Cardiac Fas-Dependent and Mitochondria-Dependent Apoptosis after Chronic Cocaine Abuse




Abstract
:1. Introduction
2. Results and Discussion
2.1. Body Weight and Cardiac Characteristics
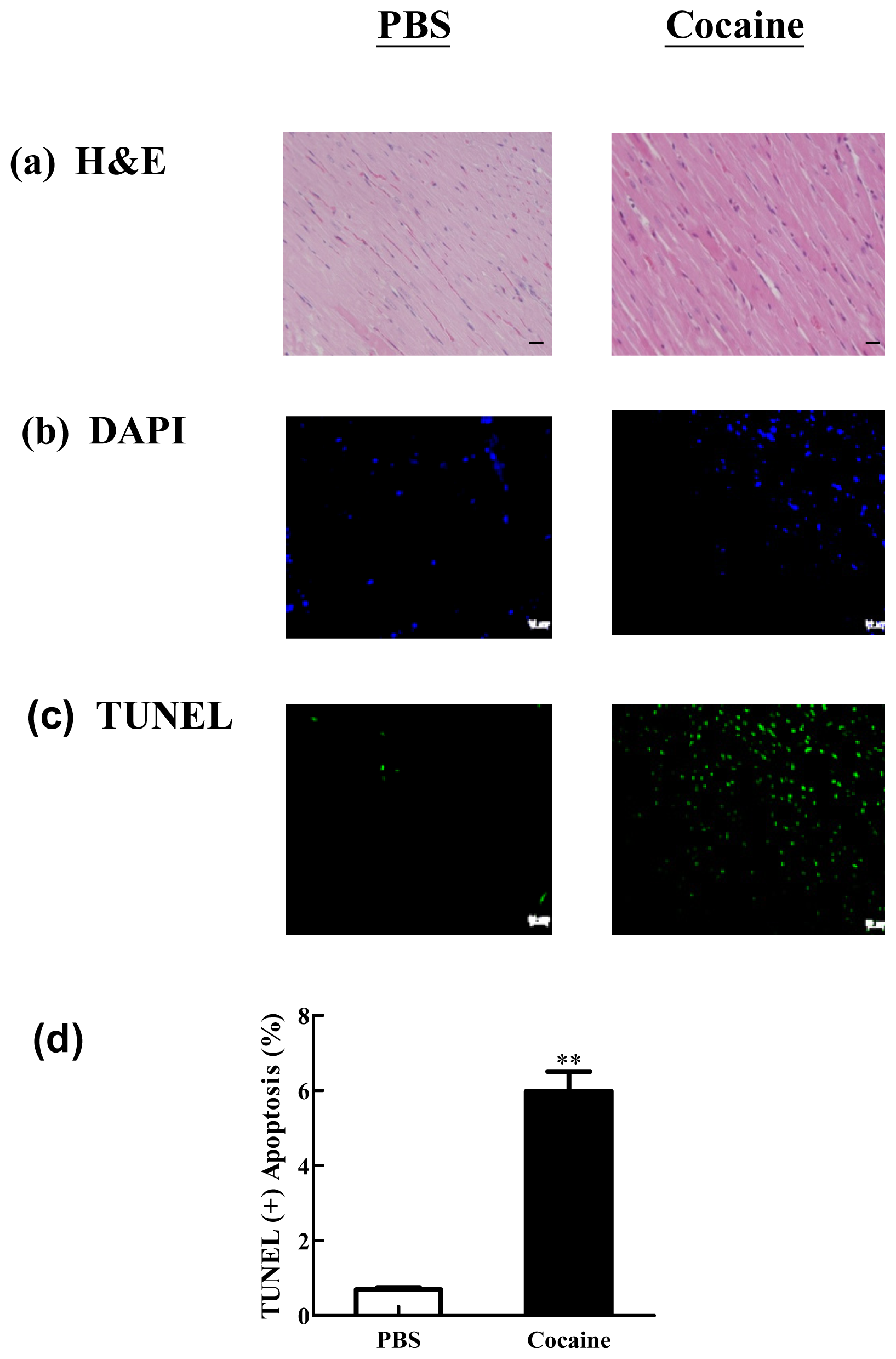
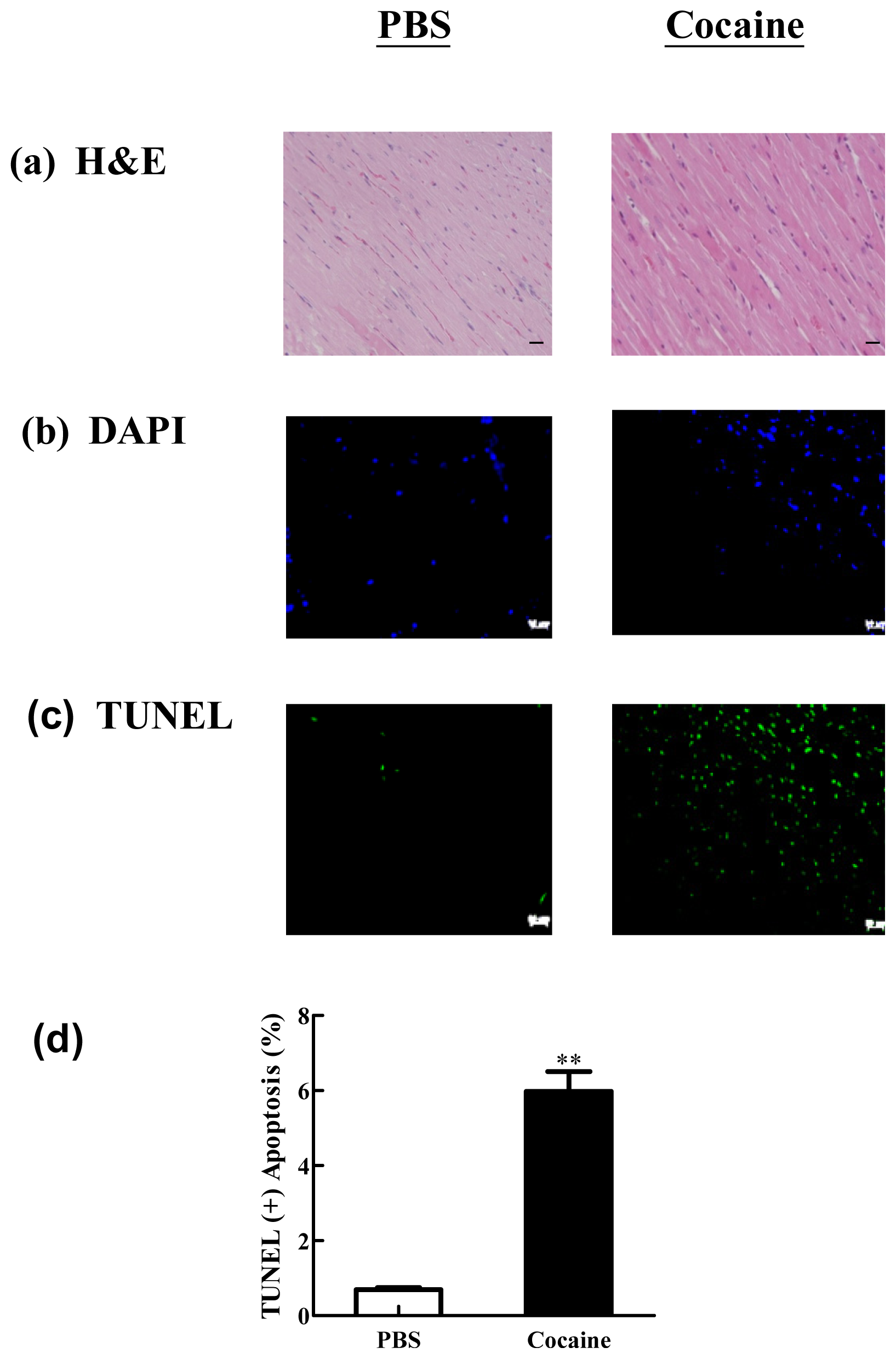
2.2. Cardiac Histopathological and Apoptotic Cells Changes
2.3. Upstream Components of Cardiac Fas Receptor Dependent Apoptotic Pathways
2.4. Main Intracellular Molecule Signaling Mediator from Fas to Mitochondrial Pathway
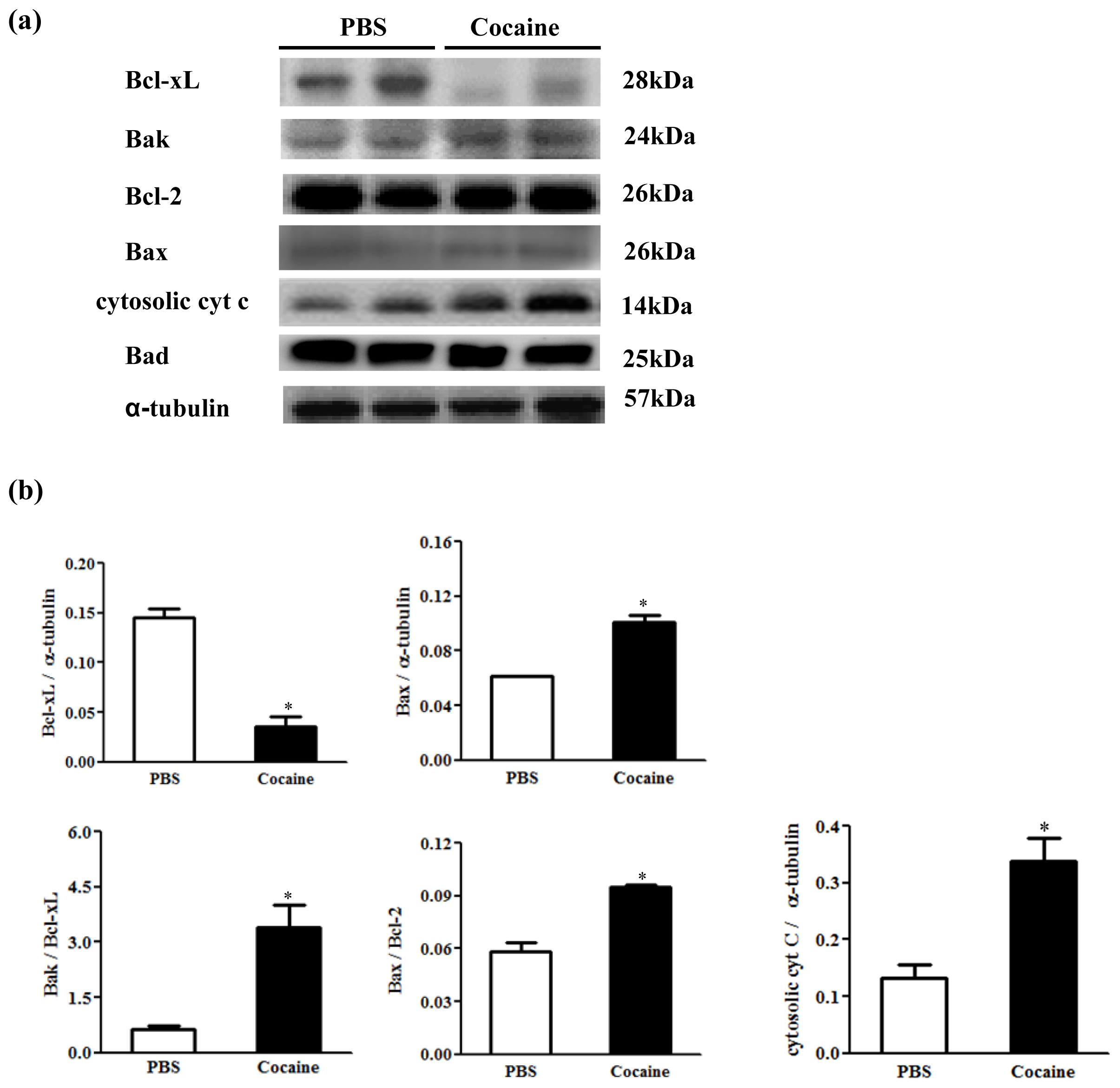
2.5. Upstream Components of Cardiac Mitochondria-Dependent Apoptotic Pathways
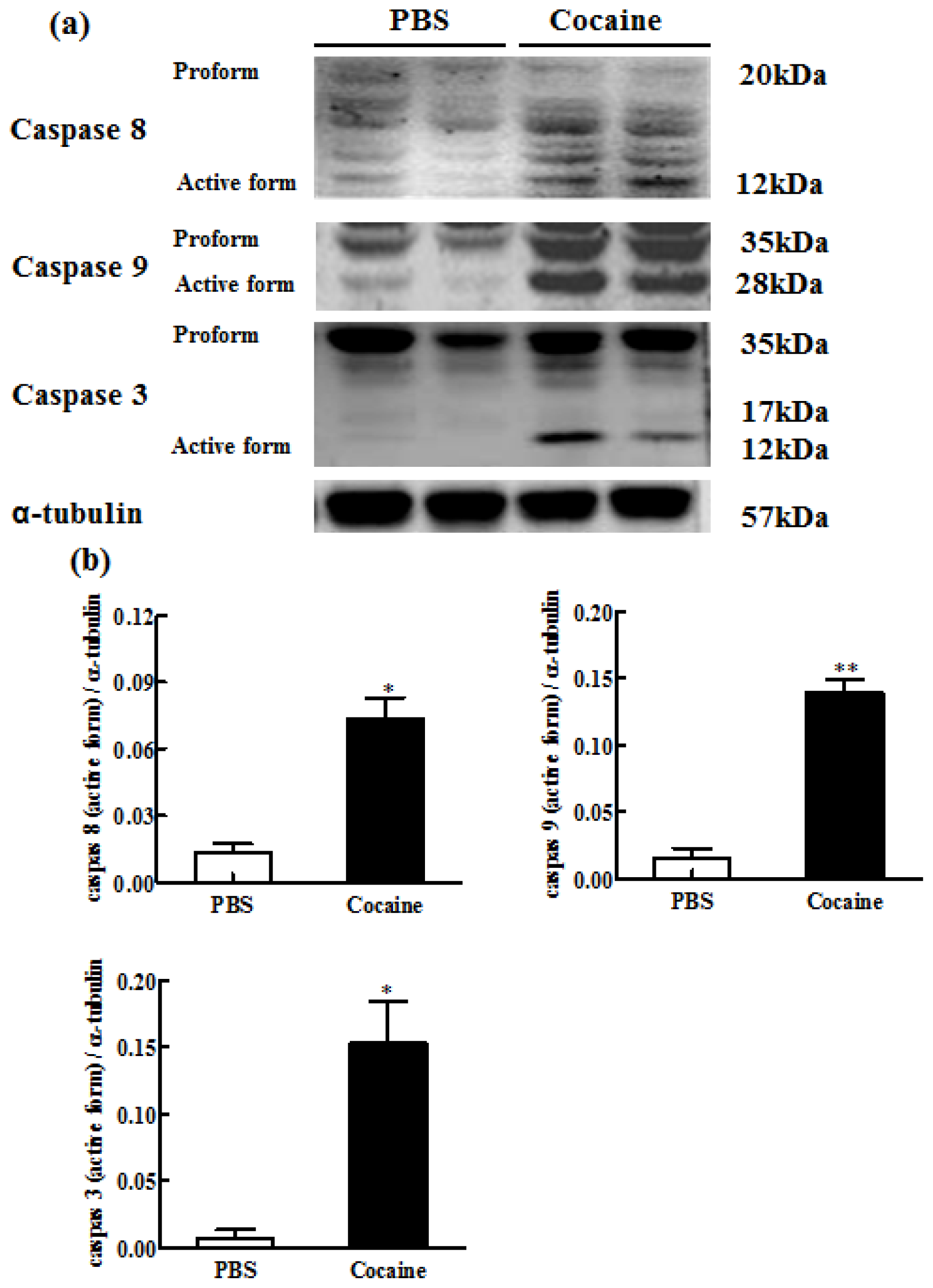
2.6. Downstream Components of Cardiac Fas-Dependent and Mitochondria-Dependent Apoptotic Pathways
3. Experimental Section
3.1. Animals
3.2. Chronic Cocaine Exposure
3.3. Cardiac Characteristics
3.4. Hematoxylin-Eosin Staining
3.5. Terminal Deoxynucleotidyl Transferase Biotin-dUTP Nick End Labeling (TUNEL) Assay and 4′,6-Diamidino-2-phenylindole (DAPI) Staining
3.6. Tissue Extraction
3.7. Separation of Cytosolic and Mitochondrial Fractions
3.8. Electrophoresis and Western Blot
3.9. Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| H&E | Hematoxylin and eosin |
| DAPI | 4′,6-diamidino-2-phenylindole |
| TUNEL | Terminal deoxynucleotidyltransferase UTP Nick End Labeling |
