Synergistic Enhancement of Cancer Therapy Using a Combination of Ceramide and Docetaxel

Abstract
:
1. Introduction
2. Results
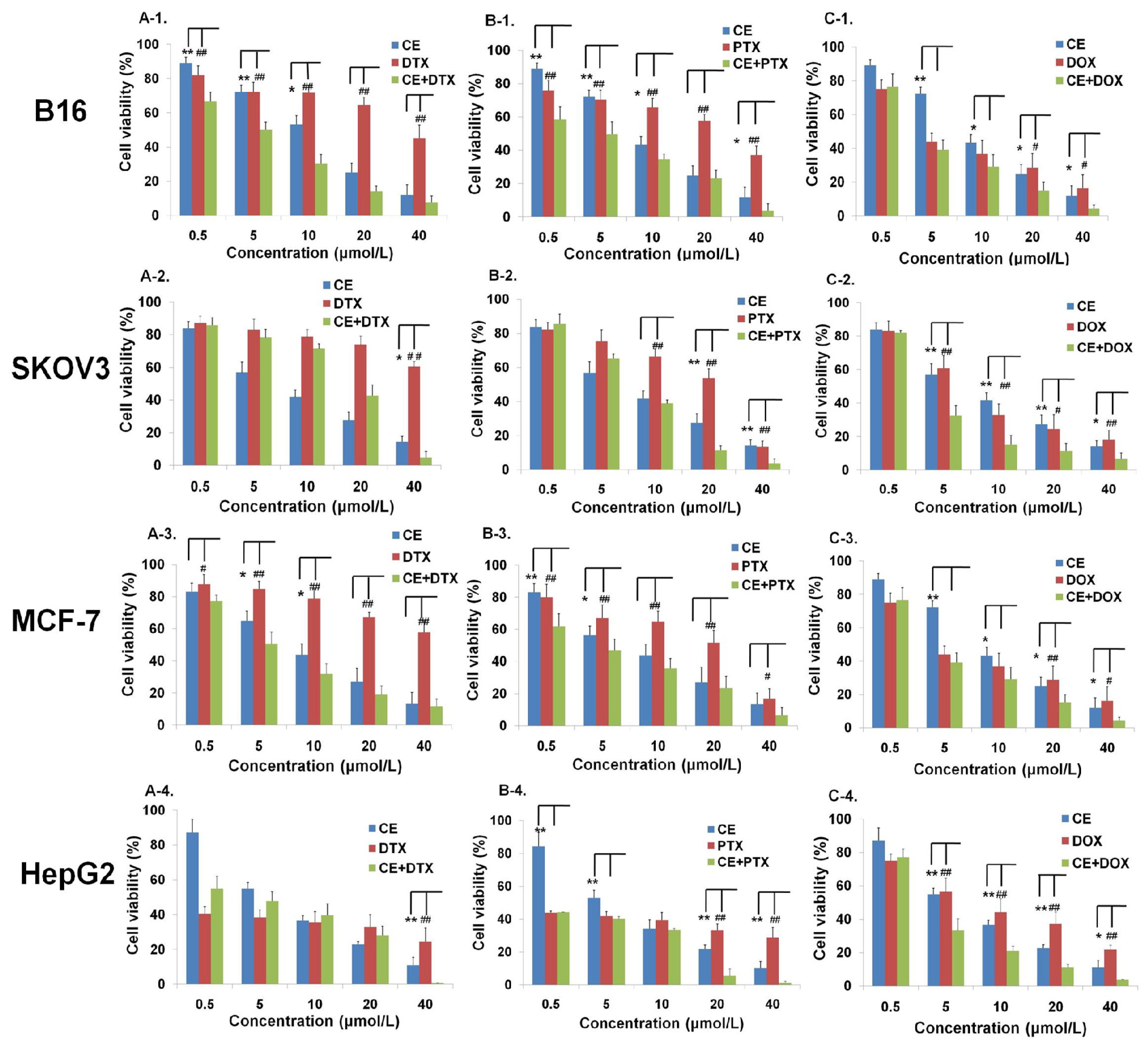
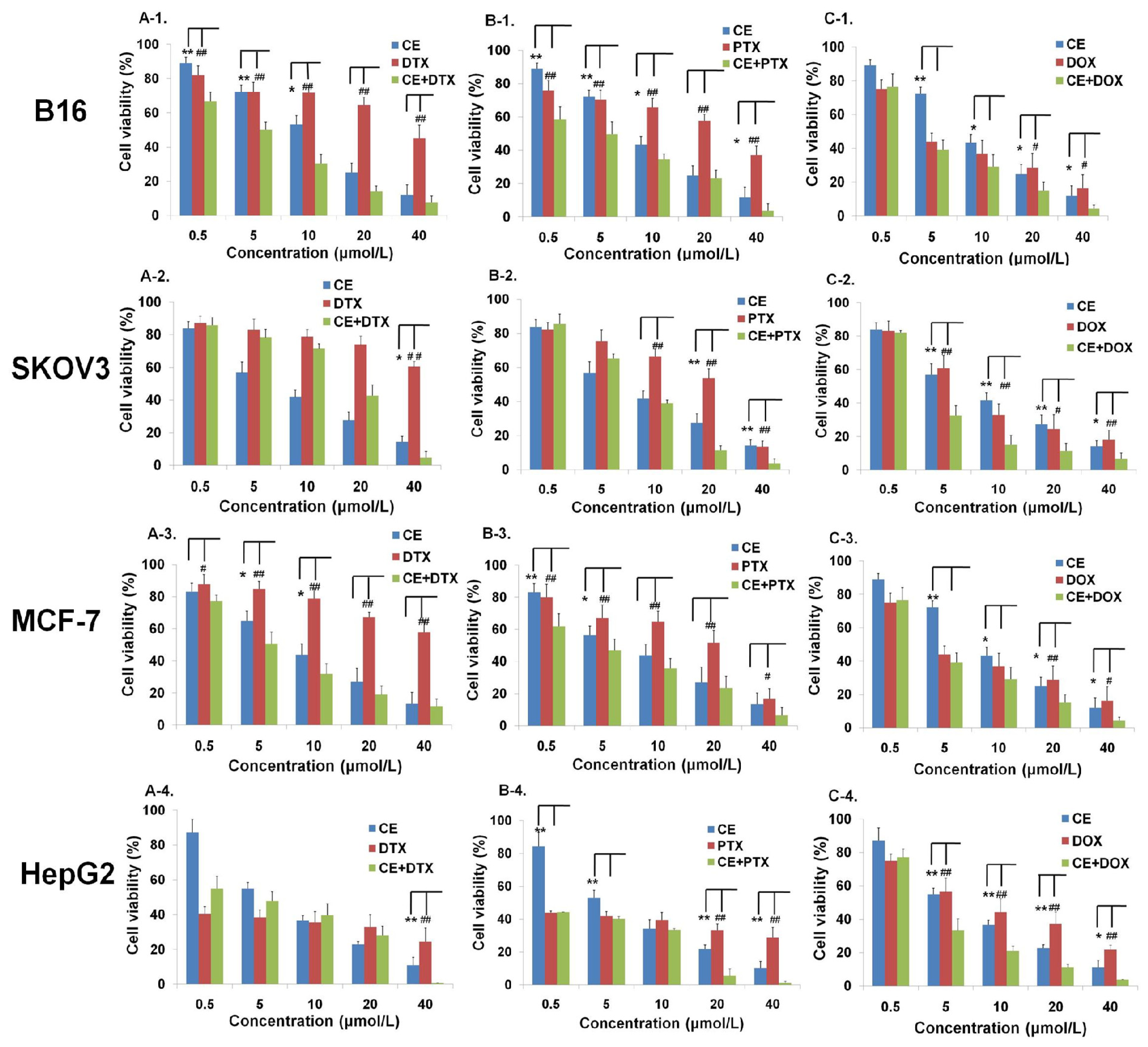
2.1. Effects of CE Combination on