predicting risk of type 2 diabetes among men. Am. J. Clin. Nutr 2005, 81, 555–563. [Google Scholar]
- Kim, J.Y.; van de Wall, E.; Laplante, M.; Azzara, A.; Trujillo, M.E.; Hofmann, S.M.; Schraw, T.; Durand, J.L.; Li, H.; Li, G.; et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J. Clin. Investig 2007, 117, 2621–2637. [Google Scholar]
- Weyer, C.; Foley, J.E.; Bogardus, C.; Tataranni, P.A.; Pratley, R.E. Enlarged subcutaneous abdominal adipocyte size, but not obesity itself, predicts type II diabetes independent of insulin resistance. Diabetologia 2000, 43, 1498–1506. [Google Scholar]
- Boden, G. Role of fatty acids in the pathogenesis of insulin resistance and NIDDM. Diabetes 1997, 46, 3–10. [Google Scholar]
- Kelley, D.E.; Mokan, M.; Simoneau, J.A.; Mandarino, L.J. Interaction between glucose and free fatty acid metabolism in human skeletal muscle. J. Clin. Investig 1993, 92, 91–98. [Google Scholar]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig 2006, 116, 3015–3025. [Google Scholar]
- Suganami, T.; Nishida, J.; Ogawa, Y. A paracrine loop between adipocytes and macrophages aggravates inflammatory changes: Role of free fatty acids and tumor necrosis factor alpha. Arterioscler. Thromb. Vasc. Biol 2005, 25, 2062–2068. [Google Scholar]
- Byrne, C.D.; Maison, P.; Halsall, D.; Martensz, N.; Hales, C.N.; Wareham, N.J. Cross-sectional but not longitudinal associations between non-esterified fatty acid levels and glucose intolerance and other features of the metabolic syndrome. Diabet. Med 1999, 16, 1007–1015. [Google Scholar]
- Cho, S.J.; Jung, U.J.; Choi, M.S. Differential effects of low-dose resveratrol on adiposity and hepatic steatosis in diet-induced obese mice. Br. J. Nutr 2012, 108, 2166–2175. [Google Scholar]
- Charles, M.A.; Fontbonne, A.; Thibult, N.; Claude, J.R.; Warnet, J.M.; Rosselin, G.; Ducimetière, P.; Eschwège, E. High plasma nonesterified fatty acids are predictive of cancer mortality but not of coronary heart disease mortality: Results from the paris prospective study. Am. J. Epidemiol 2001, 153, 292–298. [Google Scholar]
- Reeds, D.N.; Stuart, C.A.; Perez, O.; Klein, S. Adipose tissue, hepatic, and skeletal muscle insulin sensitivity in extremely obese subjects with acanthosis nigricans. Metabolism 2006, 55, 1658–1663. [Google Scholar]
- Jernas, M.; Palming, J.; Sjoholm, K.; Jennische, E.; Svensson, P.A.; Gabrielsson, B.G.; Levin, M.; Sjogren, A.; Rudemo, M.; Lystig, T.C.; et al. Separation of human adipocytes by size: Hypertrophic fat cells display distinct gene expression. FASEB J 2006, 20, 1540–1542. [Google Scholar]
- Skurk, T.; Alberti-Huber, C.; Herder, C.; Hauner, H. Relationship between adipocyte size and adipokine expression and secretion. J. Clin. Endocrinol. Metab 2007, 92, 1023–1033. [Google Scholar]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-α: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar]
- Amrani, A.; Jafarian-Tehrani, M.; Mormède, P.; Durant, S.; Pleau, J.M.; Haour, F.; Dardenne, M.; Homo-Delarche, F. Interleukin-1 effect on glycemia in the non-obese diabetic mouse at the pre-diabetic stage. J. Endocrinol 1996, 148, 139–148. [Google Scholar]
- Sartipy, P.; Loskutoff, D.J. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2003, 100, 7265–7270. [Google Scholar]
- Rotter, V.; Nagaev, I.; Smith, U. Interleukin-6 (IL-6) induces insulin resistance in 3T3-L1 adipocytes and is, like IL-8 and tumor necrosis factor-alpha, overexpressed in human fat cells from insulin-resistant subjects. J. Biol. Chem 2003, 278, 45777–45784. [Google Scholar]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig 2003, 112, 1796–1808. [Google Scholar]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig 2007, 117, 175–184. [Google Scholar]
- Jiao, P.; Chen, Q.; Shah, S.; Du, J.; Tao, B.; Tzameli, I.; Yan, W.; Xu, H. Obesity-related upregulation of monocyte chemotactic factors in adipocytes: Involvement of nuclear factor-kappaB and c-Jun NH2-terminal kinase pathways. Diabetes 2009, 58, 104–115. [Google Scholar]
- Lee, Y.H.; Thacker, R.I.; Hall, B.E.; Kong, R.; Granneman, J.G. Exploring the activated adipogenic niche: Interactions of macrophages and adipocyte progenitors. Cell Cycle 2014, 13, 184–190. [Google Scholar]
- Nomiyama, T.; Perez-Tilve, D.; Ogawa, D.; Gizard, F.; Zhao, Y.; Heywood, E.B.; Jones, K.L.; Kawamori, R.; Cassis, L.A.; Tschöp, M.H.; et al. Osteopontin mediates obesity-induced adipose tissue macrophage infiltration and insulin resistance in mice. J. Clin. Investig 2007, 117, 2877–2888. [Google Scholar]
- Weisberg, S.P.; Hunter, D.; Huber, R.; Lemieux, J.; Slaymaker, S.; Vaddi, K.; Charo, I.; Leibel, R.L.; Ferrante, A.W., Jr. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J. Clin. Investig 2006, 116, 115–124. [Google Scholar]
- Nara, N.; Nakayama, Y.; Okamoto, S.; Tamura, H.; Kiyono, M.; Muraoka, M.; Tanaka, K.; Taya, C.; Shitara, H.; Ishii, R.; et al. Disruption of CXC motif chemokine ligand-14 in mice ameliorates obesity-induced insulin resistance. J. Biol. Chem 2007, 282, 30794–30803. [Google Scholar]
- Feng, B.; Jiao, P.; Nie, Y.; Kim, T.; Jun, D.; van Rooijen, N.; Yang, Z.; Xu, H. Clodronate liposomes improve metabolic profile and reduce visceral adipose macrophage content in diet-induced obese mice. PLoS One 2011, 6, e24358. [Google Scholar]
- Clément, K.; Viguerie, N.; Poitou, C.; Carette, C.; Pelloux, V.; Curat, C.A.; Sicard, A.; Rome, S.; Benis, A.; Zucker, J.D.; et al. Weight loss regulates inflammation-related genes in white adipose tissue of obese subjects. FASEB J 2004, 18, 1657–1669. [Google Scholar]
- Cancello, R.; Henegar, C.; Viguerie, N.; Taleb, S.; Poitou, C.; Rouault, C.; Coupaye, M.; Pelloux, V.; Hugol, D.; Bouillot, J.L.; et al. Reduction of macrophage infiltration and chemoattractant gene expression changes in white adipose tissue of morbidly obese subjects after surgery-induced weight loss. Diabetes 2005, 54, 2277–2286. [Google Scholar]
- Schipper, H.S.; Prakken, B.; Kalkhoven, E.; Boes, M. Adipose tissue-resident immune cells: Key players in immunometabolism. Trends. Endocrinol. Metab 2012, 23, 407–415. [Google Scholar]
- Arita, Y.; Kihara, S.; Ouchi, N.; Takahashi, M.; Maeda, K.; Miyagawa, J.; Hotta, K.; Shimomura, I.; Nakamura, T.; Miyaoka, K.; et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem. Biophys. Res. Commun 1999, 257, 79–83. [Google Scholar]
- Lillioja, S.; Mott, D.M.; Spraul, M.; Ferraro, R.; Foley, J.E.; Ravussin, E.; Knowler, W.C.; Bennett, P.H.; Bogardus, C. Insulin resistance and insulin secretory dysfunction as precursors of non-insulin-dependent diabetes mellitus. N. Engl. J. Med 1993, 329, 1988–1992. [Google Scholar]
- Kahn, B.B.; Flier, J.S. Obesity and insulin resistance. J. Clin. Investig 2000, 106, 473–481. [Google Scholar]
- Schenk, S.; Saberi, M.; Olefsky, J.M. Insulin sensitivity: Modulation by nutrients and inflammation. J. Clin. Investig 2008, 118, 2992–3002. [Google Scholar]
- Holland, W.L.; Brozinick, J.T.; Wang, L.P.; Hawkins, E.D.; Sargent, K.M.; Liu, Y.; Narra, K.; Hoehn, K.L.; Knotts, T.A.; Siesky, A.; et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab 2007, 5, 167–179. [Google Scholar]
- Dressler, K.A.; Mathias, S.; Kolesnick, R.N. Tumor necrosis factor-alpha activates the sphingomyelin signal transduction pathway in a cell-free system. Science 1992, 255, 1715–1718. [Google Scholar]
- Teruel, T.; Hernandez, R.; Lorenzo, M. Ceramide mediates insulin resistance by tumor necrosis factor-alpha in brown adipocytes by maintaining Akt in an inactive dephosphorylated state. Diabetes 2001, 50, 2563–2571. [Google Scholar]