| PBS | phosphate buffered saline, vehicle-treated group |
| Cocaine | cocaine-treated group |
| TNF-α | Tumor necrosis factor-alpha |
| Fas | Fas death receptor |
| Fas L | Fas ligand |
| FADD | Fas-associated death domain |
| Bid | Bcl-2 homology domain 3 (BH3) interacting domain death agonist |
| t-Bid | truncated Bid |
| Bcl-xL | B-cell lymphoma-extra large |
| Bak | Bcl-2 homologous antagonist/killer |
| Bcl-2 | B-cell lymphoma 2 |
| Bax | Bcl-2–associated X protein |
| Cyt c | cytochrome c |
| Bad | Bcl-2–associated death promoter |
| Cas | caspase |
References
- Zhang, L.; Xiao, Y.; He, J. Cocaine and apoptosis in myocardial cells. Anat. Rec 1999, 257, 208–216. [Google Scholar]
- Schwartz, B.G.; Rezkalla, S.; Kloner, R.A. Cardiovascular effects of cocaine. Circulation 2010, 122, 2558–2569. [Google Scholar]
- Phillips, K.; Luk, A.; Soor, G.S.; Abraham, J.R.; Leong, S.; Butany, J. Cocaine cardiotoxicity: A review of the pathophysiology, pathology, and treatment options. Am. J. Cardiovasc. Drugs 2009, 9, 177–196. [Google Scholar]
- Knuepfer, M.M. Cardiovascular disorders associated with cocaine use: Myths and truths. Pharmacol. Ther 2003, 97, 181–222. [Google Scholar]
- Brickner, M.E.; Willard, J.E.; Eichhorn, E.J.; Black, J.; Grayburn, P.A. Left ventricular hypertrophy associated with chronic cocaine abuse. Circulation 1991, 84, 1130–1135. [Google Scholar]