Cell Proliferation (MTT Assay)
2.2. Determination of the Optimal Dose Schedule of CE + DTX on B16 and MCF-7 Cells
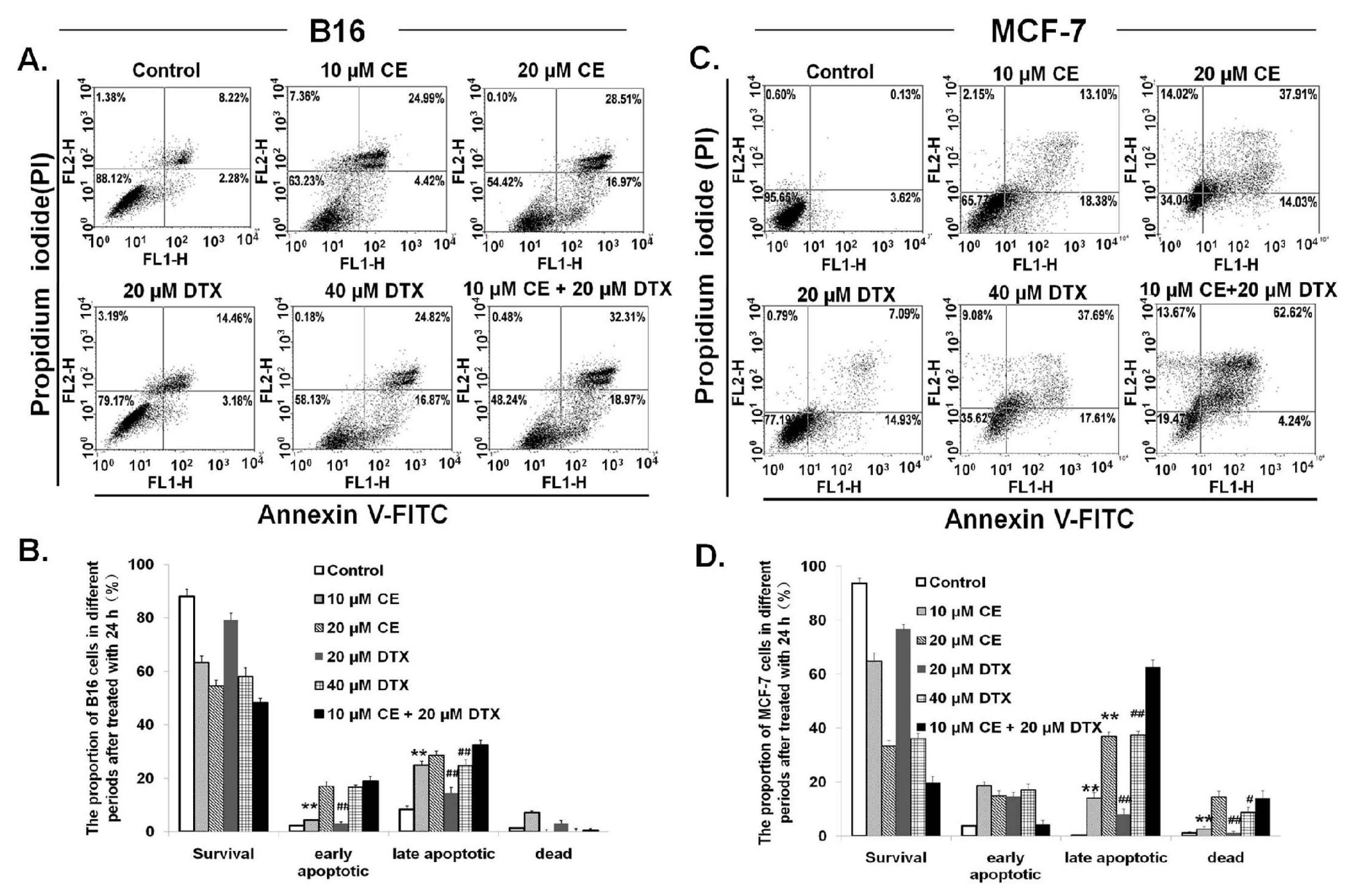
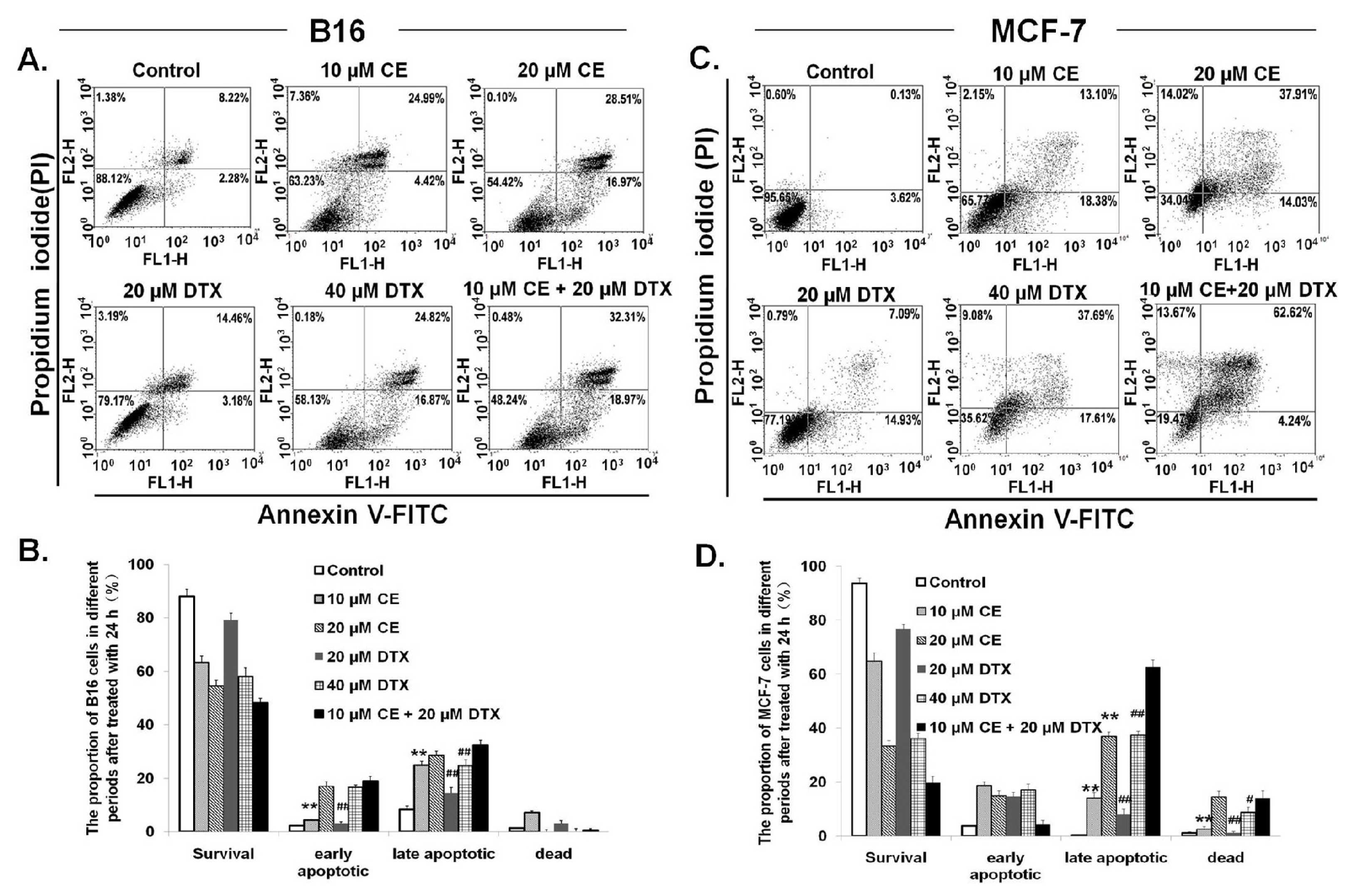
2.3. Induction of Apoptosis on B16 and MCF-7 Cells by CE + DTX