- Haus, J.M.; Kashyap, S.R.; Kasumov, T.; Zhang, R.; Kelly, K.R.; Defronzo, R.A.; Kirwan, J.P. Plasma ceramides are elevated in obese subjects with type 2 diabetes and correlate with the severity of insulin resistance. Diabetes 2009, 58, 337–343. [Google Scholar]
- Holland, W.L.; Bikman, B.T.; Wang, L.P.; Yuguang, G.; Sargent, K.M.; Bulchand, S.; Knotts, T.A.; Shui, G.; Clegg, D.J.; Wenk, M.R.; et al. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J. Clin. Investig 2011, 121, 1858–1870. [Google Scholar]
- Horowitz, J.F.; Klein, S. Whole body and abdominal lipolytic sensitivity to epinephrine is suppressed in upper body obese women. Am. J. Physiol. Endocrinol. Metab 2000, 278, E1144–E1152. [Google Scholar]
- Bajaj, M.; Suraamornkul, S.; Romanelli, A.; Cline, G.W.; Mandarino, L.J.; Shulman, G.I.; DeFronzo, R.A. Effect of a sustained reduction in plasma free fatty acid concentration on intramuscular long-chain fatty Acyl-CoAs and insulin action in type 2 diabetic patients. Diabetes 2005, 54, 3148–3153. [Google Scholar]
- Santomauro, A.T.; Boden, G.; Silva, M.E.; Rocha, D.M.; Santos, R.F.; Ursich, M.J.; Strassmann, P.G.; Wajchenberg, B.L. Overnight lowering of free fatty acids with Acipimox improves insulin resistance and glucose tolerance in obese diabetic and nondiabetic subjects. Diabetes 1999, 48, 1836–1841. [Google Scholar]
- Girousse, A.; Tavernier, G.; Valle, C.; Moro, C.; Mejhert, N.; Dinel, A.L.; Houssier, M.; Roussel, B.; Besse-Patin, A.; Combes, M.; et al. Partial inhibition of adipose tissue lipolysis improves glucose metabolism and insulin sensitivity without alteration of fat mass. PLoS Biol 2013, 11, e1001485. [Google Scholar]
- Kosteli, A.; Sugaru, E.; Haemmerle, G.; Martin, J.F.; Lei, J.; Zechner, R.; Ferrante, A.W., Jr. Weight loss and lipolysis promote a dynamic immune response in murine adipose tissue. J. Clin. Investig 2010, 120, 3466–3479. [Google Scholar]
- Apovian, C.M.; Bigornia, S.; Mott, M.; Meyers, M.R.; Ulloor, J.; Gagua, M.; McDonnell, M.; Hess, D.; Joseph, L.; Gokce, N. Adipose macrophage infiltration is associated with insulin resistance and vascular endothelial dysfunction in obese subjects. Arterioscler. Thromb. Vasc. Biol 2008, 28, 1654–1659. [Google Scholar]
- Anderson, E.K.; Gutierrez, D.A.; Hasty, A.H. Adipose tissue recruitment of leukocytes. Curr. Opin. Lipidol 2010, 21, 172–177. [Google Scholar]
- Fain, J.N. Release of inflammatory mediators by human adipose tissue is enhanced in obesity and primarily by the nonfat cells: A review. Mediat. Inflamm 2010, 2010, 513948. [Google Scholar]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol 2011, 11, 85–97. [Google Scholar]
- Huber, J.; Kiefer, F.W.; Zeyda, M.; Ludvik, B.; Silberhumer, G.R.; Prager, G.; Zlabinger, G.J.; Stulnig, T.M. CC chemokine and CC chemokine receptor profiles in visceral and subcutaneous adipose tissue are altered in human obesity. J. Clin. Endocrinol. Metab 2008, 93, 3215–3221. [Google Scholar]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte chemoattractant protein-1 (MCP-1): An overview. J. Interferon Cytokine Res 2009, 29, 313–326. [Google Scholar]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Investig 2006, 116, 1494–1505. [Google Scholar]
- Kamei, N.; Tobe, K.; Suzuki, R.; Ohsugi, M.; Watanabe, T.; Kubota, N.; Ohtsuka-Kowatari, N.; Kumagai, K.; Sakamoto, K.; Kobayashi, M.; et al. Overexpression of monocyte chemoattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistance. J. Biol. Chem 2006, 281, 26602–26614. [Google Scholar]
- Lumeng, C.N.; DelProposto, J.B.; Westcott, D.J.; Saltiel, A.R. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes 2008, 57, 3239–3246. [Google Scholar]
- Meijer, K.; de Vries, M.; Al-Lahham, S.; Bruinenberg, M.; Weening, D.; Dijkstra, M.; Kloosterhuis, N.; van der Leij, R.J.; van der Want, H.; Kroesen, B.J.; et al. Human primary adipocytes exhibit immune cell function: Adipocytes prime inflammation independent of macrophages. PLoS One 2011, 6, e17154. [Google Scholar]
- Simeoni, E.; Hoffmann, M.M.; Winkelmann, B.R.; Ruiz, J.; Fleury, S.; Boehm, B.O.; März, W.; Vassalli, G. Association between the A-2518G polymorphism in the monocyte chemoattractant protein-1 gene and insulin resistance and Type 2 diabetes mellitus. Diabetologia 2004, 47, 1574–1580. [Google Scholar]
- Karadeniz, M.; Erdogan, M.; Cetinkalp, S.; Berdeli, A.; Eroglu, Z.; Ozgen, A.G. Monocyte chemoattractant protein-1 (MCP-1) 2518G/A gene polymorphism in Turkish type 2 diabetes patients with nephropathy. Endocrine 2010, 37, 513–517. [Google Scholar]
- Jing, Y.; Zhu, D.; Bi, Y.; Yang, D.; Hu, Y.; Shen, S. Monocyte chemoattractant protein 1–2518 A/G polymorphism and susceptibility to type 2 diabetes in a Chinese population. Clin. Chim. Acta 2011, 412, 466–469. [Google Scholar]
- Zietz, B.; Büchler, C.; Herfarth, H.; Müller-Ladner, U.; Spiegel, D.; Schölmerich, J.; Schäffler, A. Caucasian patients with type 2 diabetes mellitus have elevated levels of monocyte chemoattractant protein-1 that are not influenced by the −2518 A→G promoter polymorphism. Diabetes Obes. Metab 2005, 7, 570–578. [Google Scholar]
- Katakami, N.; Matsuhisa, M.; Kaneto, H.; Matsuoka, T.A.; Imamura, K.; Ishibashi, F.; Kanda, T.; Kawai, K.; Osonoi, T.; Kashiwagi, A.; et al. Monocyte chemoattractant protein-1 (MCP-1) gene polymorphism as a potential risk factor for diabetic retinopathy in Japanese patients with type 2 diabetes. Diabetes Res. Clin. Pract 2010, 89, e9–e12. [Google Scholar]
- Keophiphath, M.; Rouault, C.; Divoux, A.; Clement, K.; Lacasa, D. CCL5 promotes macrophage recruitment and survival in human adipose tissue. Arterioscler. Thromb. Vasc. Biol 2009, 30, 39–45. [Google Scholar]
- Chavey, C.; Lazennec, G.; Lagarrigue, S.; Clape, C.; Iankova, I.; Teyssier, J.; Annicotte, J.S.; Schmidt, J.; Mataki, C.; Yamamoto, H.; et al. CXC ligand 5 is an adipose-tissue derived factor that links obesity to insulin resistance. Cell Metab 2009, 9, 339–349. [Google Scholar]
- Tourniaire, F.; Romier-Crouzet, B.; Lee, J.H.; Marcotorchino, J.; Gouranton, E.; Salles, J.; Malezet, C.; Astier, J.; Darmon, P.; Blouin, E.; et al. Chemokine Expression in Inflamed Adipose Tissue Is Mainly Mediated by NF-κB. PLoS One 2013, 8, e66515. [Google Scholar]
- Ruan, H.; Lodish, H.F. Insulin resistance in adipose tissue: Direct and indirect effects of tumor necrosis factor-α. Cytokine Growth Factor Rev 2003, 14, 447–455. [Google Scholar]
- Uysal, K.T.; Wiesbrock, S.M.; Marino, M.W.; Hotamisligil, G.S. Protection from obesity-induced insulin resistance in mice lacking TNF-α function. Nature 1997, 389, 610–614. [Google Scholar]
- Miyazaki, Y.; Pipek, R.; Mandarino, L.J.; DeFronzo, R.A. Tumor necrosis factor α and insulin resistance in obese type 2 diabetic patients. Int. J. Obes. Relat. Metab. Disord 2003, 27, 88–94. [Google Scholar]
- Wascher, T.C.; Lindeman, J.H.; Sourij, H.; Kooistra, T.; Pacini, G.; Roden, M. Chronic TNF-α neutralization does not improve insulin resistance or endothelial function in “healthy” men with metabolic syndrome. Mol. Med 2011, 17, 189–193. [Google Scholar]
- Bernstein, L.E.; Berry, J.; Kim, S.; Canavan, B.; Grinspoon, S.K. Effects of etanercept in patients with the metabolic syndrome. Arch. Intern. Med 2006, 166, 902–908. [Google Scholar]
- McArdle, M.A.; Finucane, O.M.; Connaughton, R.M.; McMorrow, A.M.; Roche, H.M. Mechanisms of obesity-induced inflammation and insulin resistance: Insights into the emerging role of nutritional strategies. Front. Endocrinol. (Lausanne) 2013, 4, 52. [Google Scholar]
- Illei, G.G.; Lipsky, P.E. Novel, antigen-specific therapeutic approaches to autoimmuneinflammatory diseases. Curr. Opin. Immunol 2000, 12, 712–718. [Google Scholar]