- Wiener, R.S.; Lockhart, J.T.; Schwartz, R.G. Dilated cardiomyopathy and cocaine abuse. Report of two cases. Am. J. Med 1986, 81, 699–701. [Google Scholar]
- Bertolet, B.D.; Freund, G.; Martin, C.A.; Perchalski, D.L.; Williams, C.M.; Pepine, C.J. Unrecognized left ventricular dysfunction in an apparently healthy cocaine abuse population. Clin. Cardiol 1990, 13, 323–328. [Google Scholar]
- Chokshi, S.K.; Moore, R.; Pandian, N.G.; Isner, J.M. Reversible cardiomyopathy associated with cocaine intoxication. Ann. Int. Med 1989, 111, 1039–1040. [Google Scholar]
- Haunstetter, A.; Izumo, S. Apoptosis: Basic mechanisms and implications for cardiovascular disease. Circ. Res 1998, 82, 1111–1129. [Google Scholar]
- Lee, S.D.; Chu, C.H.; Huang, E.J.; Lu, M.C.; Liu, J.Y.; Liu, C.J.; Hsu, H.H.; Lin, J.A.; Kuo, W.W.; Huang, C.Y. Roles of insulin-like growth factor II in cardiomyoblast apoptosis and in hypertensive rat heart with abdominal aorta ligation. Am. J. Physiol. Endocrinol. Metab 2006, 291, E306–E314. [Google Scholar]
- Narula, J.; Pandey, P.; Arbustini, E.; Haider, N.; Narula, N.; Kolodgie, F.D.; dal Bello, B.; Semigran, M.J.; Bielsa-Masdeu, A.; Dec, G.W.; et al. Apoptosis in heart failure: Release of cytochrome c from mitochondria and activation of caspase-3 in human cardiomyopathy. Proc. Natl. Acad. Sci. USA 1999, 96, 8144–8149. [Google Scholar]