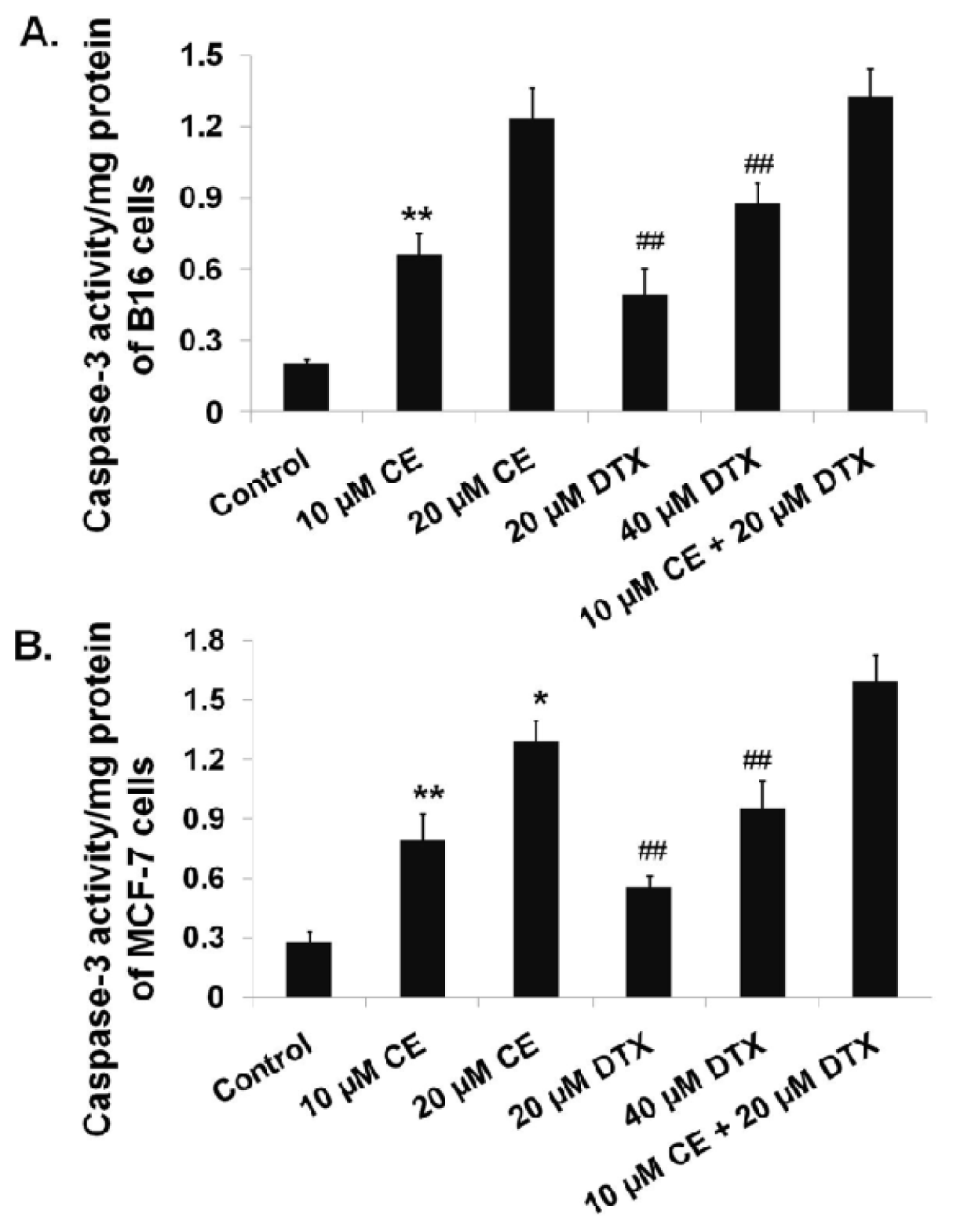
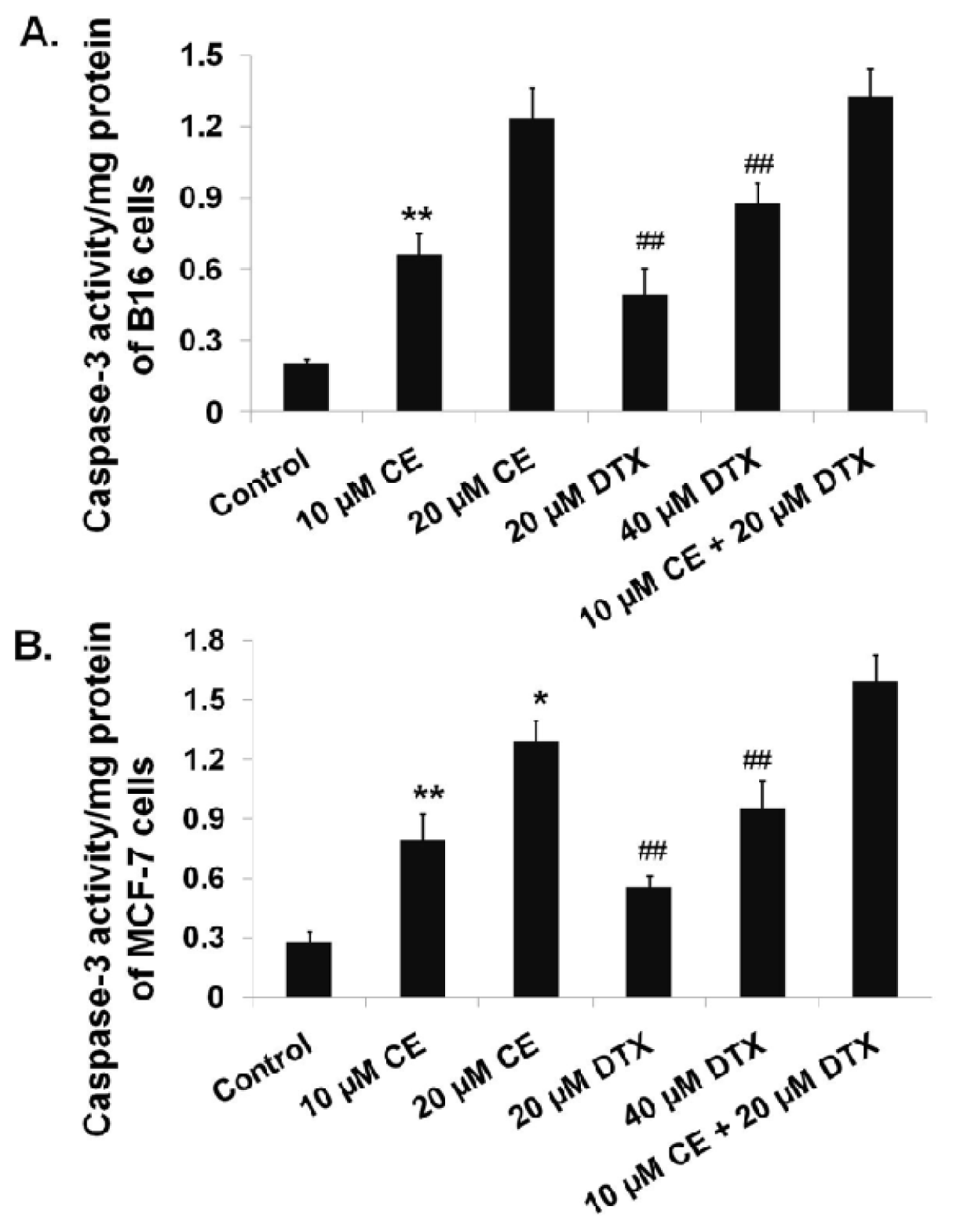
2.4. Activation of