Cetuximab-Induced MET Activation Acts as a Novel Resistance Mechanism in Colon Cancer Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
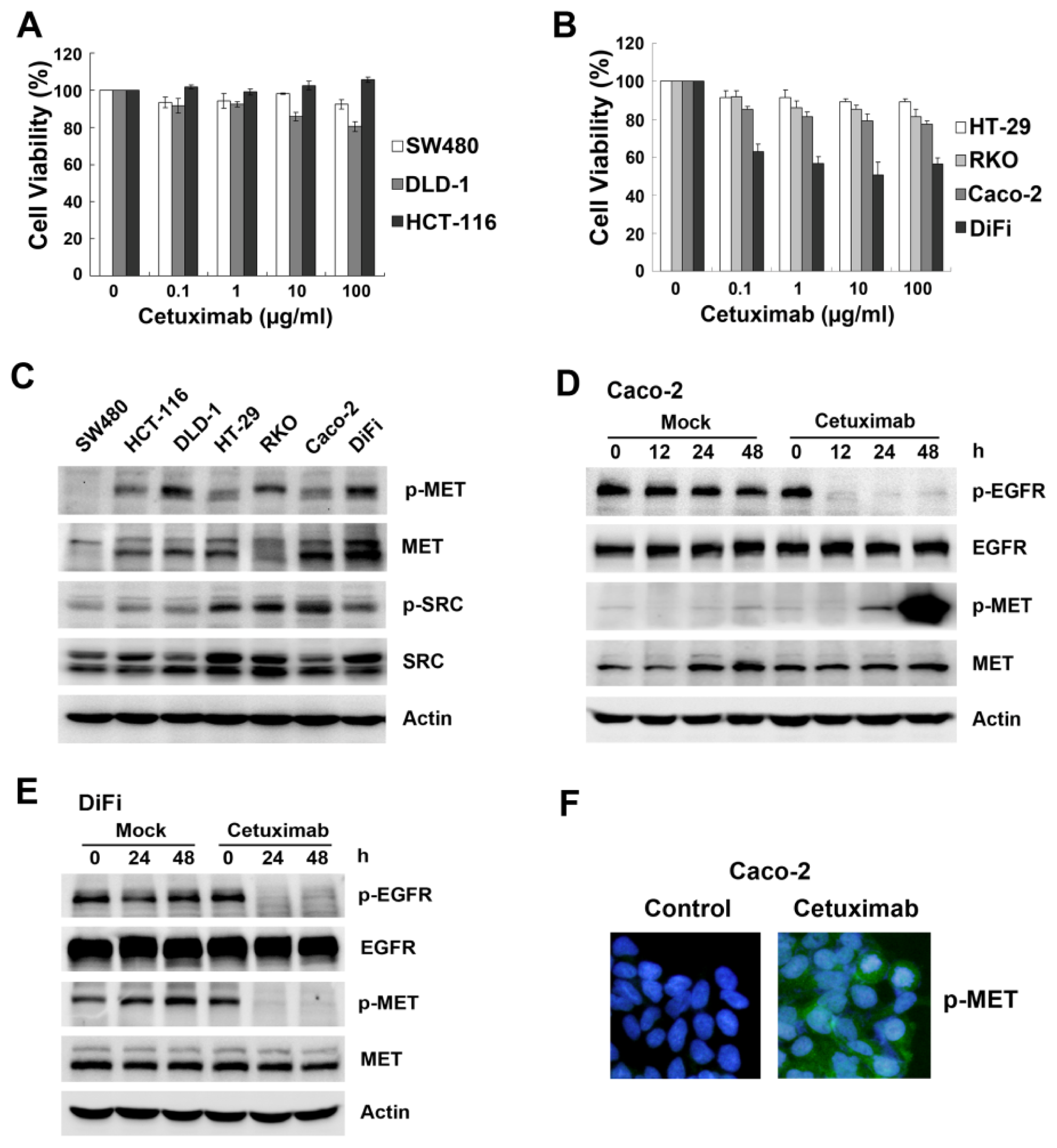
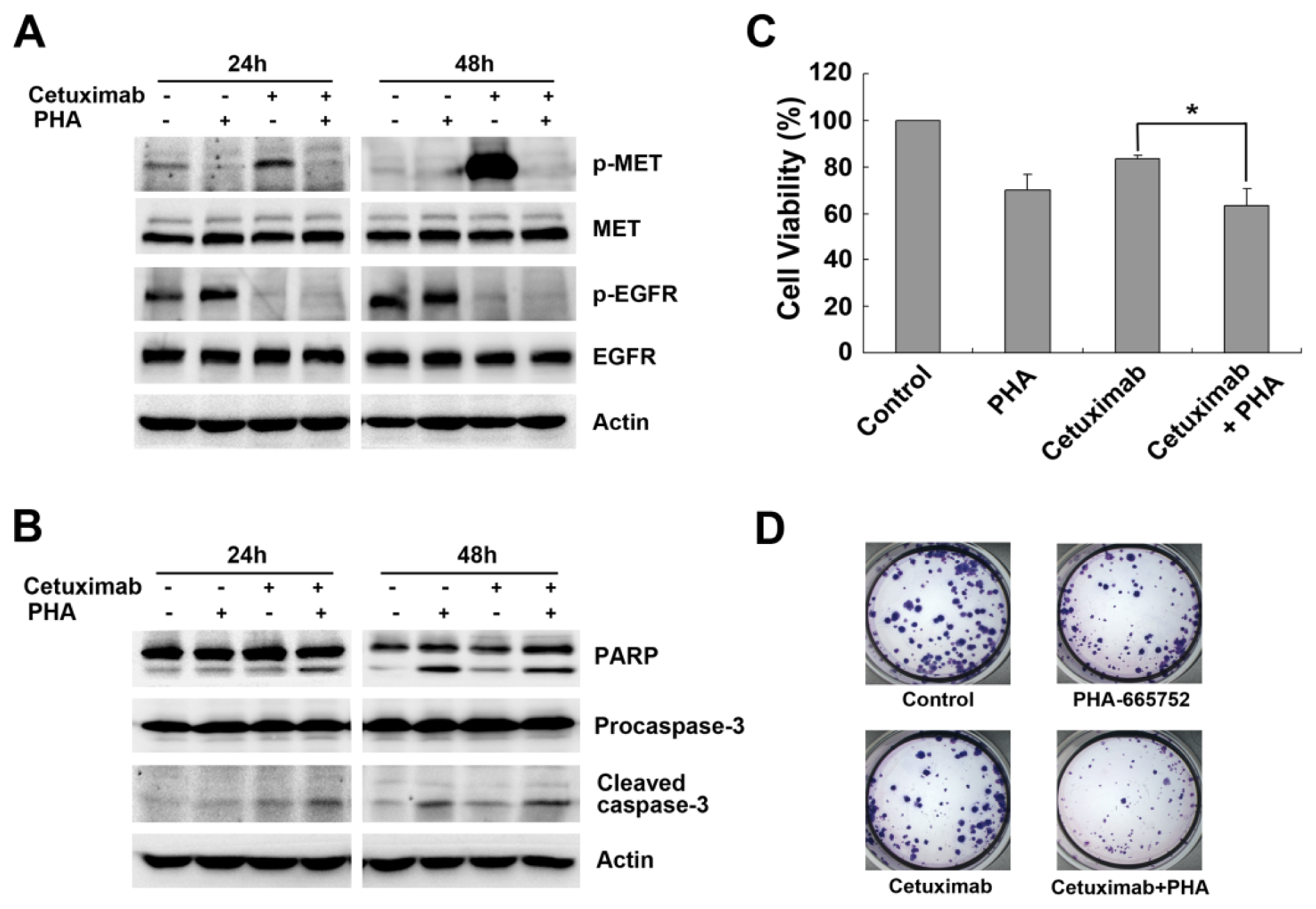
2.1. Cetuximab Induces MET Activation in Cetuximab-Insensitive Caco-2 Cells
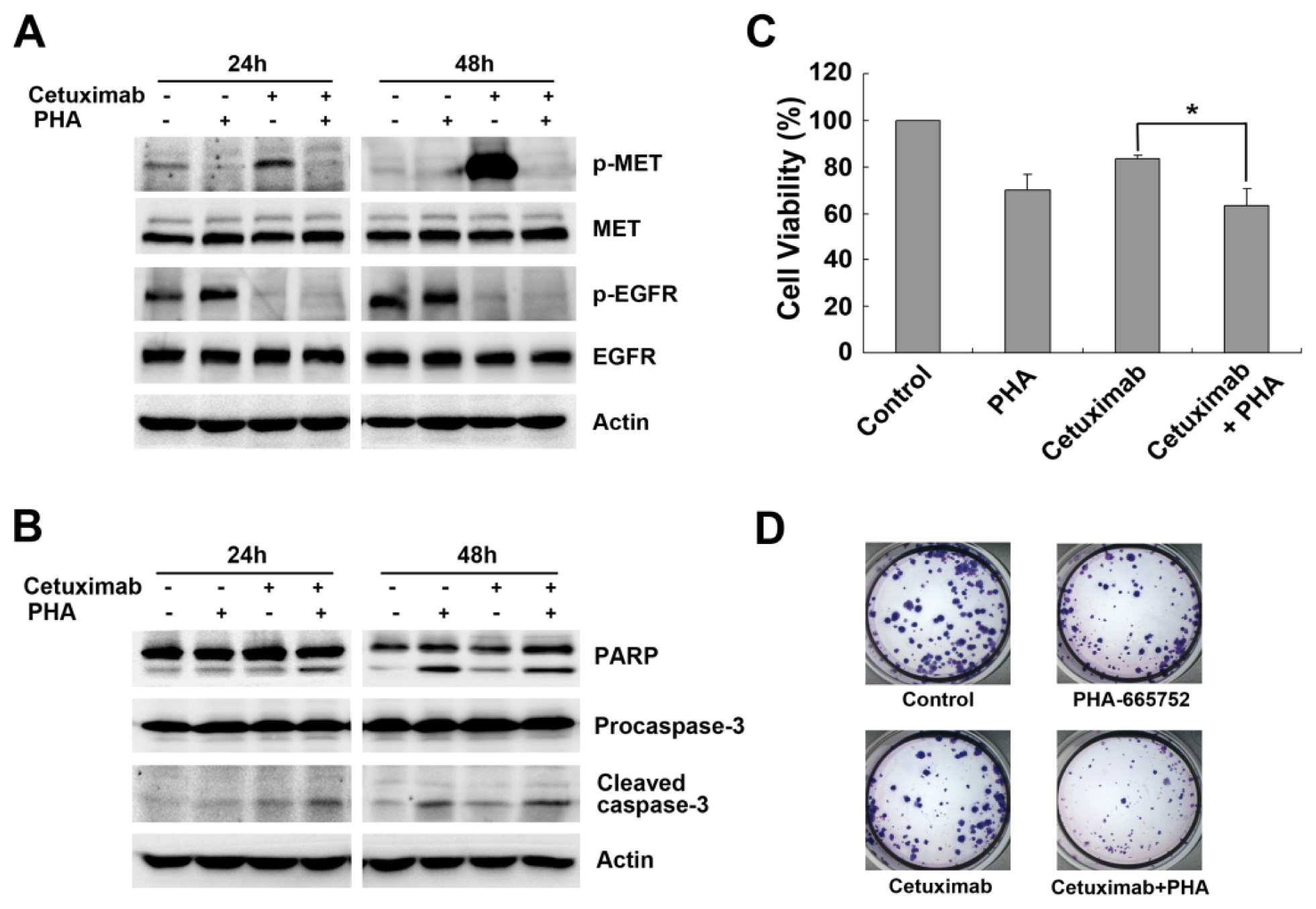
2.2. Cetuximab-Induced MET Activation Contributes to Cetuximab Resistance
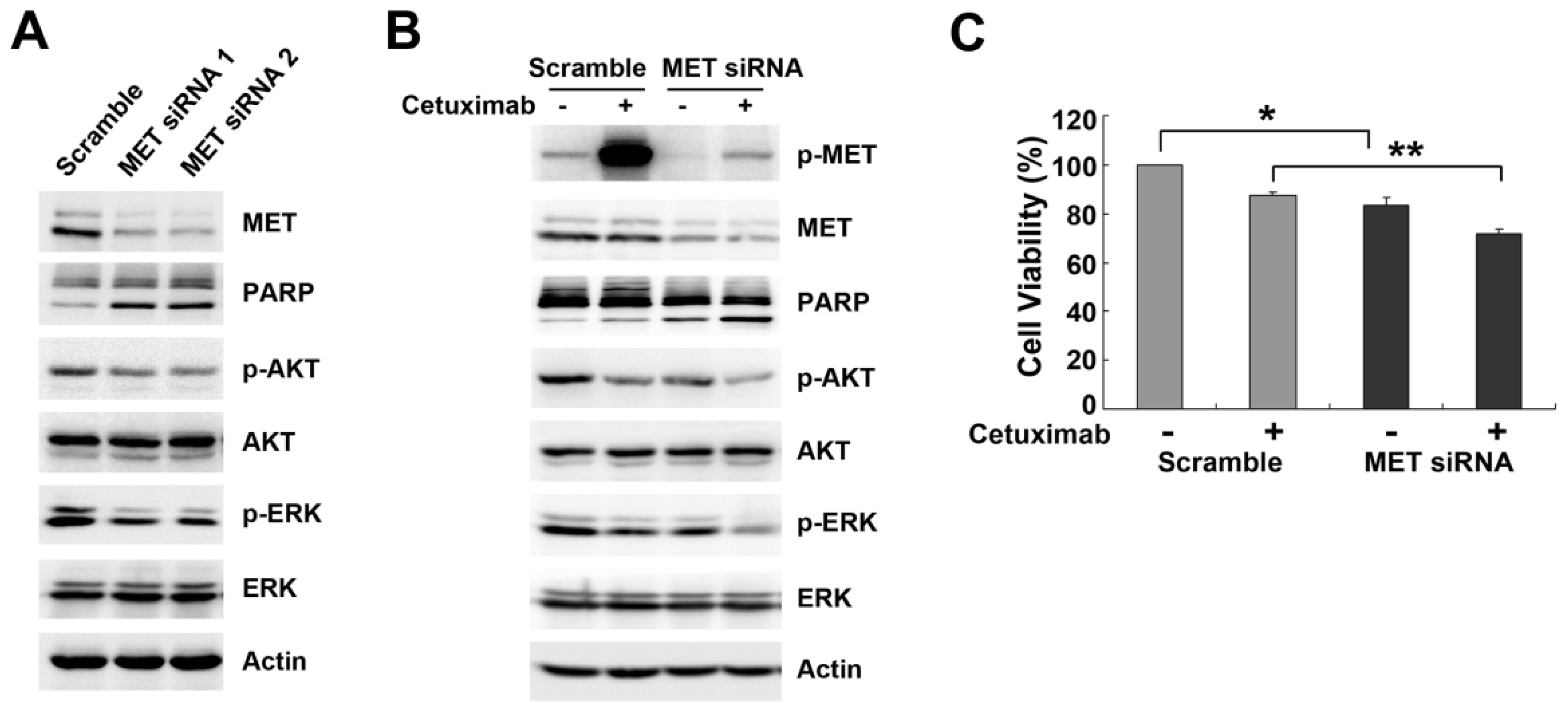
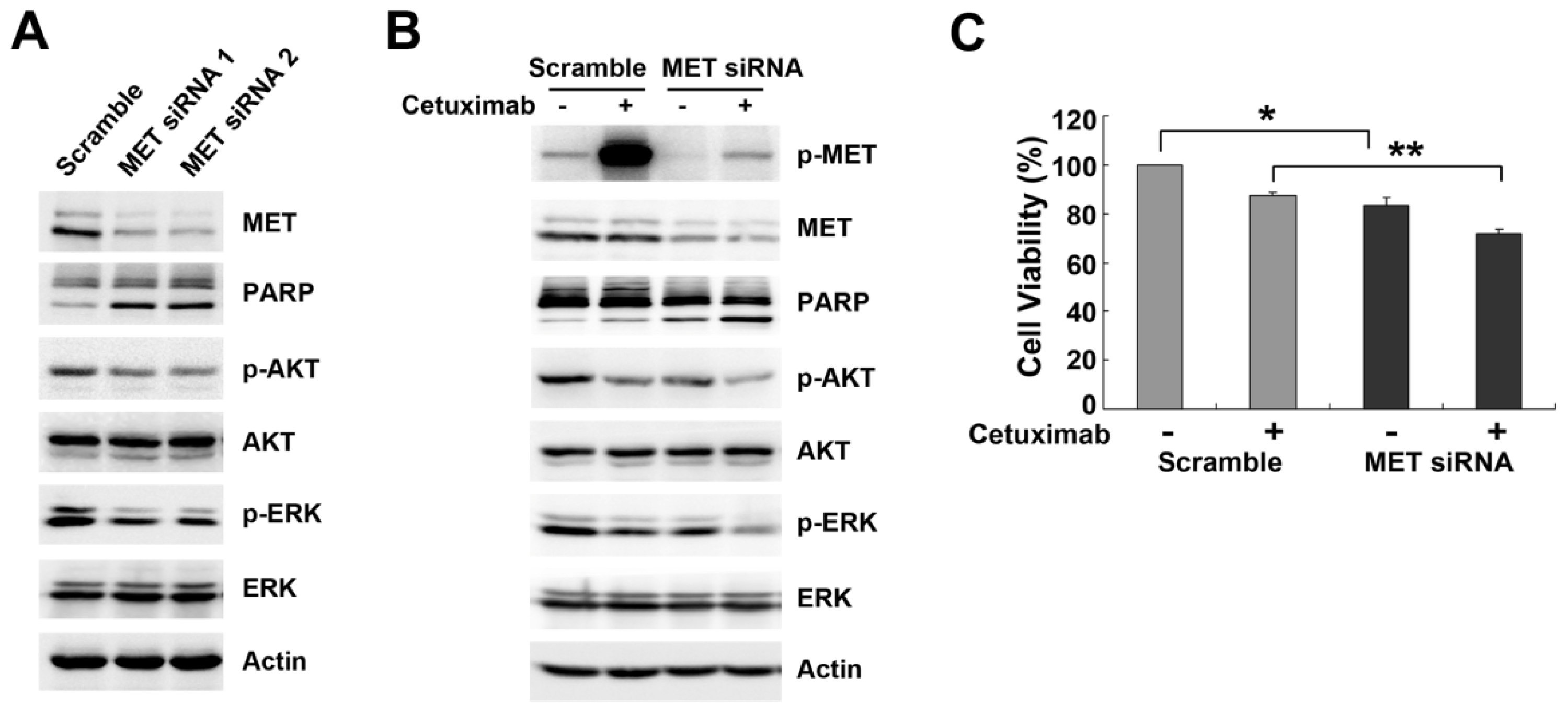
2.3. Downregulation of MET by siRNA Restores Cetuximab-Induced Cell Proliferation Inhibition
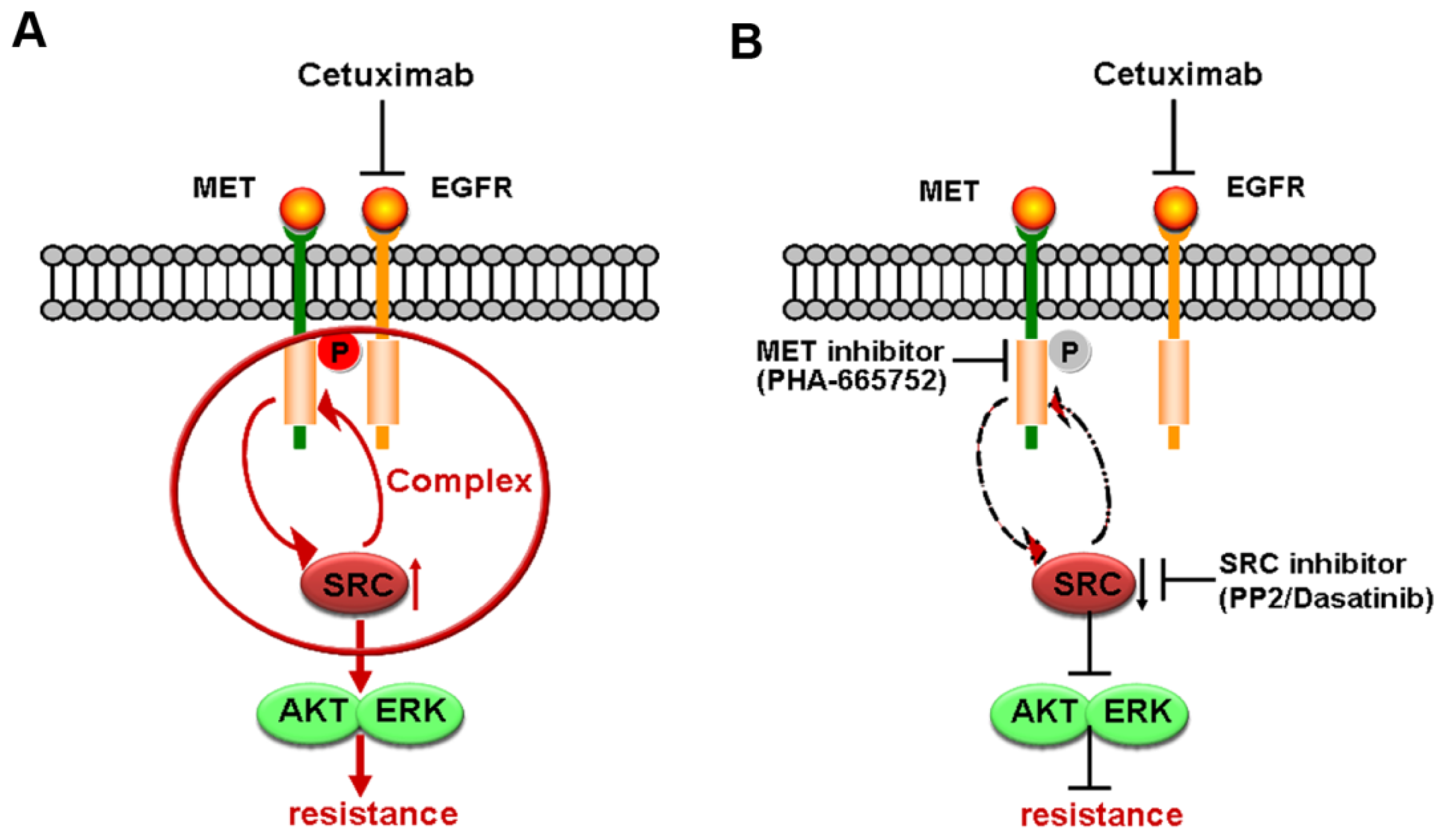
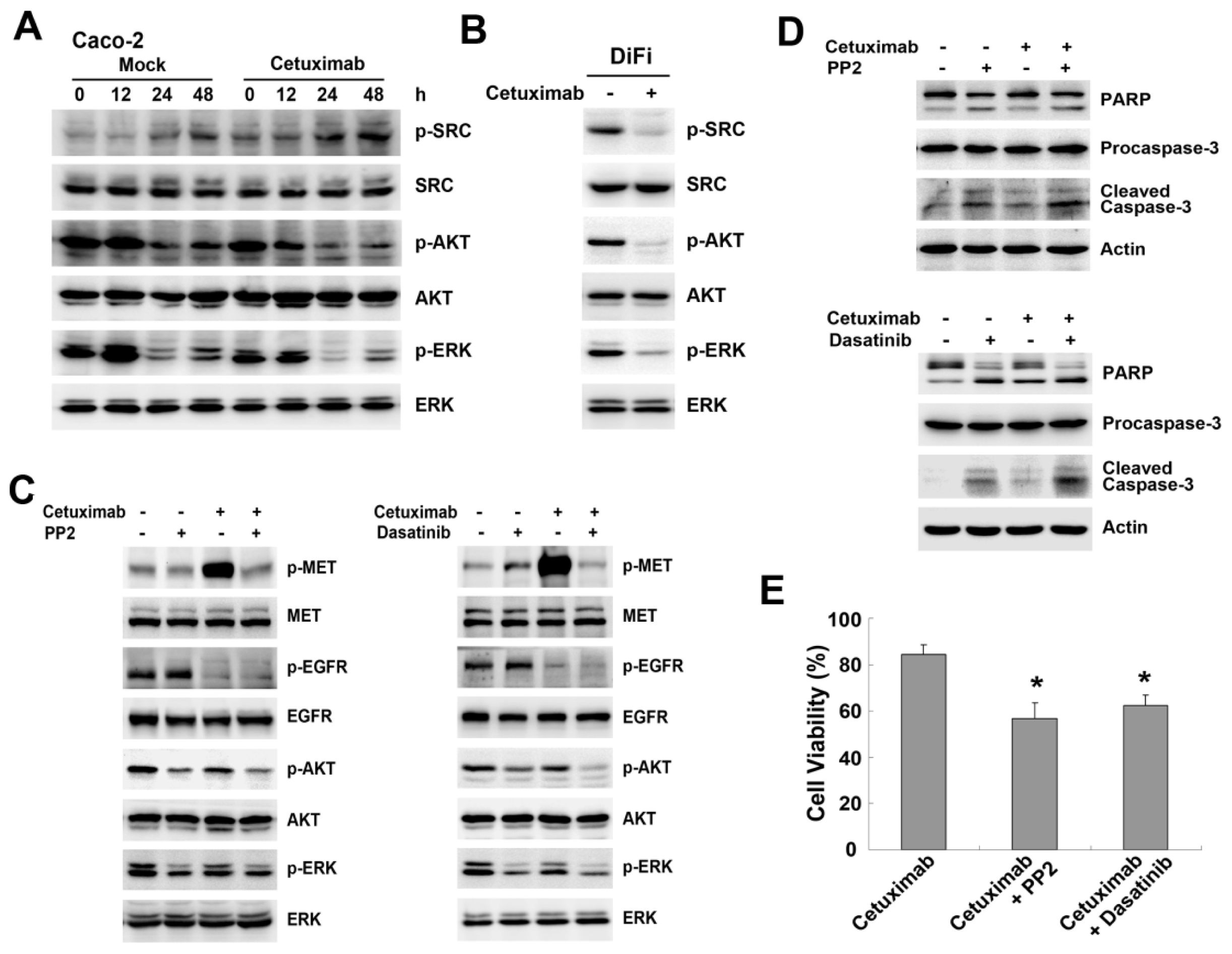
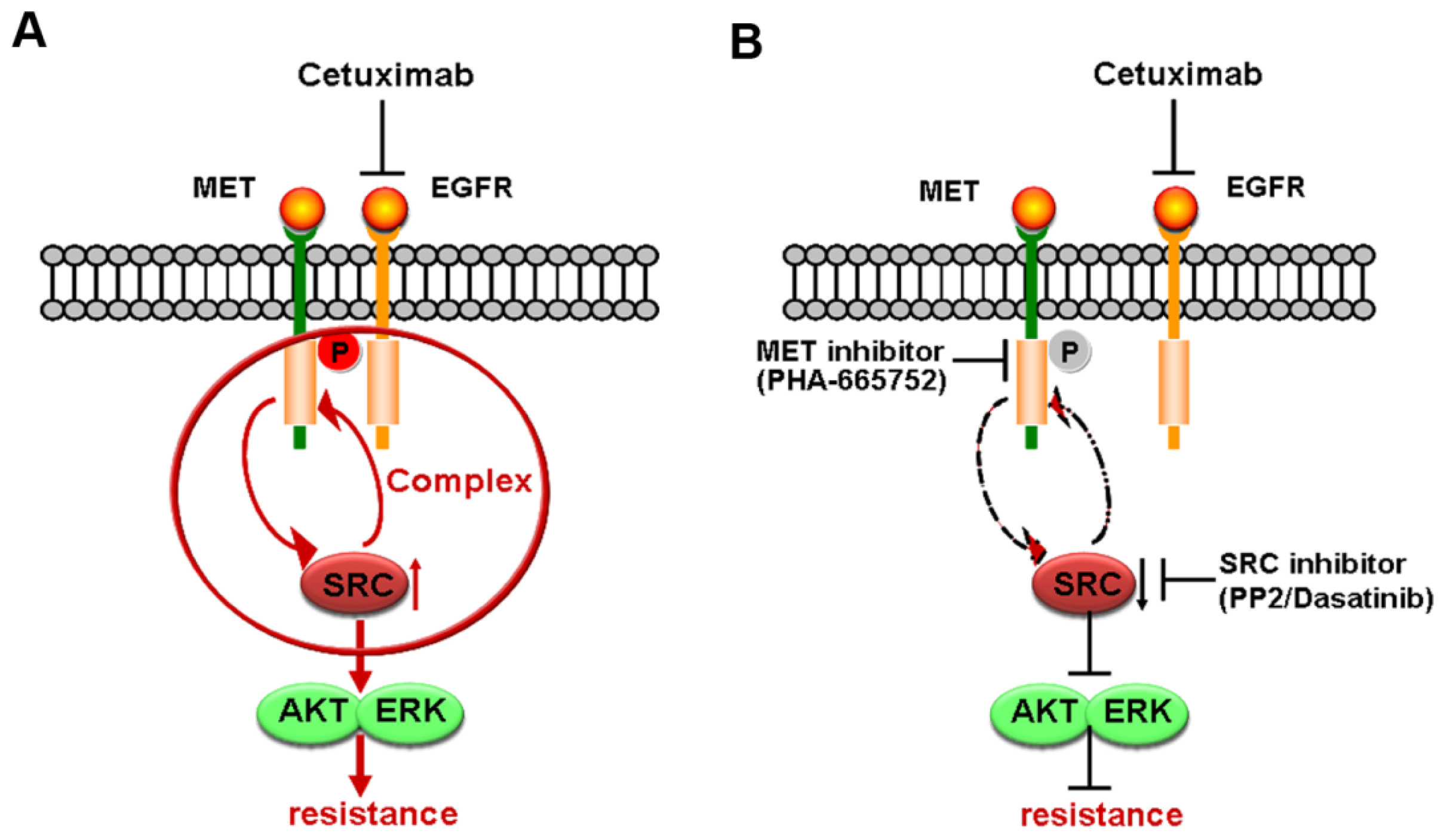
2.4. SRC Mediates Cetuximab-Induced MET Activation
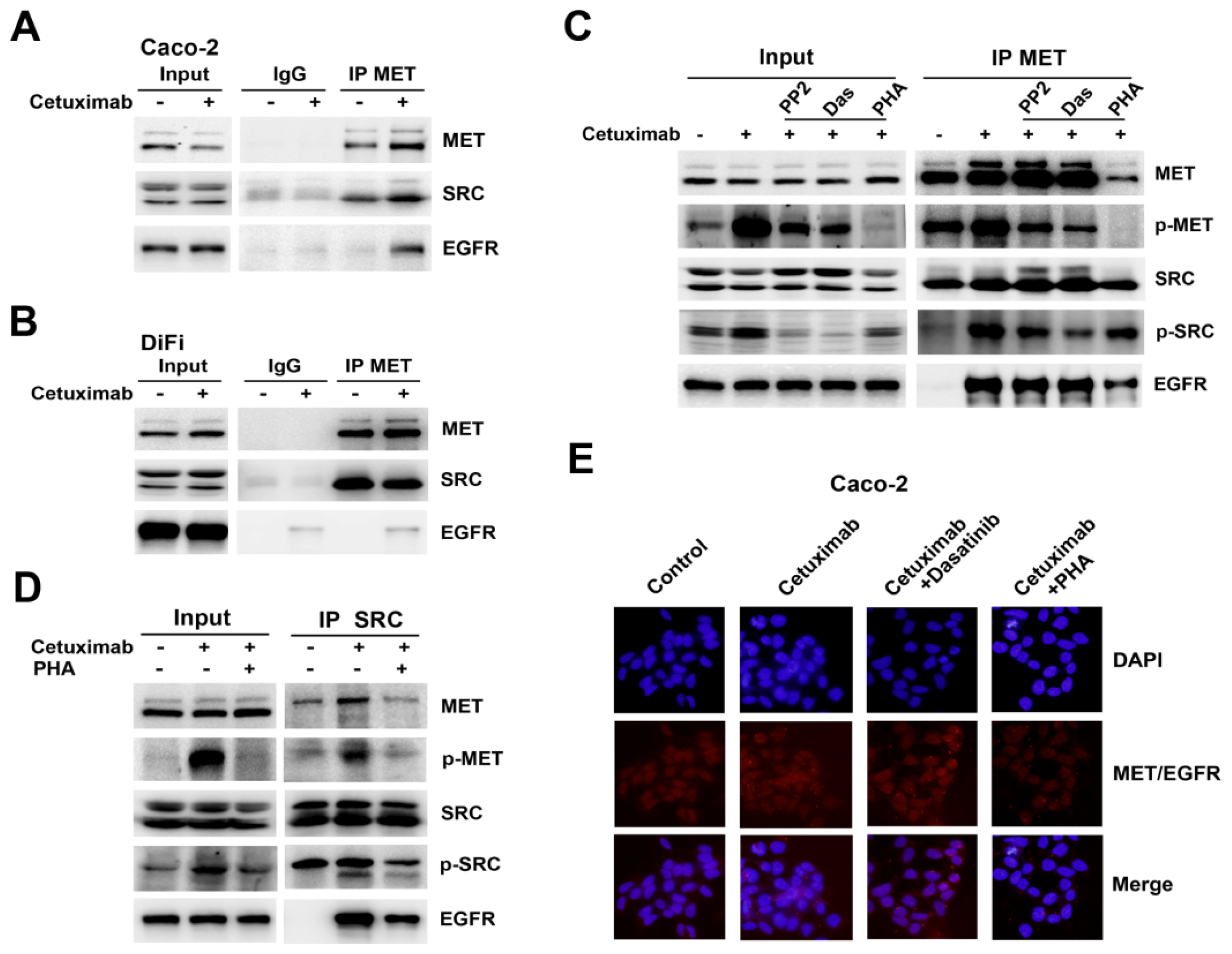
2.5. Formation of MET/SRC/EGFR Complex Induced by Cetuximab Is Required for MET Activation
3. Discussion
4. Experimental Section
4.1. Cell Culture and Reagents
4.2. Cell Viability Assay
4.3. Western Blotting