- Narula, J.; Haider, N.; Arbustini, E.; Chandrashekhar, Y. Mechanisms of disease: Apoptosis in heart failure—Seeing hope in death. Nat. Clin. Pract. Cardiovasc. Med 2006, 3, 681–688. [Google Scholar]
- Sinha-Hikim, I.; Shen, R.; Nzenwa, I.; Gelfand, R.; Mahata, S.K.; Sinha-Hikim, A.P. Minocycline suppresses oxidative stress and attenuates fetal cardiac myocyte apoptosis triggered by in utero cocaine exposure. Apoptosis 2011, 16, 563–573. [Google Scholar]
- Bishopric, N.H.; Andreka, P.; Slepak, T.; Webster, K.A. Molecular mechanisms of apoptosis in the cardiac myocyte. Curr. Opin. Pharmacol 2001, 1, 141–150. [Google Scholar]
- Barnhart, B.C.; Alappat, E.C.; Peter, M.E. The CD95 type I/type II model. Semin. Immunol 2003, 15, 185–193. [Google Scholar]
- Bang, S.; Jeong, E.J.; Kim, I.K.; Jung, Y.K.; Kim, K.S. Fas- and tumor necrosis factor-mediated apoptosis uses the same binding surface of FADD to trigger signal transduction. A typical model for convergent signal transduction. J. Biol. Chem 2000, 275, 36217–36222. [Google Scholar]
- Jeremias, I.; Stahnke, K.; Debatin, K.M. CD95/Apo-1/Fas: Independent cell death induced by doxorubicin in normal cultured cardiomyocytes. Cancer Immunol. Immunother 2005, 54, 655–662. [Google Scholar]
- Adams, J.M.; Cory, S. Life-or-death decisions by the Bcl-2 protein family. Trends Biochem. Sci 2001, 26, 61–66. [Google Scholar]