Caspase-3 on B16 and MCF-7 Cells by CE + DTX
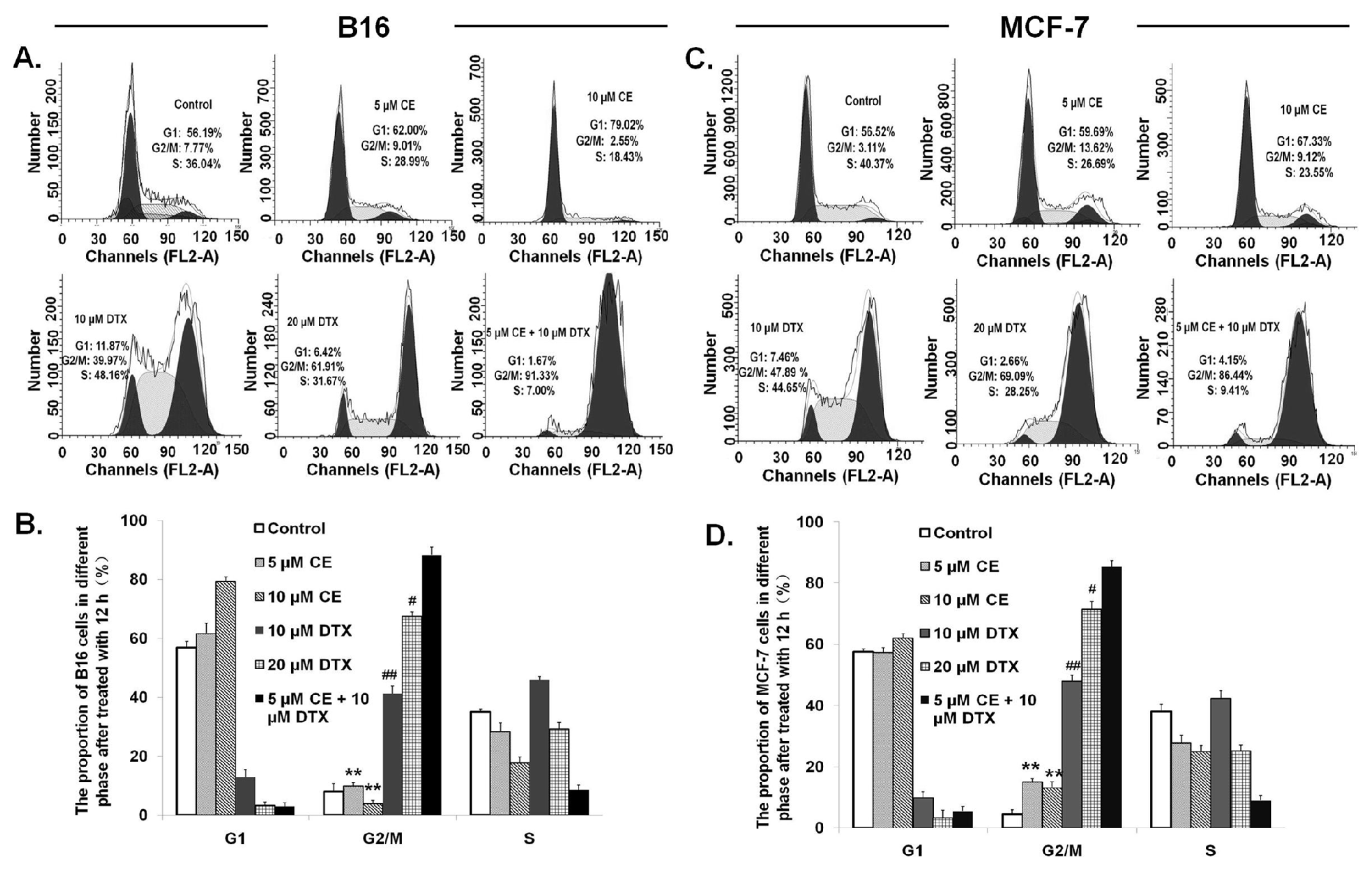
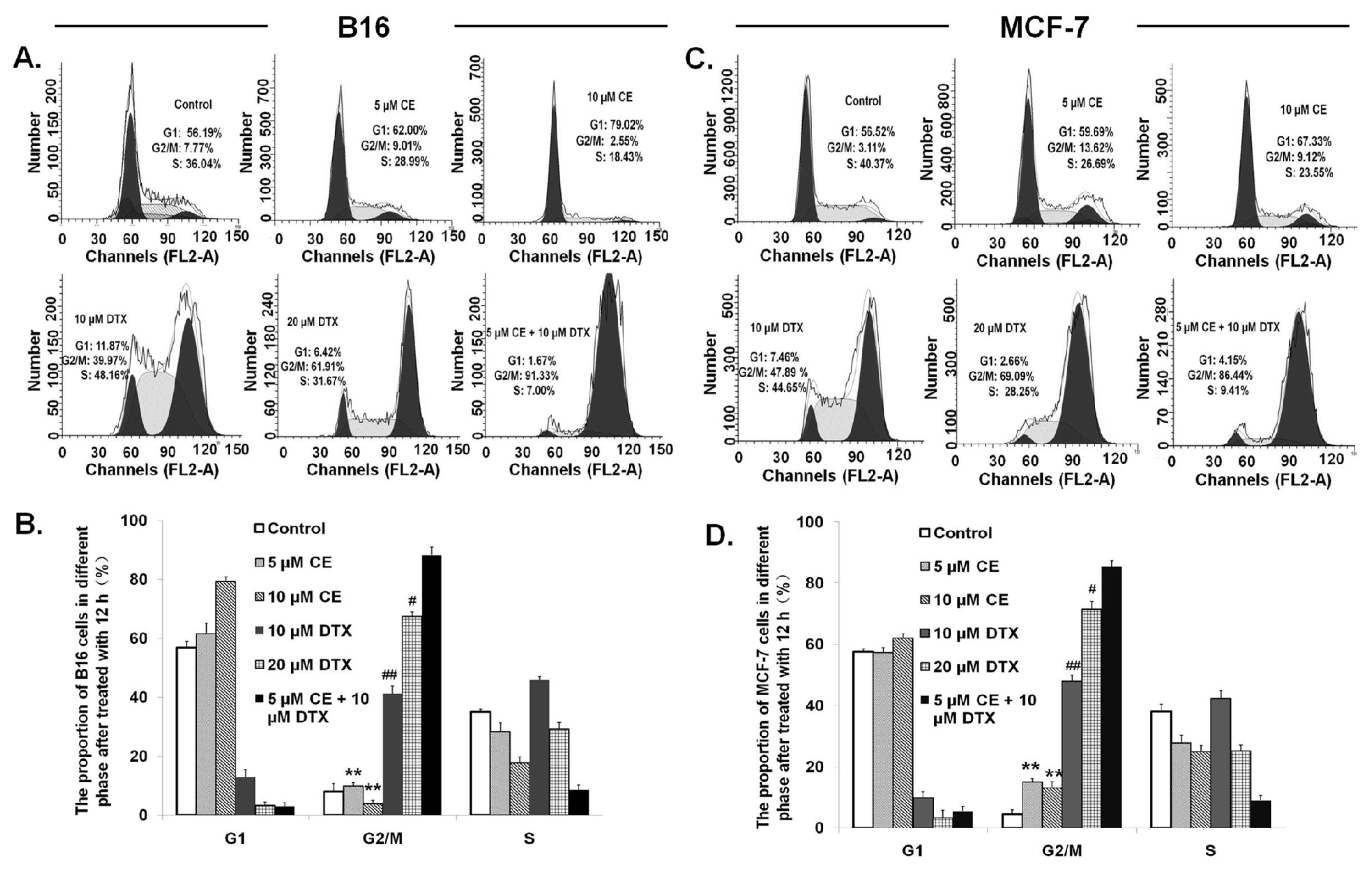