4.4. Fluorescence Microscopy
4.5. Clonogenic Assay
4.6. Small Interfering RNA Transfections
4.7. Co-Immunoprecipitation
4.8. In Situ Proximity Ligation Assay
4.9. Statistical Analysis
5. Conclusions
Supplementary Information
ijms-15-05838-s001.pdfAcknowledgments
Conflicts of Interest
References
- Bokemeyer, C.; van Cutsem, E.; Rougier, P.; Ciardiello, F.; Heeger, S.; Schlichting, M.; Celik, I.; Köhne, C.H. Addition of cetuximab to chemotherapy as first-line treatment for KRAS wild-type metastatic colorectal cancer: Pooled analysis of the CRYSTAL and OPUS randomised clinical trials. Eur. J. Cancer 2012, 48, 1466–1475. [Google Scholar]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med 2008, 359, 1757–1765. [Google Scholar]
- Douillard, J.Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N. Engl. J. Med 2013, 369, 1023–1034. [Google Scholar]
- De Roock, W.; Claes, B.; Bernasconi, D.; de Schutter, J.; Biesmans, B.; Fountzilas, G.; Kalogeras, K.T.; Kotoula, V.; Papamichael, D.; Laurent-Puig, P.; et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: A retrospective consortium analysis. Lancet Oncol 2010, 11, 753–762. [Google Scholar]