- Hodge-Dufour, J.; Marino, M.W.; Horton, M.R.; Jungbluth, A.; Burdick, R.D.; Strieter, R.M.; Noble, P.W.; Hunter, C.A.; Pure, E. Inhibition of interferon gamma induced interleukin 12 production—A potential mechanism for the anti-inflammatory activities of tumor necrosis factor. Proc. Natl. Acad. Sci. USA 1998, 95, 13806–13811. [Google Scholar]
- O’Rourke, R.W.; White, A.E.; Metcalf, M.D.; Winters, B.R.; Diggs, B.S.; Zhu, X.; Marks, D.L. Systemic inflammation and insulin sensitivity in obese IFN-γ knockout mice. Metabolism 2012, 61, 1152–1161. [Google Scholar]
- Sultan, A.; Strodthoff, D.; Robertson, A.K.; Paulsson-Berne, G.; Fauconnier, J.; Parini, P.; Rydén, M.; Thierry-Mieg, N.; Johansson, M.E.; Chibalin, A.V.; et al. T cell-mediated inflammation in adipose tissue does not cause insulin resistance in hyperlipidemic mice. Circ. Res 2009, 104, 961–968. [Google Scholar]
- Fernandez-Real, J.M.; Ricart, W. Insulin resistance and chronic cardiovascular inflammatory syndrome. Endocr. Rev 2003, 24, 278–301. [Google Scholar]
- Mohamed-Ali, V.; Goodrick, S.; Rawesh, A.; Katz, D.R.; Miles, J.M.; Yudkin, J.S.; Klein, S.; Coppack, S.W. Subcutaneous adipose tissue releases interleukin-6, but not tumor necrosis factor-alpha in vivo. J. Clin. Endocrinol. Metab 1997, 82, 4196–4200. [Google Scholar]
- Sopasakis, V.R.; Sandqvist, M.; Gustafson, B.; Hammarstedt, A.; Schmelz, M.; Yang, X.; Jansson, P.A.; Smith, U. High local concentrations and effects on differentiation implicate interleukin-6 as a paracrine regulator. Obes. Res 2004, 12, 454–460. [Google Scholar]
- Bastard, J.P.; Maachi, M.; van Nhieu, J.T.; Jardel, C.; Bruckert, E.; Grimaldi, A.; Robert, J.J.; Capeau, J.; Hainque, B. Adipose tissue IL-6 content correlates with resistance to insulin activation of glucose uptake both in vivo and in vitro. J. Clin. Endocrinol. Metab. 2002, 87, 2084–2089. [Google Scholar]
- Pradhan, A.D.; Manson, J.E.; Rifai, N.; Buring, J.E.; Ridker, P.M. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA 2001, 286, 327–334. [Google Scholar]
- Tsigos, C.; Papanicolaou, D.A.; Kyrou, I.; Defensor, R.; Mitsiadis, C.S.; Chrousos, G.P. Dose-dependent effects of recombinant human interleukin-6 on glucose regulation. J. Clin. Endocrinol. Metab 1997, 82, 4167–4170. [Google Scholar]
- Wallenius, V.; Wallenius, K.; Ahren, B.; Rudling, M.; Carlsten, H.; Dickson, S.L.; Ohlsson, C.; Jansson, J.O. Interleukin-6-deficient mice develop mature-onset obesity. Nat. Med 2002, 8, 75–79. [Google Scholar]
- Di Gregorio, G.B.; Hensley, L.; Lu, T.; Ranganathan, G.; Kern, P.A. Lipid and carbohydrate metabolism in mice with a targeted mutation in the IL-6 gene: Absence of development of age-related obesity. Am. J. Physiol. Endocrinol. Metab 2004, 287, E182–E187. [Google Scholar]
- Starkie, R.; Ostrowski, S.R.; Jauffred, S.; Febbraio, M.; Pedersen, B.K. Exercise and IL-6 infusion inhibit endotoxin-induced TNF-α production in humans. FASEB J 2003, 17, 884–886. [Google Scholar]
- Carey, A.L.; Steinberg, G.R.; Macaulay, S.L.; Thomas, W.G.; Holmes, A.G.; Ramm, G.; Prelovsek, O.; Hohnen-Behrens, C.; Watt, M.J.; James, D.E.; et al. Interleukin-6 increases insulin-stimulated glucose disposal in humans and glucose uptake and fatty acid oxidation in vitro via AMP-activated protein kinase. Diabetes 2006, 55, 2688–2697. [Google Scholar]
- Wood, I.S.; Wang, B.; Jenkins, J.R.; Trayhurn, P. The pro-inflammatory cytokine IL-18 is expressed in human adipose tissue and strongly upregulated by TNFalpha in human adipocytes. Biochem. Biophys. Res. Commun 2005, 337, 422–429. [Google Scholar]
- Esposito, K.; Pontillo, A.; Ciotola, M.; di Palo, C.; Grella, E.; Nicoletti, G.; Giugliano, D. Weight loss reduces interleukin-18 levels in obese women. J. Clin. Endocrinol. Metab 2002, 87, 3864–3866. [Google Scholar]
- Tan, H.W.; Liu, X.; Bi, X.P.; Xing, S.S.; Li, L.; Gong, H.P.; Zhong, M.; Wang, Z.H.; Zhang, Y.; Zhang, W. IL-18 overexpression promotes vascular inflammation and remodeling in a rat model of metabolic syndrome. Atherosclerosis 2010, 208, 350–357. [Google Scholar]
- Netea, M.G.; Joosten, L.A.; Lewis, E.; Jensen, D.R.; Voshol, P.J.; Kullberg, B.J.; Tack, C.J.; van Krieken, H.; Kim, S.H.; Stalenhoef, A.F.; et al. Deficiency of interleukin-18 in mice leads to hyperphagia, obesity and insulin resistance. Nat. Med 2006, 12, 650–656. [Google Scholar]
- Friedman, J.M.; Halaas, J.L. Leptin and the regulation of body weight in mammals. Nature 1998, 395, 763–770. [Google Scholar]
- Considine, R.V.; Sinha, M.K.; Heiman, M.L.; Kriauciunas, A.; Stephens, T.W.; Nyce, M.R.; Ohannesian, J.P.; Marco, C.C.; McKee, L.J.; Bauer, T.L.; et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N. Engl. J. Med 1996, 334, 292. [Google Scholar]
- Kouidhi, S.; Jarboui, S.; Clerget Froidevaux, M.S.; Abid, H.; Demeneix, B.; Zaouche, A.; Benammar Elgaaied, A.; Guissouma, H. Relationship between subcutaneous adipose tissue expression of leptin and obesity in Tunisian patients. Tunis. Med 2010, 88, 569–572. [Google Scholar]
- Marroquí, L.; Gonzalez, A.; Ñeco, P.; Caballero-Garrido, E.; Vieira, E.; Ripoll, C.; Nadal, A.; Quesada, I. Role of leptin in the pancreatic β-cell: Effects and signaling pathways. J. Mol. Endocrinol 2012, 49, R9–R17. [Google Scholar]
- Müller, G.; Ertl, J.; Gerl, M.; Preibisch, G. Leptin impairs metabolic actions of insulin in isolated rat adipocytes. J. Biol. Chem 1997, 272, 10585–10593. [Google Scholar]
- Pérez, C.; Fernández-Galaz, C.; Fernández-Agulló, T.; Arribas, C.; Andrés, A.; Ros, M.; Carrascosa, J.M. Leptin impairs insulin signaling in rat adipocytes. Diabetes 2004, 53, 347–353. [Google Scholar]
- Paz-Filho, G.; Mastronardi, C.; Franco, C.B.; Wang, K.B.; Wong, M.L.; Licinio, J. Leptin: Molecular mechanisms, systemic pro-inflammatory effects, and clinical implications. Arq. Bras. Endocrinol. Metabol 2012, 56, 597–607. [Google Scholar]
- Lord, G.M.; Matarese, G.; Howard, J.K.; Baker, R.J.; Bloom, S.R.; Lechler, R.I. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature 1998, 394, 897–901. [Google Scholar]
- Grunfeld, C.; Zhao, C.; Fuller, J.; Pollack, A.; Moser, A.; Friedman, J.; Feingold, K.R. Endotoxin and cytokines induce expression of leptin, the ob gene product, in hamsters. J. Clin. Investig 1996, 97, 2152–2157. [Google Scholar]
- Satoh, H.; Nguyen, M.T.; Miles, P.D.; Imamura, T.; Usui, I.; Olefsky, J.M. Adenovirus-mediated chronic “hyper-resistinemia” leads to in vivo insulin resistance in normal rats. J. Clin. Investig 2004, 114, 224–231. [Google Scholar]
- Qi, Y.; Nie, Z.; Lee, Y.S.; Singhal, N.S.; Scherer, P.E.; Lazar, M.A.; Ahima, R.S. Loss of resistin improves glucose homeostasis in leptin deficiency. Diabetes 2006, 55, 3083–3090. [Google Scholar]
- Banerjee, R.R.; Rangwala, S.M.; Shapiro, J.S.; Rich, A.S.; Rhoades, B.; Qi, Y.; Wang, J.; Rajala, M.W.; Pocai, A.; Scherer, P.E.; et al. Regulation of fasted blood glucose by resistin. Science 2004, 303, 1195–1198. [Google Scholar]
- Steppan, C.M.; Wang, J.; Whiteman, E.L.; Birnbaum, M.J.; Lazar, M.A. Activation of SOCS-3 by resistin. Mol. Cell. Biol 2005, 25, 1569–1575. [Google Scholar]
- Vidal-Puig, A.; O’Rahilly, S. Resistin: A new link between obesity and insulin resistance? Clin. Endocrinol 2001, 55, 437–438. [Google Scholar]
- McTernan, C.L.; McTernan, P.G.; Harte, A.L.; Levick, P.L.; Barnett, A.H.; Kumar, S. Resistin, central obesity, and type 2 diabetes. Lancet 2002, 359, 46–47. [Google Scholar]