- Kubasiak, L.A.; Hernandez, O.M.; Bishopric, N.H.; Webster, K.A. Hypoxia and acidosis activate cardiac myocyte death through the Bcl-2 family protein BNIP3. Proc. Natl. Acad. Sci. USA 2002, 99, 12825–12830. [Google Scholar]
- Brown, G.C.; Borutaite, V. Nitric oxide, cytochrome c and mitochondria. Biochem. Soc. Symp 1999, 66, 17–25. [Google Scholar]
- Aggarwal, B.B.; Bhardwaj, U.; Takada, Y. Regulation of TRAIL-induced apoptosis by ectopic expression of antiapoptotic factors. Vitam. Horm 2004, 67, 453–483. [Google Scholar]
- Gross, A.; Yin, X.M.; Wang, K.; Wei, M.C.; Jockel, J.; Milliman, C.; Erdjument-Bromage, H.; Tempst, P.; Korsmeyer, S.J. Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-XL prevents this release but not tumor necrosis factor-R1/Fas death. J. Biol. Chem 1999, 274, 1156–1163. [Google Scholar]
- Lange, R.A.; Hillis, L.D. Cardiovascular complications of cocaine use. N. Engl. J. Med 2001, 345, 351–358. [Google Scholar]
- Xiao, D.; Zhang, L. Upregulation of Bax and Bcl-2 following prenatal cocaine exposure induces apoptosis in fetal rat brain. Int. J. Med. Sci 2008, 5, 295–302. [Google Scholar]
- Wang, J.F.; Ren, X.; DeAngelis, J.; Min, J.; Zhang, Y.; Hampton, T.G.; Amende, I.; Morgan, J.P. Differential patterns of cocaine-induced organ toxicity in murine heart vs. liver. Exp. Biol. Med 2001, 226, 52–60. [Google Scholar]