2.5. Cell Cycle Effect of CE + DTX
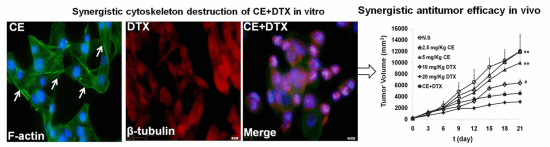
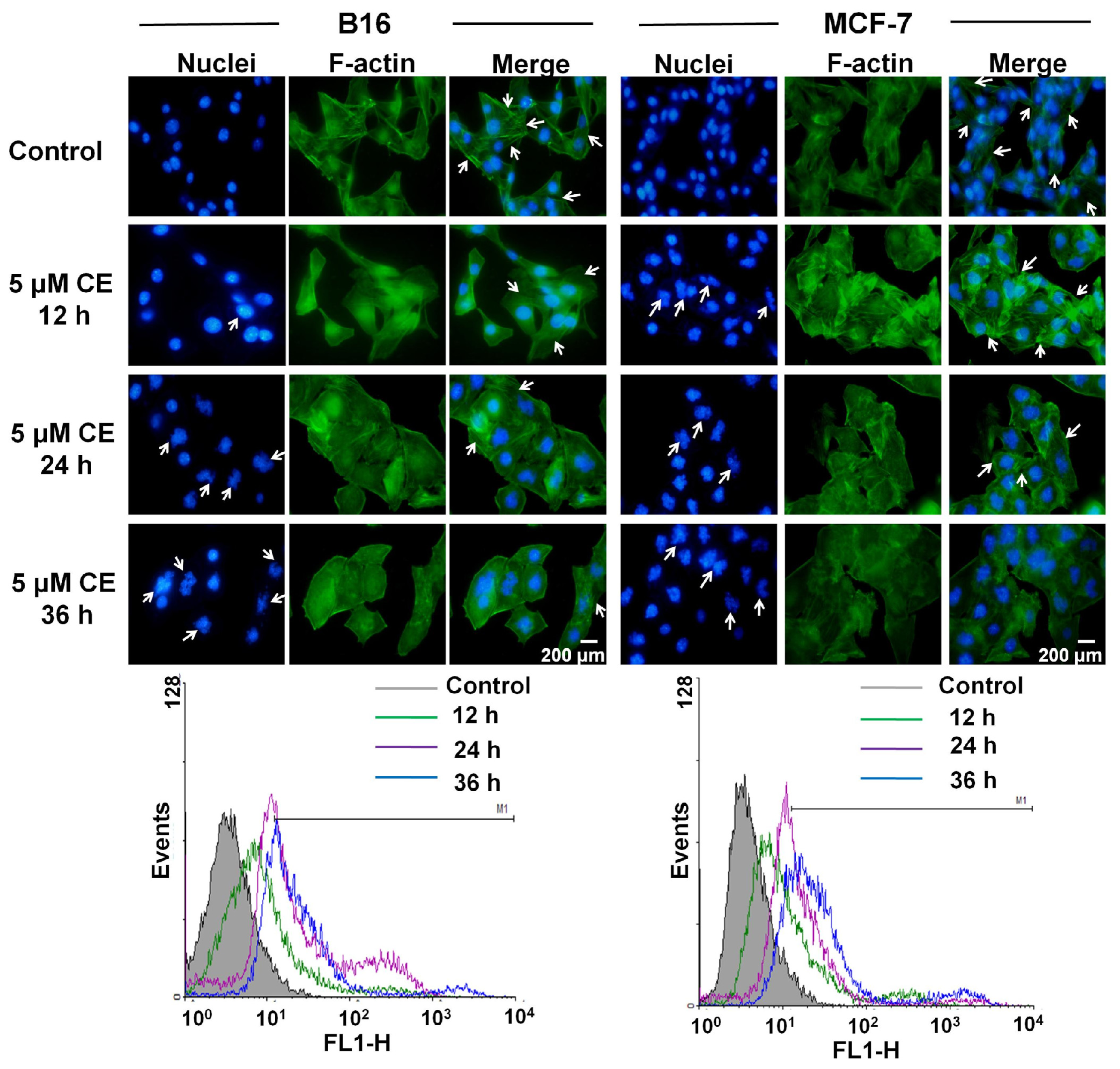