- McTernan, P.G.; McTernan, C.L.; Chetty, R.; Jenner, K.; Fisher, F.M.; Lauer, M.N.; Crocker, J.; Barnett, A.H.; Kumar, S. Increased resistin gene and protein expression in human abdominal adipose tissue. J. Clin. Endocrinol. Metab 2002, 87, 2407–2410. [Google Scholar]
- Wang, H.; Chu, W.S.; Hemphill, C.; Elbein, S.C. Human resistin gene: Molecular scanning and evaluation of association with insulin sensitivity and type 2 diabetes in Caucasians. J. Clin. Endocrinol. Metab 2002, 87, 2520–2524. [Google Scholar]
- Osawa, H.; Yamada, K.; Onuma, H.; Murakami, A.; Ochi, M.; Kawata, H.; Nishimiya, T.; Niiya, T.; Shimizu, I.; Nishida, W.; et al. The G/G genotype of a resistin single-nucleotide polymorphism at −420 increases type 2 diabetes mellitus susceptibility by inducing promoter activity through specific binding of Sp1/3. Am. J. Hum. Genet 2004, 75, 678–686. [Google Scholar]
- Kielstein, J.T.; Becker, B.; Graf, S.; Brabant, G.; Haller, H.; Fliser, D. Increased resistin blood levels are not associated with insulin resistance in patients with renal disease. Am. J. Kidney Dis 2003, 42, 62–66. [Google Scholar]
- Patel, L.; Buckels, A.C.; Kinghorn, I.J.; Murdock, P.R.; Holbrook, J.D.; Plumpton, C.; Macphee, C.H.; Smith, S.A. Resistin is expressed in human macrophages and directly regulated by PPARγ activators. Biochem. Biophys. Res. Commun 2003, 300, 472–476. [Google Scholar]
- Qatanani, M.; Szwergold, N.R.; Greaves, D.R.; Ahima, R.S.; Lazar, M.A. Macrophage-derived human resistin exacerbates adipose tissue inflammation and insulin resistance in mice. J. Clin. Investig 2009, 119, 531–539. [Google Scholar]
- Mertens, I.; van Gaal, L.F. Obesity, haemostasis and the fibrinolytic system. Obes. Rev 2002, 3, 85–101. [Google Scholar]
- Juhan-Vague, I.; Alessi, M.C.; Mavri, A.; Morange, P.E. Plasminogen activator inhibitor-1, inflammation, obesity, insulin resistance and vascular risk. J. Thromb. Haemost 2003, 1, 1575–1579. [Google Scholar]
- Ma, L.J.; Mao, S.L.; Taylor, K.L.; Kanjanabuch, T.; Guan, Y.; Zhang, Y.; Brown, N.J.; Swift, L.L.; McGuinness, O.P.; Wasserman, D.H.; et al. Prevention of obesity and insulin resistance in mice lacking plasminogen activator inhibitor 1. Diabetes 2004, 53, 336–346. [Google Scholar]
- Liang, X.; Kanjanabuch, T.; Mao, S.L.; Hao, C.M.; Tang, Y.W.; Declerck, P.J.; Hasty, A.H.; Wasserman, D.H.; Fogo, A.B.; Ma, L.J. Plasminogen activator inhibitor-1 modulates adipocyte differentiation. Am. J. Physiol. Endocrinol. Metab 2006, 290, E103–E113. [Google Scholar]
- Xu, X.; Wang, H.; Wang, Z.; Xiao, W. Plasminogen activator inhibitor-1 promotes inflammatory process induced by cigarette smoke extraction or lipopolysaccharides in alveolar epithelial cells. Exp. Lung Res 2009, 35, 795–805. [Google Scholar]
- Samal, B.; Sun, Y.; Stearns, G.; Xie, C.; Suggs, S.; McNiece, I. Cloning and characterization of the cDNA encoding a novel human pre-B-cell colony-enhancing factor. Mol. Cell. Biol 1994, 14, 1431–1437. [Google Scholar]
- Fukuhara, A.; Matsuda, M.; Nishizawa, M.; Segawa, K.; Tanaka, M.; Kishimoto, K.; Matsuki, Y.; Murakami, M.; Ichisaka, T.; Murakami, H.; et al. Visfatin: A protein secreted by visceral fat that mimics the effects of insulin. Science 2005, 307, 426–430. [Google Scholar]
- Revollo, J.R.; Körner, A.; Mills, K.F.; Satoh, A.; Wang, T.; Garten, A.; Dasgupta, B.; Sasaki, Y.; Wolberger, C.; Townsend, R.R.; et al. Nampt/PBEF/Visfatin regulates insulin secretion in β cells as a systemic NAD biosynthetic enzyme. Cell Metab 2007, 6, 363–375. [Google Scholar]
- Pagano, C.; Pilon, C.; Olivieri, M.; Mason, P.; Fabris, R.; Serra, R.; Milan, G.; Rossato, M.; Federspil, G.; Vettor, R. Reduced plasma visfatin/pre B-cell colony-enhancing factor in obesity is not related to insulin resistance in humans. J. Clin. Endocrinol. Metab 2006, 91, 3165–3170. [Google Scholar]
- Berndt, J.; Klöting, N.; Kralisch, S.; Kovacs, P.; Fasshauer, M.; Schön, M.R.; Stumvollx, M.; Blüher, M. Plasma visfatin concentrations and fat-depot specific mRNA expression in humans. Diabetes 2005, 54, 2911–2916. [Google Scholar]
- Fain, J.N.; Sacks, H.S.; Buehrer, B.; Bahouth, S.W.; Garrett, E.; Wolf, R.Y.; Carter, R.A.; Tichansky, D.S.; Madan, A.K. Identification of omentin mRNA in human epicardial adipose tissue: Comparison to omentin in subcutaneous, internal mammary artery periadventitial and visceral abdominal depots. Int. J. Obes 2008, 32, 810–815. [Google Scholar]
- Chang, Y.C.; Chang, T.J.; Lee, W.J.; Chuang, L.M. The relationship of visfatin/pre-B-cell colony-enhancing factor/nicotinamide phosphoribosyltransferase in adipose tissue with inflammation, insulin resistance, and plasma lipids. Metabolism 2010, 59, 93–99. [Google Scholar]
- Haider, D.G.; Schindler, K.; Schaller, G.; Prager, G.; Wolzt, M.; Ludvik, B. Increased plasma visfatin concentrations in morbidly obese subjects are reduced after gastric banding. J. Clin. Endocrinol. Metab 2006, 91, 1578–1581. [Google Scholar]
- El-Mesallamy, H.O.; Kassem, D.H.; El-Demerdash, E.; Amin, A.I. Vaspin and visfatin/Nampt are interesting interrelated adipokines playing a role in the pathogenesis of type 2 diabetes mellitus. Metabolism 2011, 60, 63–70. [Google Scholar]
- Varma, V.; Yao-Borengasser, A.; Rasouli, N.; Bodles, A.M.; Phanavanh, B.; Lee, M.J.; Starks, T.; Kern, L.M.; Spencer, H.J., 3rd; McGehee, R.E., Jr.; et al. Human visfatin expression: Relationship to insulin sensitivity, intramyocellular lipids, and inflammation. J. Clin. Endocrinol. Metab 2007, 92, 666–672. [Google Scholar]
- Kim da, S.; Kang, S.; Moon, N.R.; Park, S. Central visfatin potentiates glucose-stimulated insulin secretion and β-cell mass without increasing serum visfatin levels in diabetic rats. Cytokine 2014, 65, 159–166. [Google Scholar]
- Oki, K.; Yamane, K.; Kamei, N.; Nojima, H.; Kohno, N. Circulating visfatin level is correlated with inflammation, but not with insulin resistance. Clin. Endocrinol 2007, 67, 796–800. [Google Scholar]
- Quadro, L.; Blaner, W.S.; Salchow, D.J.; Vogel, S.; Piantedosi, R.; Gouras, P.; Freeman, S.; Cosma, M.P.; Colantuoni, V.; Gottesman, M.E. Impaired retinal function and vitamin A availability in mice lacking retinol-binding protein. EMBO J 1999, 18, 4633–4644. [Google Scholar]
- Yang, Q.; Graham, T.E.; Mody, N.; Preitner, F.; Peroni, O.D.; Zabolotny, J.M.; Kotani, K.; Quadro, L.; Kahn, B.B. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature 2005, 436, 356–362. [Google Scholar]
- Klöting, N.; Graham, T.E.; Berndt, J.; Kralisch, S.; Kovacs, P.; Wason, C.J.; Fasshauer, M.; Schön, M.R.; Stumvoll, M.; Blüher, M.; et al. Serum retinol-binding protein is more highly expressed in visceral than in subcutaneous adipose tissue and is a marker of intra-abdominal fat mass. Cell Metab 2007, 6, 79–87. [Google Scholar]
- Ost, A.; Danielsson, A.; Liden, M.; Eriksson, U.; Nystrom, F.H.; Stralfors, P. Retinol-binding protein-4 attenuates insulin-induced phosphorylation of IRS1 and ERK1/2 in primary human adipocytes. FASEB J 2007, 21, 3696–3704. [Google Scholar]
- Graham, T.E.; Yang, Q.; Bluher, M.; Hammarstedt, A.; Ciaraldi, T.P.; Henry, R.R.; Wason, C.J.; Oberbach, A.; Jansson, P.A.; Smith, U.; et al. Retinol-binding protein 4 and insulin resistance in lean, obese, and diabetic subjects. N. Engl. J. Med 2006, 354, 2552–2563. [Google Scholar]
- Gavi, S.; Stuart, L.M.; Kelly, P.; Melendez, M.M.; Mynarcik, D.C.; Gelato, M.C.; McNurlan, M.A. Retinol-binding protein 4 is associated with insulin resistance and body fat distribution in nonobese subjects without type 2 diabetes. J. Clin. Endocrinol. Metab 2007, 92, 1886–1890. [Google Scholar]