- Vergeade, A.; Mulder, P.; Vendeville-Dehaudt, C.; Estour, F.; Fortin, D.; Ventura-Clapier, R.; Thuillez, C.; Monteil, C. Mitochondrial impairment contributes to cocaine-induced cardiac dysfunction: Prevention by the targeted antioxidant MitoQ. Free Radic. Biol. Med 2010, 49, 748–756. [Google Scholar]
- Meijer, M.K.; Spruijt, B.M.; van Zutphen, L.F.; Baumans, V. Effect of restraint and injection methods on heart rate and body temperature in mice. Lab. Anim 2006, 40, 382–391. [Google Scholar]
- Virmani, R.; Robinowitz, M.; Smialek, J.E.; Smyth, D.F. Cardiovascular effects of cocaine: An autopsy study of 40 patients. Am. Heart J 1988, 115, 1068–1076. [Google Scholar]
- He, J.; Xiao, Y.; Casiano, C.A.; Zhang, L. Role of mitochondrial cytochrome c in cocaine-induced apoptosis in coronary artery endothelial cells. J. Pharmacol. Exp. Ther 2000, 295, 896–903. [Google Scholar]
- Li, G.; Xiao, Y.; Zhang, L. Cocaine induces apoptosis in fetal rat myocardial cells through the p38 mitogen-activated protein kinase and mitochondrial/cytochrome c pathways. J. Pharmacol. Exp. Ther 2005, 312, 112–119. [Google Scholar]
- Xiao, Y.; He, J.; Gilbert, R.D.; Zhang, L. Cocaine induces apoptosis in fetal myocardial cells through a mitochondria-dependent pathway. J. Pharmacol. Exp. Ther 2000, 292, 8–14. [Google Scholar]