- Jhawer, M.; Goel, S.; Wilson, A.J.; Montagna, C.; Ling, Y.H.; Byun, D.S.; Nasser, S.; Arango, D.; Shin, J.; Klampfer, L.; et al. PIK3CA mutation/PTEN expression status predicts response of colon cancer cells to the epidermal growth factor receptor inhibitor cetuximab. Cancer Res 2008, 68, 1953–1961. [Google Scholar]
- Wheeler, D.L.; Dunn, E.F.; Harari, P.M. Understanding resistance to EGFR inhibitors-impact on future treatment strategies. Nat. Rev. Clin. Oncol 2010, 7, 493–507. [Google Scholar]
- Corso, S.; Giordano, S. Cell-autonomous and non-cell-autonomous mechanisms of HGF/MET-driven resistance to targeted therapies: From basic research to a clinical perspective. Cancer Discov 2013, 3, 978–992. [Google Scholar]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar]
- Gusenbauer, S.; Vlaicu, P.; Ullrich, A. HGF induces novel EGFR functions involved in resistance formation to tyrosine kinase inhibitors. Oncogene 2013, 32, 3846–3856. [Google Scholar]
- Liska, D.; Chen, C.T.; Bachleitner-Hofmann, T.; Christensen, J.G.; Weiser, M.R. HGF rescues colorectal cancer cells from EGFR inhibition via MET activation. Clin. Cancer Res 2011, 17, 472–482. [Google Scholar]
- Cipriani, N.A.; Abidoye, O.O.; Vokes, E.; Salgia, R. MET as a target for treatment of chest tumors. Lung Cancer 2009, 63, 169–179. [Google Scholar]
- Morgillo, F.; Woo, J.K.; Kim, E.S.; Hong, W.K.; Lee, H.Y. Heterodimerization of insulin-like growth factor receptor/epidermal growth factor receptor and induction of survivin expression counteract the antitumor action of erlotinib. Cancer Res 2006, 66, 10100–10111. [Google Scholar]
- Yang, L.; Li, J.; Ran, L.; Pan, F.; Zhao, X.; Ding, Z.; Chen, Y.; Peng, Q.; Liang, H. Phosphorylated insulin-like growth factor 1 receptor is implicated in resistance to the cytostatic effect of gefitinib in colorectal cancer cells. J. Gastrointest. Surg 2011, 15, 942–957. [Google Scholar]