- Balagopal, P.; Graham, T.E.; Kahn, B.B.; Altomare, A.; Funanage, V.; George, D. Reduction of elevated serum retinol binding protein in obese children by lifestyle intervention: Association with subclinical inflammation. J. Clin. Endocrinol. Metab 2007, 92, 1971–1974. [Google Scholar]
- Nair, A.K.; Sugunan, D.; Kumar, H.; Anilkumar, G. Case-control analysis of SNPs in GLUT4, RBP4 and STRA6: Association of SNPs in STRA6 with type 2 diabetes in a South Indian population. PLoS One 2010, 5, e11444. [Google Scholar]
- Munkhtulga, L.; Nakayama, K.; Utsumi, N.; Yanagisawa, Y.; Gotoh, T.; Omi, T.; Kumada, M.; Erdenebulgan, B.; Zolzaya, K.; Lkhagvasuren, T.; et al. Identification of a regulatory SNP in the retinol binding protein 4 gene associated with type 2 diabetes in Mongolia. Hum. Genet 2007, 120, 879–888. [Google Scholar]
- Munkhtulga, L.; Nagashima, S.; Nakayama, K.; Utsumi, N.; Yanagisawa, Y.; Gotoh, T.; Omi, T.; Kumada, M.; Zolzaya, K.; Lkhagvasuren, T.; et al. Regulatory SNP in the RBP4 gene modified the expression in adipocytes and associated with BMI. Obesity 2010, 18, 1006–1014. [Google Scholar]
- Yao-Borengasser, A.; Varma, V.; Bodles, A.M.; Rasouli, N.; Phanavanh, B.; Lee, M.J.; Starks, T.; Kern, L.M.; Spencer, H.J., III; Rashidi, A.A.; et al. Retinol binding protein 4 expression in humans: Relationship to insulin resistance, inflammation, and response to pioglitazone. J. Clin. Endocrinol. Metab 2007, 92, 2590–2597. [Google Scholar]
- Ulgen, F.; Herder, C.; Kuhn, M.C.; Willenberg, H.S.; Schott, M.; Scherbaum, W.A.; Schinner, S. Association of serum levels of retinol-binding protein 4 with male sex but not with insulin resistance in obese patients. Arch. Physiol. Biochem 2010, 116, 57–62. [Google Scholar]
- Kos, K.; Wong, S.; Tan, B.; Kerrigan, D.; Randeva, H.; Pinkney, J.; Wilding, J. Human RBP4 adipose tissue expression is gender specific and influenced by leptin. Clin. Endocrinol. (Oxf.) 2011, 74, 197–205. [Google Scholar]
- Tabata, M.; Kadomatsu, T.; Fukuhara, S.; Miyata, K.; Ito, Y.; Endo, M.; Urano, T.; Zhu, H.J.; Tsukano, H.; Tazume, H.; et al. Angiopoietin-like protein 2 promotes chronic adipose tissue inflammation and obesity-related systemic insulin resistance. Cell Metab 2009, 10, 178–188. [Google Scholar]
- Doi, Y.; Ninomiya, T.; Hirakawa, Y.; Takahashi, O.; Mukai, N.; Hata, J.; Iwase, M.; Kitazono, T.; Oike, Y.; Kiyohara, Y. Angiopoietin-like protein 2 and risk of type 2 diabetes in a general Japanese population: The Hisayama study. Diabetes Care 2013, 36, 98–100. [Google Scholar]
- Berg, A.H.; Combs, T.P.; Du, X.; Brownlee, M.; Scherer, P.E. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat. Med 2001, 7, 947–953. [Google Scholar]
- Diez, J.J.; Iglesias, P. The role of the novel adipocyte-derived hormone adiponectin in human disease. Eur. J. Endocrinol 2003, 148, 293–300. [Google Scholar]
- Kern, P.A.; di Gregorio, G.B.; Lu, T.; Rassouli, N.; Ranganathan, G. Adiponectin expression from human adipose tissue: Relation to obesity, insulin resistance, and tumor necrosis factor-alpha expression. Diabetes 2003, 52, 1779–1785. [Google Scholar]
- Maeda, N.; Shimomura, I.; Kishida, K.; Nishizawa, H.; Matsuda, M.; Nagaretani, H.; Furuyama, N.; Kondo, H.; Takahashi, M.; Arita, Y.; et al. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat. Med 2002, 8, 731–737. [Google Scholar]
- Kadowaki, T.; Yamauchi, T.; Kubota, N.; Hara, K.; Ueki, K.; Tobe, K. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J. Clin. Investig 2006, 116, 1784–1792. [Google Scholar]
- Ouchi, N.; Higuchi, A.; Ohashi, K.; Oshima, Y.; Gokce, N.; Shibata, R.; Akasaki, Y.; Shimono, A.; Walsh, K. Sfrp5 is an anti-inflammatory adipokine that modulates metabolic dysfunction in obesity. Science 2010, 329, 454–457. [Google Scholar]
- Hu, Z.; Deng, H.; Qu, H. Plasma SFRP5 levels are decreased in Chinese subjects with obesity and type 2 diabetes and negatively correlated with parameters of insulin resistance. Diabetes Res. Clin. Pract 2013, 99, 391–395. [Google Scholar]
- Hu, W.; Li, L.; Yang, M.; Luo, X.; Ran, W.; Liu, D.; Xiong, Z.; Liu, H.; Yang, G. Circulating Sfrp5 is a signature of obesity-related metabolic disorders and is regulated by glucose and liraglutide in humans. J. Clin. Endocrinol. Metab 2013, 98, 290–298. [Google Scholar]
- Tan, X.; Wang, X.; Chu, H.; Liu, H.; Yi, X.; Xiao, Y. SFRP5 correlates with obesity and metabolic syndrome and increases after weight loss in children. Clin. Endocrinol. (Oxf.) 2013. [Google Scholar] [CrossRef]
- Carstensen, M.; Herder, C.; Kempf, K.; Erlund, I.; Martin, S.; Koenig, W.; Sundvall, J.; Bidel, S.; Kuha, S.; Roden, M.; et al. Sfrp5 correlates with insulin resistance and oxidative stress. Eur. J. Clin. Investig 2013, 43, 350–357. [Google Scholar]
- Saleh, J.; Sniderman, A.D.; Cianflone, K. Regulation of Plasma fatty acid metabolism. Clin. Chim. Acta 1999, 286, 163–180. [Google Scholar]
- Clemente-Postigo, M.; Queipo-Ortuno, M.I.; Fernandez-Garcia, D.; Gomez-Huelgas, R.; Tinahones, F.J.; Cardona, F. Adipose tissue gene expression of factors related to lipid processing in obesity. PLoS One 2011, 6, e24783. [Google Scholar]
- Klop, B.; Jukema, J.W.; Rabelink, T.J.; Castro Cabezas, M. A physician’s guide for the management of hypertriglyceridemia: The etiology of hypertriglyceridemia determines treatment strategy. Panminerva Med 2012, 54, 91–103. [Google Scholar]
- Klop, B.; Elte, J.W.; Cabezas, M.C. Dyslipidemia in obesity: Mechanisms and potential targets. Nutrients 2013, 5, 1218–1240. [Google Scholar]
- Guendouzi, K.; Jaspard, B.; Barbaras, R.; Motta, C.; Vieu, C.; Marcel, Y.; Chap, H.; Perret, B.; Collet, X. Biochemical and physical properties of remnant-HDL2 and of pre beta 1-HDL produced by hepatic lipase. Biochemistry 1999, 38, 2762–2768. [Google Scholar]
- Friedewald, W.T.; Levy, R.I.; Fredrickson, D.S. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin. Chem 1972, 18, 499–502. [Google Scholar]
- Contois, J.H.; Warnick, G.R.; Sniderman, A.D. Reliability of low-density lipoprotein cholesterol, non-high-density lipoprotein cholesterol, and apolipoprotein B measurement. J. Clin. Lipidol 2011, 5, 264–272. [Google Scholar]
- Ito, Y.; Fujimura, M.; Ohta, M.; Hirano, T. Development of a homogeneous assay for measurement of small dense LDL cholesterol. Clin. Chem 2011, 57, 57–65. [Google Scholar]
- St-Pierre, A.C.; Cantin, B.; Dagenais, G.R.; Mauriege, P.; Bernard, P.-M.; Despres, J.P.; Lamarche, B. Low-density lipoprotein subfractions and the long-term risk of ischemic heart disease in men. Arterioscler. Thromb. Vasc. Biol 2005, 25, 553–559. [Google Scholar]
- Rizzo, M.; Pernice, V.; Frasheri, A.; di Lorenzo, G.; Rini, G.B.; Spinas, G.A.; Berneis, K. Small, dense low-density lipoproteins (LDL) are predictors of cardio- and cerebro-vascular events in subjects with the metabolic syndrome. Clin. Endocrinol. (Oxf.) 2009, 70, 870–875. [Google Scholar] [Green Version]
- Engfeldt, P.; Arner, P. Lipolysis in human adipocytes, effects of cell size, age and of regional differences. Horm. Metab. Res 1988, 19, 26–29. [Google Scholar]
- Veilleux, A.; Caron-Jobin, M.; Noël, S.; Laberge, P.Y.; Tchernof, A. Visceral adipocyte hypertrophy is associated with dyslipidemia independent of body composition and fat distribution in women. Diabetes 2011, 60, 1504–1511. [Google Scholar]
- Sam, S.; Haffner, S.; Davidson, M.H.; D’Agostino, R.B., Sr.; Feinstein, S.; Kondos, G.; Perez, A.; Mazzone, T. Relationship of abdominal visceral and subcutaneous adipose tissue with lipoprotein particle number and size in type 2 diabetes. Diabetes 2008, 57, 2022–2027. [Google Scholar]