- Liou, C.M.; Tsai, S.C.; Kuo, C.H.; Williams, T.; Ting, H.; Lee, S.D. Chronic methamphetamine exposure induces cardiac fas-dependent and mitochondria-dependent apoptosis. Cardiovasc. Toxicol 2013. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cardiac characteristics | PBS | Cocaine |
|---|---|---|
| Number of animals | 8 | 8 |
| Body weight (BW), gm | 340.70 ± 33.40 | 319.50 ± 11.80 |
| Whole heart weight (wet WHW), gm | 1.51 ± 0.18 | 1.31 ± 0.18 |
| WHW/BW (×104) | 44.76 ± 7.14 | 41.21 ± 5.80 |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liou, C.-M.; Tsai, S.-C.; Kuo, C.-H.; Ting, H.; Lee, S.-D. Cardiac Fas-Dependent and Mitochondria-Dependent Apoptosis after Chronic Cocaine Abuse. Int. J. Mol. Sci. 2014, 15, 5988-6001. https://doi.org/10.3390/ijms15045988
Liou C-M, Tsai S-C, Kuo C-H, Ting H, Lee S-D. Cardiac Fas-Dependent and Mitochondria-Dependent Apoptosis after Chronic Cocaine Abuse. International Journal of Molecular Sciences. 2014; 15(4):5988-6001. https://doi.org/10.3390/ijms15045988
Chicago/Turabian StyleLiou, Cher-Ming, Shiow-Chwen Tsai, Chia-Hua Kuo, Hua Ting, and Shin-Da Lee. 2014. "Cardiac Fas-Dependent and Mitochondria-Dependent Apoptosis after Chronic Cocaine Abuse" International Journal of Molecular Sciences 15, no. 4: 5988-6001. https://doi.org/10.3390/ijms15045988