- Dulak, A.M.; Gubish, C.T.; Stabile, L.P.; Henry, C.; Siegfried, J.M. HGF-independent potentiation of EGFR action by c-Met. Oncogene 2011, 30, 3625–3635. [Google Scholar]
- Mueller, K.L.; Hunter, L.A.; Ethier, S.P.; Boerner, J.L. Met and c-Src cooperate to compensate for loss of epidermal growth factor receptor kinase activity in breast cancer cells. Cancer Res 2008, 68, 3314–3322. [Google Scholar]
- Zhang, S.; Huang, W.C.; Li, P.; Guo, H.; Poh, S.B.; Brady, S.W.; Xiong, Y.; Tseng, L.M.; Li, S.H.; Ding, Z.; et al. Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat. Med 2011, 17, 461–469. [Google Scholar]
- Dunn, E.F.; Iida, M.; Myers, R.A.; Campbell, D.A.; Hintz, K.A.; Armstrong, E.A.; Li, C.; Wheeler, D.L. Dasatinib sensitizes KRAS mutant colorectal tumors to cetuximab. Oncogene 2011, 30, 561–574. [Google Scholar]
- Krumbach, R.; Schüler, J.; Hofmann, M.; Giesemann, T.; Fiebig, H.H.; Beckers, T. Primary resistance to cetuximab in a panel of patient-derived tumour xenograft models: Activation of MET as one mechanism for drug resistance. Eur. J. Cancer 2011, 47, 1231–1243. [Google Scholar]