- Johnson, J.A.; Fried, S.K.; Pi-Sunyer, F.X.; Albu, J.B. Impaired insulin action in subcutaneous adipocytes from women with visceral obesity. Am. J. Physiol. Endocrinol. Metab 2001, 280, E40–E49. [Google Scholar]
- Despres, J.P.; Ferland, M.; Moorjani, S.; Nadeau, A.; Tremblay, A.; Lupien, P.J.; Theriault, G.; Bouchard, C. Role of hepatic-triglyceride lipase activity in the association between intra-abdominal fat and plasma HDL cholesterol in obese women. Arteriosclerosis 1989, 9, 485–492. [Google Scholar]
- Carr, M.C.; Hokanson, J.E.; Zambon, A.; Deeb, S.S.; Barrett, P.H.; Purnell, J.Q.; Brunzell, J.D. The contribution of intraabdominal fat to gender differences in hepatic lipase activity and low/high density lipoprotein heterogeneity. J. Clin. Endocrinol. Metab 2001, 86, 2831–2837. [Google Scholar]
- Cancello, R.; Tordjman, J.; Poitou, C.; Guilhem, G.; Bouillot, J.L.; Hugol, D.; Coussieu, C.; Basdevant, A.; Bar Hen, A.; Bedossa, P.; et al. Increased infiltration of macrophages in omental adipose tissue is associated with marked hepatic lesions in morbid human obesity. Diabetes 2006, 55, 1554–1561. [Google Scholar]
- Esteve, E.; Ricart, W.; Fernández-Real, J.M. Dyslipidemia and inflammation: An evolutionary conserved mechanism. Clin. Nutr 2005, 24, 16–31. [Google Scholar]
- Yang, Y.; Ju, D.; Zhang, M.; Yang, G. Interleukin-6 stimulates lipolysis in porcine adipocytes. Endocrine 2008, 33, 261–269. [Google Scholar]
- Hardardóttir, I.; Doerrler, W.; Feingold, K.R.; Grünfeld, C. Cytokines stimulate lipolysis and decrease lipoprotein lipase activity in cultured fat cells by a prostaglandin independent mechanism. Biochem. Biophys. Res. Commun 1992, 186, 237–243. [Google Scholar]
- Hardardottir, I.; Moser, A.H.; Memon, R.; Grunfeld, C.; Feingold, K.R. Effects of TNF, IL-1, and the combination of both cytokines on cholesterol metabolism in Syrian hamsters. Lymphokine Cytokine Res 1994, 13, 161–166. [Google Scholar]
- Kawakami, M.; Murase, T.; Ogawa, H.; Ishibashi, S.; Mori, N.; Takaku, F.; Shibata, S. Human recombinant TNF suppresses lipoprotein lipase activity and stimulates lipolysis in 3T3-L1 cells. J. Biochem 1987, 10, 331–338. [Google Scholar]
- Greenberg, A.S.; Nordan, R.P.; McIntosh, J.; Calvo, J.C.; Scow, R.O.; Jablons, D. Interleukin 6 reduces lipoprotein lipase activity in adipose tissue of mice in vivo and in 3T3-L1 adipocytes (a possible role for interleukin 6 in cancer cachexia). Cancer Res 1992, 52, 4113–4116. [Google Scholar]
- Rouzer, C.A.; Cerami, A. Hypertriglyceridemia associated with Trypanosoma brucei brucei infection in rabbits: Role of defective triglyceride removal. Mol. Biochem. Parasitol 1980, 2, 31–38. [Google Scholar]
- Jovinge, S.; Hamsten, A.; Tornvall, P.; Proudler, A.; Båvenholm, P.; Ericsson, C.G.; Godsland, I.; de Faire, U.; Nilsson, J. Evidence for a role of tumor necrosis factor alpha in disturbances of triglyceride and glucose metabolism predisposing to coronary heart disease. Metabolism 1998, 47, 113–118. [Google Scholar]
- Feingold, K.R.; Serio, M.K.; Adi, S.; Moser, A.H.; Grunfeld, C. Tumor necrosis factor stimulates hepatic lipid synthesis and secretion. Endocrinology 1989, 124, 2336–2342. [Google Scholar]
- Kawakami, M.; Cerami, A. Studies of endotoxin-induced decrease in lipoprotein lipase activity. J. Exp. Med 1981, 154, 631–639. [Google Scholar]
- Qin, B.; Anderson, R.A.; Adeli, K. Tumor necrosis factor-alpha directly stimulates the overproduction of hepatic apolipoprotein B100-containing VLDL via impairment of hepatic insulin signaling. Am. J. Physiol. Gastrointest. Liver Physiol 2008, 294, G1120–G1129. [Google Scholar]
- Mohrschladt, M.F.; Weverling-Rijnsburger, A.W.; de Man, F.H.; Stoeken, D.J.; Sturk, A.; Smelt, A.H.; Westendorp, R.G. Hyperlipoproteinemia affects cytokine production in whole blood samples ex vivo. The influence of lipid-lowering therapy. Atherosclerosis 2000, 148, 413–419. [Google Scholar]
- Jonkers, I.J.; Mohrschladt, M.F.; Westendorp, R.G.; van der Laarse, A.; Smelt, A.H. Severe hypertriglyceridemia with insulin resistance is associated with systemic inflammation (reversal with bezafibrate therapy in a randomized controlled trial). Am. J. Med 2002, 112, 275–280. [Google Scholar]
- Nappo, F.; Esposito, K.; Cioffi, M.; Giugliano, G.; Molinari, A.M.; Paolisso, G.; Marfella, R.; Giugliano, D. Postprandial endothelial activation in healthy subjects and in type 2 diabetic patients (role of fat and carbohydrate meals). J. Am. Coll. Cardiol 2002, 39, 1145–1150. [Google Scholar]
- Van Exel, E.; Gussekloo, J.; de Craen, A.J.; Frolich, M.; Bootsma-van der Wiel, A.; Westendorp, R.G. Leiden 85 Plus Study. Low production capacity of interleukin-10 associates with the metabolic syndrome and type 2 diabetes: The Leiden 85-Plus Study. Diabetes 2002, 51, 1088–1092. [Google Scholar]
- Grunfeld, C.; Dinarello, C.A.; Feingold, K.R. Tumor necrosis factor-alpha, interleukin-1, and interferon alpha stimulate triglyceride synthesis in HepG2 cells. Metabolism 1991, 40, 894–898. [Google Scholar]
- Fernandez-Real, J.M.; Gutierrez, C.; Ricart, W.; Castineira, M.J.; Vendrell, J.; Richart, C. Plasma levels of the soluble fraction of tumor necrosis factor receptors 1 and 2 are independent determinants of plasma cholesterol and LDL-cholesterol concentrations in healthy subjects. Atherosclerosis 1999, 146, 321–327. [Google Scholar]
- Skoog, T.; Dichtl, W.; Boquist, S.; Skoglund-Andersson, C.; Karpe, F.; Tang, R.; Bond, M.G.; de Faire, U.; Nilsson, J.; Eriksson, P.; et al. Plasma tumour necrosis factor-alpha and early carotid atherosclerosis in healthy middle-aged men. Eur. Heart J 2002, 23, 376–383. [Google Scholar]
- Mizia-Stec, K.; Zahorska-Markiewicz, B.; Mandecki, T.; Janowska, J.; Szulc, A.; Jastrzekbska-Maj, E.; Gasior, Z. Hyperlipidaemias and serum cytokines in patients with coronary artery disease. Acta Cardiol 2003, 58, 9–15. [Google Scholar]
- Saez, Y.; Vacas, M.; Santos, M.; Saez de Lafuente, J.P.; Sagastagoitia, J.D.; Molinero, E.; Iriarte, J.A. Relation of high-density lipoprotein cholesterol and apoprotein A1 levels with presence and severity of coronary obstruction. ISRN Vasc. Med 2012, 2012, 1–5. [Google Scholar]
- Ettinger, W.H.; Miller, L.A.; Smith, T.K.; Parks, J.S. Effect of interleukin-1 alpha on lipoprotein lipids in cynomolgus monkeys (comparison to tumor necrosis factor). Biochim. Biophys. Acta 1992, 1128, 186–192. [Google Scholar]
- Ettinger, W.H.; Miller, L.D.; Albers, J.J.; Smith, T.K.; Parks, J.S. Lipopolysaccharide and tumor necrosis factor cause a fall in plasma concentration of lecithin (cholesterol acyltransferase in cynomolgus monkeys). J. Lipid Res 1990, 31, 1099–1107. [Google Scholar]
- Memon, R.A.; Grunfeld, C.; Moser, A.H.; Feingold, K.R. Tumor necrosis factor mediates the effects of endotoxin on cholesterol and triglyceride metabolism in mice. Endocrinology 1993, 132, 2246–2253. [Google Scholar]
- Feingold, K.R.; Pollock, A.S.; Moser, A.H.; Shigenaga, J.K.; Grunfeld, C. Discordant regulation of proteins of cholesterol metabolism during the acute phase response. J. Lipid Res 1995, 36, 1474–1482. [Google Scholar]
- Ruan, X.Z.; Varghese, Z.; Fernando, R.; Moorhead, J.F. Cytokine regulation of low-density lipoprotein receptor gene transcription in human mesangial cells. Nephrol. Dial. Transplant 1998, 13, 1391–1397. [Google Scholar]
- Ruan, X.Z.; Varghese, Z.; Powis, S.H.; Moorhead, J.F. Dysregulation of LDL receptor under the influence of inflammatory cytokines (a new pathway for foam cell formation). Kidney Int 2001, 60, 1716–1725. [Google Scholar]
- Ruan, X.Z.; Moorhead, J.F.; Fernando, R.; Wheeler, D.C.; Powis, S.H.; Varghese, Z. Regulation of lipoprotein trafficking in the kidney (role of inflammatory mediators and transcription factors). Biochem. Soc. Trans 2004, 32, 88–91. [Google Scholar]
- Bartolome, N.; Rodriguez, L.; Martinez, M.J.; Ochoa, B.; Chico, Y. Upregulation of apolipoprotein B secretion, but not lipid, by tumor necrosis factor-alpha in rat hepatocyte cultures in the absence of extracellular fatty acids. Ann. N. Y. Acad. Sci 2007, 1096, 55–69. [Google Scholar]
- Homma, Y. Predictors of atherosclerosis. J. Atheroscler. Thromb 2004, 11, 265–270. [Google Scholar]
- Crowl, R.M.; Stoller, T.J.; Conroy, R.R.; Stoner, C.R. Induction of phospholipase A2 gene expression in human hepatoma cells by mediators of the acute phase response. J. Biol. Chem 1991, 266, 2647–2651. [Google Scholar]
- Kugiyama, K.; Ota, Y.; Takazoe, K.; Moriyama, Y.; Kawano, H.; Miyao, Y.; Sakamoto, T.; Soejima, H.; Ogawa, H.; Doi, H.; et al. Circulating levels of secretory type II phospholipase A(2) predict coronary events in patients with coronary artery disease. Circulation 1999, 100, 1280–1284. [Google Scholar]
- O’Brien, K.D.; Chait, A. Serum amyloid A: The “other” inflammatory protein. Curr. Atheroscler. Rep 2006, 8, 62–68. [Google Scholar]
- Poitou, C.; Viguerie, N.; Cancello, R.; de Matteis, R.; Cinti, S.; Stich, V.; Coussieu, C.; Gauthier, E.; Courtine, M.; Zucker, J.D.; et al. Serum amyloid A: Production by human white adipocyte and regulation by obesity and nutrition. Diabetologia 2005, 48, 519–528. [Google Scholar]
- Fasshauer, M.; Klein, J.; Kralisch, S.; Klier, M.; Lossner, U.; Bluher, M.; Paschke, R. Serum amyloid A3 expression is stimulated by dexamethasone and interleukin-6 in 3T3–L1 adipocytes. J. Endocrinol 2004, 183, 561–567. [Google Scholar]
- Yang, R.Z.; Lee, M.J.; Hu, H.; Pollin, T.I.; Ryan, A.S.; Nicklas, B.J.; Snitker, S.; Horenstein, R.B.; Hull, K.; Goldberg, N.H.; et al. Acute-phase serum amyloid A: An inflammatory adipokine and potential link between obesity and its metabolic complications. PLoS Med 2006, 3, e287. [Google Scholar]
- Chen, C.H.; Wang, P.H.; Liu, B.H.; Hsu, H.H.; Mersmann, H.J.; Ding, S.T. Serum amyloid A protein regulates the expression of porcine genes related to lipid metabolism. J. Nutr 2008, 138, 674–679. [Google Scholar]
- Cai, L.; de Beer, M.C.; de Beer, F.C.; van der Westhuyzen, D.R. Serum amyloid A is a ligand for scavenger receptor class B type I and inhibits high density lipoprotein binding and selective lipid uptake. J. Biol. Chem 2005, 280, 2954–2961. [Google Scholar]
- Rigotti, A.; Miettinen, H.E.; Krieger, M. The role of the high-density lipoprotein receptor SR-BI in the lipid metabolism of endocrine and other tissues. Endocr. Rev 2003, 24, 357–387. [Google Scholar]
- Lewis, K.E.; Kirk, E.A.; McDonald, T.O.; Wang, S.; Wight, T.N.; O’Brien, K.D.; Chait, A. Increase in serum amyloid a evoked by dietary cholesterol is associated with increased atherosclerosis in mice. Circulation 2004, 110, 540–545. [Google Scholar]
- Ouchi, N.; Kihara, S.; Arita, Y.; Maeda, K.; Kuriyama, H.; Okamoto, Y.; Hotta, K.; Nishida, M.; Takahashi, M.; Nakamura, T.; et al. Novel modulator for endothelial adhesion molecules: Adipocyte-derived plasma protein adiponectin. Circulation 1999, 100, 2473–2476. [Google Scholar]
- Matsubara, M.; Maruoka, S.; Katayose, S. Decreased plasma adiponectin concentrations in women with dyslipidemia. J. Clin. Endocrinol. Metab 2002, 87, 2764–2769. [Google Scholar]
- Okamoto, Y.; Kihara, S.; Funahashi, T.; Matsuzawa, Y.; Libby, P. Adiponectin: A key adipocytokine in metabolic syndrome. Clin. Sci. (Lond.) 2006, 110, 267–278. [Google Scholar]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med 2002, 8, 1288–1295. [Google Scholar]
- Berneis, K.K.; Krauss, R.M. Metabolic origins and clinical significance of LDL heterogeneity. J. Lipid Res 2002, 43, 1363–1379. [Google Scholar]
- Schneider, J.G.; von Eynatten, M.; Schiekofer, S.; Nawroth, P.P.; Dugi, K.A. Low plasma adiponectin levels are associated with increased hepatic lipase activityin vivo. Diabetes Care 2005, 28, 2181–2186. [Google Scholar]
- Matsuura, F.; Oku, H.; Koseki, M.; Sandoval, J.C.; Yuasa-Kawase, M.; Tsubakio-Yamamoto, K.; Masuda, D.; Maeda, N.; Tsujii, K.; Ishigami, M.; et al. Adiponectin accelerates reverse cholesterol transport by increasing high density lipoprotein assembly in the liver. Biochem. Biophys. Res. Commun 2007, 358, 1091–1095. [Google Scholar]
- Oku, H.; Matsuura, F.; Koseki, M.; Sandoval, J.C.; Yuasa-Kawase, M.; Tsubakio-Yamamoto, K.; Masuda, D.; Maeda, N.; Ohama, T.; Ishigami, M.; et al. Adiponectin deficiency suppresses ABCA1 expression and ApoA-I synthesis in the liver. FEBS Lett 2007, 581, 5029–5033. [Google Scholar]
- Chang, C.Y.; Chen, M.J.; Yang, W.S.; Yeh, C.Y.; Ho, H.N.; Chen, S.U.; Yang, Y.S. Hypoadiponectinemia: A useful marker of dyslipidemia in women with polycystic ovary syndrome. Taiwan J. Obstet. Gynecol 2012, 51, 583–590. [Google Scholar]
- Angulo, P. Nonalcoholic fatty liver disease. N. Engl. J. Med 2002, 346, 1221–1231. [Google Scholar]
- Starley, B.Q.; Calcagno, C.J.; Harrison, S.A. Nonalcoholic fatty liver disease and hepatocellular carcinoma: A weighty connection. Hepatology 2010, 51, 1820–1832. [Google Scholar]
- Lazo, M.; Clark, J.M. The epidemiology of nonalcoholic fatty liver disease: A global perspective. Semin Liver Dis 2008, 28, 339–350. [Google Scholar]
- Tarantino, G.; Savastano, S.; Colao, A. Hepatic steatosis, low-grade chronic inflammation and hormone/growth factor/adipokine imbalance. World J. Gastroenterol 2010, 16, 4773–4783. [Google Scholar]
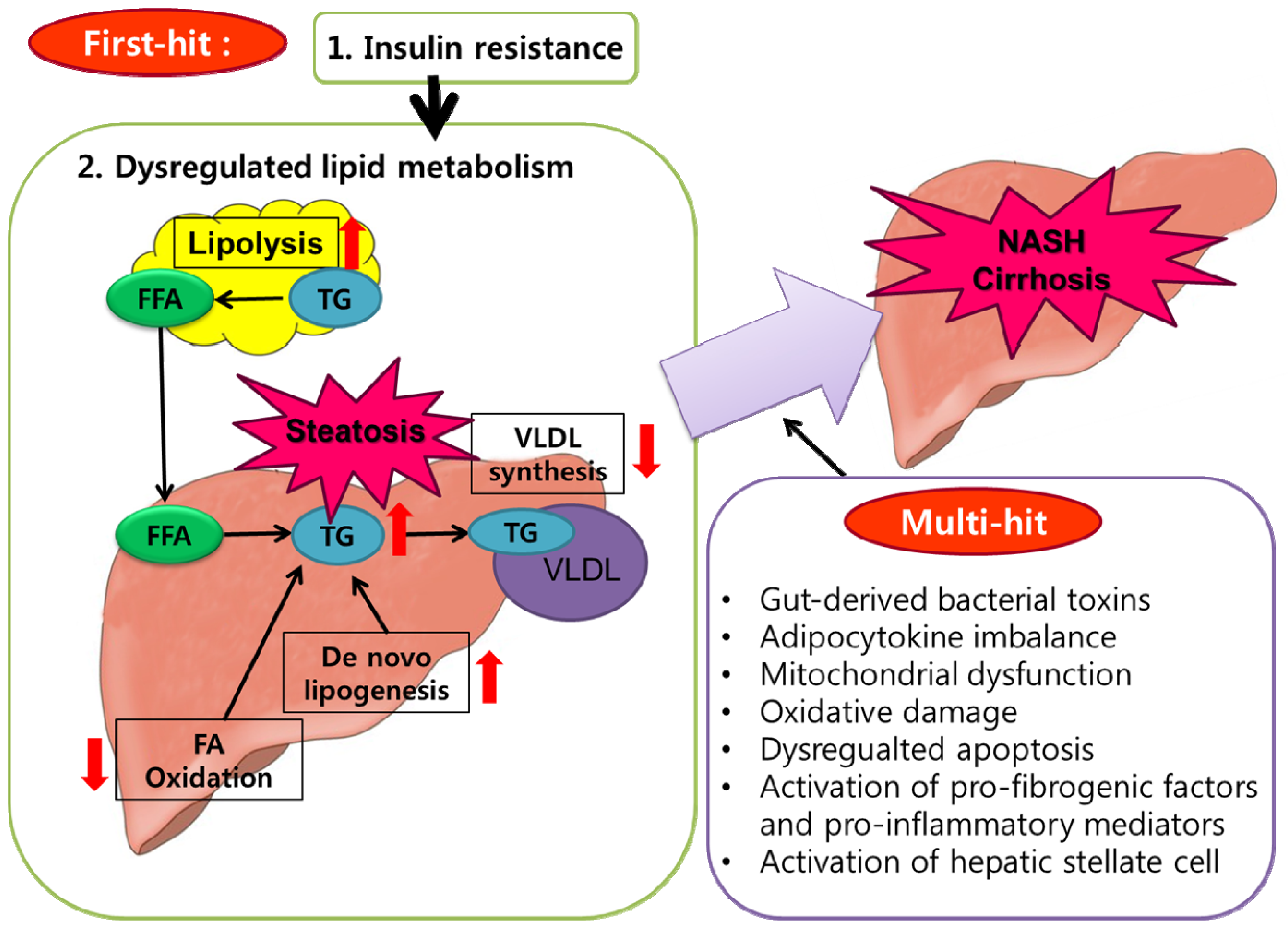
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar]
- Polyzos, S.A.; Kountouras, J.; Zavos, C.; Deretzi, G. Nonalcoholic fatty liver disease: Multimodal treatment options for a pathogenetically multiple-hit disease. J. Clin. Gastroenterol 2012, 46, 272–284. [Google Scholar]
- Bradbury, M.W.; Berk, P.D. Lipid metabolism in hepatic steatosis. Clin. Liver Dis 2004, 8, 639–671. [Google Scholar]
- Koteish, A.; Diehl, A.M. Animal models of steatosis. Semin. Liver Dis 2001, 21, 89–104. [Google Scholar]
- Lambert, J.E.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic Fatty liver disease. Gastroenterology 2014, 146, 726–735. [Google Scholar]
- Nielsen, S.; Guo, Z.; Johnson, C.M.; Hensrud, D.D.; Jensen, M.D. Splanchnic lipolysis in human obesity. J. Clin. Investig 2004, 113, 1582–1588. [Google Scholar]
- Heilbronn, L.; Smith, S.R.; Ravussin, E. Failure of fat cell proliferation, mitochondrial function and fat oxidation results in ectopic fat storage, insulin resistance and type II diabetes mellitus. Int. J. Obes. Relat. Metab. Disord 2004, 28, S12–S21. [Google Scholar]
- Roden, M. Mechanisms of disease: Hepatic steatosis in type 2 diabetes—Pathogenesis and clinical relevance. Nat. Clin. Pract. Endocrinol. Metab 2006, 2, 335–348. [Google Scholar]
- Montague, C.T.; O’Rahilly, S. The perils of portliness: Causes and consequences of visceral adiposity. Diabetes 2000, 49, 883–888. [Google Scholar]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig 2005, 115, 1343–1351. [Google Scholar]
- Parks, E.J.; Hellerstein, M.K. Thematic review series: Patient-oriented research. Recent advances in liver triacylglycerol and fatty acid metabolism using stable isotope labeling techniques. J. Lipid Res 2006, 47, 1651–1660. [Google Scholar]
- Harrison, S.A.; Day, C.P. Benefits of lifestyle modification in NAFLD. Gut 2007, 56, 1760–1769. [Google Scholar]
- Shklyaev, S.; Aslanidi, G.; Tennant, M.; Prima, V.; Kohlbrenner, E.; Kroutov, V.; Campbell-Thompson, M.; Crawford, J.; Shek, E.W.; Scarpace, P.J.; et al. Sustained peripheral expression of transgene adiponectin offsets the development of diet-induced obesity in rats. Proc. Natl. Acad. Sci. USA 2003, 100, 14217–14222. [Google Scholar]