- Wheeler, D.L.; Huang, S.; Kruser, T.J.; Nechrebecki, M.M.; Armstrong, E.A.; Benavente, S.; Gondi, V.; Hsu, K.T.; Harari, P.M. Mechanisms of acquired resistance to cetuximab: Role of HER (ErbB) family members. Oncogene 2008, 27, 3944–3956. [Google Scholar]
- Stabile, L.P.; He, G.; Lui, V.W.; Thomas, S.; Henry, C.; Gubish, C.T.; Joyce, S.; Quesnelle, K.M.; Siegfried, J.M.; Grandis, J.R. c-Src activation mediates erlotinib resistance in head and neck cancer by stimulating c-Met. Clin. Cancer Res 2013, 19, 380–392. [Google Scholar]
- Wheeler, D.L.; Iida, M.; Kruser, T.J.; Nechrebecki, M.M.; Dunn, E.F.; Armstrong, E.A.; Huang, S.; Harari, P.M. Epidermal growth factor receptor cooperates with Src family kinases in acquired resistance to cetuximab. Cancer Biol. Ther 2009, 8, 696–703. [Google Scholar]
- Mueller, K.L.; Yang, Z.Q.; Haddad, R.; Ethier, S.P.; Boerner, J.L. EGFR/Met association regulates EGFR TKI resistance in breast cancer. J. Mol. Signal 2010, 5. [Google Scholar] [CrossRef]
- Chen, C.T.; Kim, H.; Liska, D.; Gao, S.; Christensen, J.G.; Weiser, M.R. MET activation mediates resistance to lapatinib inhibition of HER2-amplified gastric cancer cells. Mol. Cancer Ther 2012, 11, 660–669. [Google Scholar]
- Bardelli, A.; Corso, S.; Bertotti, A.; Hobor, S.; Valtorta, E.; Siravegna, G.; Sartore-Bianchi, A.; Scala, E.; Cassingena, A.; Zecchin, D.; et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov 2013, 3, 658–673. [Google Scholar]
- Varkaris, A.; Gaur, S.; Parikh, N.U.; Song, J.H.; Dayyani, F.; Jin, J.K.; Logothetis, C.J.; Gallick, G.E. Ligand-independent activation of MET through IGF-1/IGF-1R signaling. Int. J. Cancer 2013, 133, 1536–1546. [Google Scholar]
- Troiani, T.; Martinelli, E.; Napolitano, S.; Vitagliano, D.; Ciuffreda, L.P.; Costantino, S.; Morgillo, F.; Capasso, A.; Sforza, V.; Nappi, A.; et al. Increased TGF-α as a mechanism of acquired resistance to the anti-EGFR inhibitor cetuximab through EGFR-MET interaction and activation of MET signaling in colon cancer cells. Clin. Cancer Res 2013, 19, 6751–6765. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Song, N.; Liu, S.; Zhang, J.; Liu, J.; Xu, L.; Liu, Y.; Qu, X. Cetuximab-Induced MET Activation Acts as a Novel Resistance Mechanism in Colon Cancer Cells. Int. J. Mol. Sci. 2014, 15, 5838-5851. https://doi.org/10.3390/ijms15045838
Song N, Liu S, Zhang J, Liu J, Xu L, Liu Y, Qu X. Cetuximab-Induced MET Activation Acts as a Novel Resistance Mechanism in Colon Cancer Cells. International Journal of Molecular Sciences. 2014; 15(4):5838-5851. https://doi.org/10.3390/ijms15045838
Chicago/Turabian StyleSong, Na, Shizhou Liu, Jingdong Zhang, Jing Liu, Ling Xu, Yunpeng Liu, and Xiujuan Qu. 2014. "Cetuximab-Induced MET Activation Acts as a Novel Resistance Mechanism in Colon Cancer Cells" International Journal of Molecular Sciences 15, no. 4: 5838-5851. https://doi.org/10.3390/ijms15045838