- Xu, A.; Wang, Y.; Keshaw, H.; Xu, L.Y.; Lam, K.S.; Cooper, G.J. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J. Clin. Investig 2003, 112, 91–100. [Google Scholar] [Green Version]
- Schindhelm, R.K.; Diamant, M.; Dekker, J.M.; Tushuizen, M.E.; Teerlink, T.; Heine, R.J. Alanine aminotransferase as a marker of non-alcoholic fatty liver disease in relation to type 2 diabetes mellitus and cardiovascular disease. Diabetes Metab. Res. Rev 2006, 22, 437–443. [Google Scholar]
- Masaki, T.; Chiba, S.; Tatsukawa, H.; Yasuda, T.; Noguchi, H.; Seike, M.; Yoshimatsu, H. Adiponectin protects LPS-induced liver injury through modulation of TNF-alpha in KK-Ay obese mice. Hepatology 2004, 40, 177–184. [Google Scholar]
- Kamada, Y.; Tamura, S.; Kiso, S.; Matsumoto, H.; Saji, Y.; Yoshida, Y.; Fukui, K.; Maeda, N.; Nishizawa, H.; Nagaretani, H.; et al. Enhanced carbon tetrachloride-induced liver fibrosis in mice lacking adiponectin. Gastroenterology 2003, 125, 1796–1807. [Google Scholar]
- Hui, J.M.; Hodge, A.; Farrell, G.C.; Kench, J.G.; Kriketos, A.; George, J. Beyond insulin resistance in NASH: TNF-alpha or adiponectin? Hepatology 2004, 40, 46–54. [Google Scholar]
- López-Bermejo, A.; Botas, P.; Funahashi, T.; Delgado, E.; Kihara, S.; Ricart, W.; Fernández-Real, J.M. Adiponectin, hepatocellular dysfunction and insulin sensitivity. Clin. Endocrinol. (Oxf.) 2004, 60, 256–263. [Google Scholar]
- Targher, G.; Bertolini, L.; Scala, L.; Poli, F.; Zenari, L.; Falezza, G. Decreased plasma adiponectin concentrations are closely associated with nonalcoholic hepatic steatosis in obese individuals. Clin. Endocrinol. (Oxf.) 2004, 61, 700–703. [Google Scholar]
- Kaser, S.; Moschen, A.; Cayon, A.; Kaser, A.; Crespo, J.; Pons-Romero, F.; Ebenbichler, C.F.; Patsch, J.R.; Tilg, H. Adiponectin and its receptors in non-alcoholic steatohepatitis. Gut 2005, 54, 117–121. [Google Scholar]
- Stefan, N.; Machicao, F.; Staiger, H.; Machann, J.; Schick, F.; Tschritter, O.; Spieth, C.; Weigert, C.; Fritsche, A.; Stumvoll, M.; et al. Polymorphisms in the gene encoding adiponectin receptor 1 are associated with insulin resistance and high liver fat. Diabetologia 2005, 48, 2282–2291. [Google Scholar]
- Minokoshi, Y.; Kim, Y.B.; Peroni, O.D.; Fryer, L.G.; Muller, C.; Carling, D.; Kahn, B.B. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 2002, 415, 339–343. [Google Scholar]
- Kakuma, T.; Lee, Y.; Higa, M.; Wang, Z.W.; Pan, W.; Shimomura, I.; Unger, R.H. Leptin, troglitazone, and the expression of sterol regulatory element binding proteins in liver and pancreatic islets. Proc. Natl. Acad. Sci. USA 2000, 18, 8536–8441. [Google Scholar]
- Lee, Y.; Yu, X.; Gonzales, F.; Mangelsdorf, D.J.; Wang, M.Y.; Richardson, C.; Witters, L.A.; Unger, R.H. PPAR alpha is necessary for the lipogenic action of hyperleptinemia on white adipose and liver tissue. Proc. Natl. Acad. Sci. USA 2002, 99, 11848–11853. [Google Scholar]
- Serin, E.; Ozer, B.; Gümürdülü, Y.; Kayaselçuk, F.; Kul, K.; Boyacioğlu, S. Serum leptin level can be a negative marker of hepatocyte damage in nonalcoholic fatty liver. J. Gastroenterol 2003, 38, 471–476. [Google Scholar]
- Poordad, F.F. The role of leptin in NAFLD contender or pretender? J. Clin. Gastroenterol 2004, 38, 841–843. [Google Scholar]
- Tobe, K.; Ogura, T.; Tsukamoto, C.; Imai, A.; Matsuura, K.; Iwasaki, Y.; Shimomura, H.; Higashi, T.; Tsuji, T. Relationship between serum leptin and fatty liver in Japanese male adolescent university students. Am. J. Gastroenterol 1999, 94, 3328–3335. [Google Scholar]
- Marra, F. Leptin and liver fibrosis: A matter of fat. Gastroenterology 2002, 122, 1529–1532. [Google Scholar]
- Cao, Q.; Mak, K.M.; Ren, C.; Lieber, C.S. Leptin stimulates tissue inhibitor of metalloproteinase-1 in human hepatic stellate cells: Respective roles of the JAK/STAT and JAK-mediated H2O2-dependent MAPK pathways. J. Biol. Chem 2004, 279, 4292–4304. [Google Scholar]
- Saxena, N.K.; Titus, M.A.; Ding, X.; Floyd, J.; Srinivasan, S.; Sitaraman, S.V.; Anania, F.A. Leptin as a novel profibrogenic cytokine in hepatic stellate cells: Mitogenesis and inhibition of apoptosis mediated by extracellular regulated kinase (Erk) and Akt phosphorylation. FASEB J 2004, 18, 1612–1614. [Google Scholar]
- Banerjee, R.R.; Lazar, M.A. Resistin: Molecular history and prognosis. J. Mol. Med 2003, 81, 218–226. [Google Scholar]
- Pagano, C.; Soardo, G.; Pilon, C.; Milocco, C.; Basan, L.; Milan, G.; Donnini, D.; Faggian, D.; Mussap, M.; Plebani, M.; et al. Increased serum resistin in nonalcoholic fatty liver disease is related to liver disease severity and not to insulin resistance. J. Clin. Endocrinol. Metab 2006, 91, 1081–1086. [Google Scholar]
- Crespo, J.; Cayon, A.; Fernandez-Gil, P.; Hernández-Guerra, M.; Mayorga, M.; Domínguez-Díez, A.; Fernández-Escalante, J.C.; Pons-Romero, F. Gene expression of tumor necrosis factor alpha and TNF-receptors, p55 and p75, in nonalcoholic steatohepatitis patients. Hepatology 2001, 34, 1158–1163. [Google Scholar]
- Ding, W.X.; Yin, X.M. Dissection of the multiple mechanisms of TNF-alpha-induced apoptosis in liver injury. J. Cell. Mol. Med 2004, 8, 445–454. [Google Scholar]
- Manco, M.; Marcellini, M.; Giannone, G.; Nobili, V. Correlation of serum TNF-alpha levels and histologic liver injury scores in pediatric nonalcoholic fatty liver disease. Am. J. Clin. Pathol 2007, 127, 954–960. [Google Scholar]
- Sanal, M.G. The blind men see the elephant-the many faces of fatty liver disease. World J. Gastroenterol 2008, 14, 831–844. [Google Scholar]
- Saleh, J.; Christou, N.; Cianflone, K. Regional specificity of ASP binding in human adipose tissue. Am. J. Physiol 1999, 276, E815–E821. [Google Scholar]
- Massiera, F.; Bloch-Faure, M.; Ceiler, D.; Murakami, K.; Fukamizu, A.; Gasc, J.M.; Quignard-Boulange, A.; Negrel, R.; Ailhaud, G.; Seydoux, J.; et al. Adipose angiotensinogen is involved in adipose tissue growth and blood pressure regulation. FASEB J 2001, 15, 2727–2729. [Google Scholar]
- Umemura, S.; Nyui, N.; Tamura, K.; Hibi, K.; Yamaguchi, S.; Nakamaru, M.; Ishigami, T.; Yabana, M.; Kihara, M.; Inoue, S.; et al. Plasma angiotensinogen concentrations in obese patients. Am. J. Hypertens 1997, 10, 629–633. [Google Scholar]
- Dixon, J.B.; Bhathal, P.S.; Jonsson, J.R.; Dixon, A.F.; Powell, E.E.; O’Brien, P.E. Pro-fibrotic polymorphisms predictive of advanced liver fibrosis in the severely obese. J. Hepatol 2003, 39, 967–971. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jung, U.J.; Choi, M.-S. Obesity and Its Metabolic Complications: The Role of Adipokines and the Relationship between Obesity, Inflammation, Insulin Resistance, Dyslipidemia and Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2014, 15, 6184-6223. https://doi.org/10.3390/ijms15046184
Jung UJ, Choi M-S. Obesity and Its Metabolic Complications: The Role of Adipokines and the Relationship between Obesity, Inflammation, Insulin Resistance, Dyslipidemia and Nonalcoholic Fatty Liver Disease. International Journal of Molecular Sciences. 2014; 15(4):6184-6223. https://doi.org/10.3390/ijms15046184
Chicago/Turabian StyleJung, Un Ju, and Myung-Sook Choi. 2014. "Obesity and Its Metabolic Complications: The Role of Adipokines and the Relationship between Obesity, Inflammation, Insulin Resistance, Dyslipidemia and Nonalcoholic Fatty Liver Disease" International Journal of Molecular Sciences 15, no. 4: 6184-6223. https://doi.org/10.3390/ijms15046184