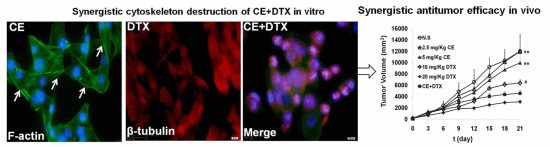
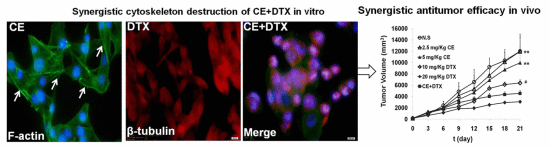
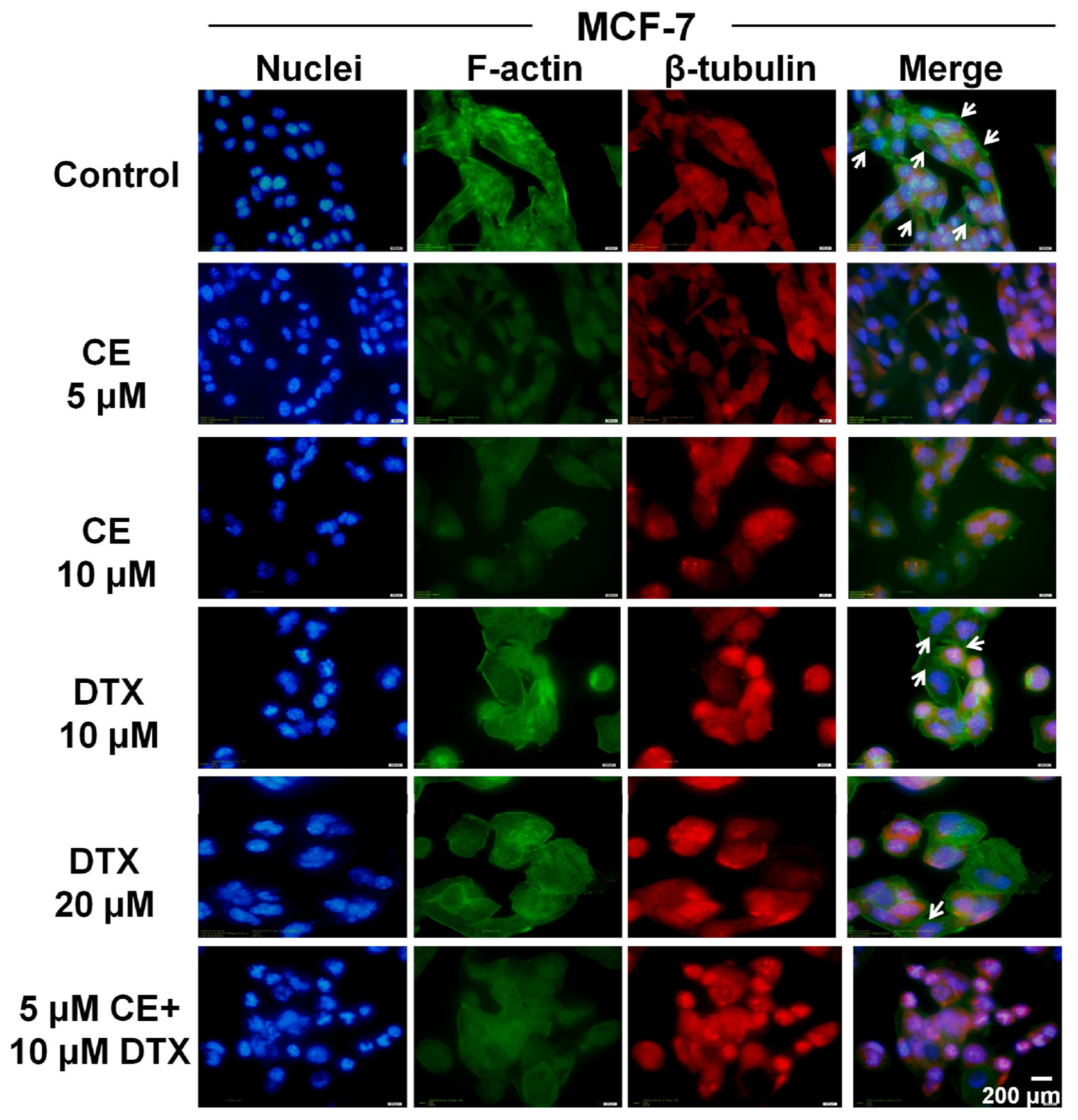
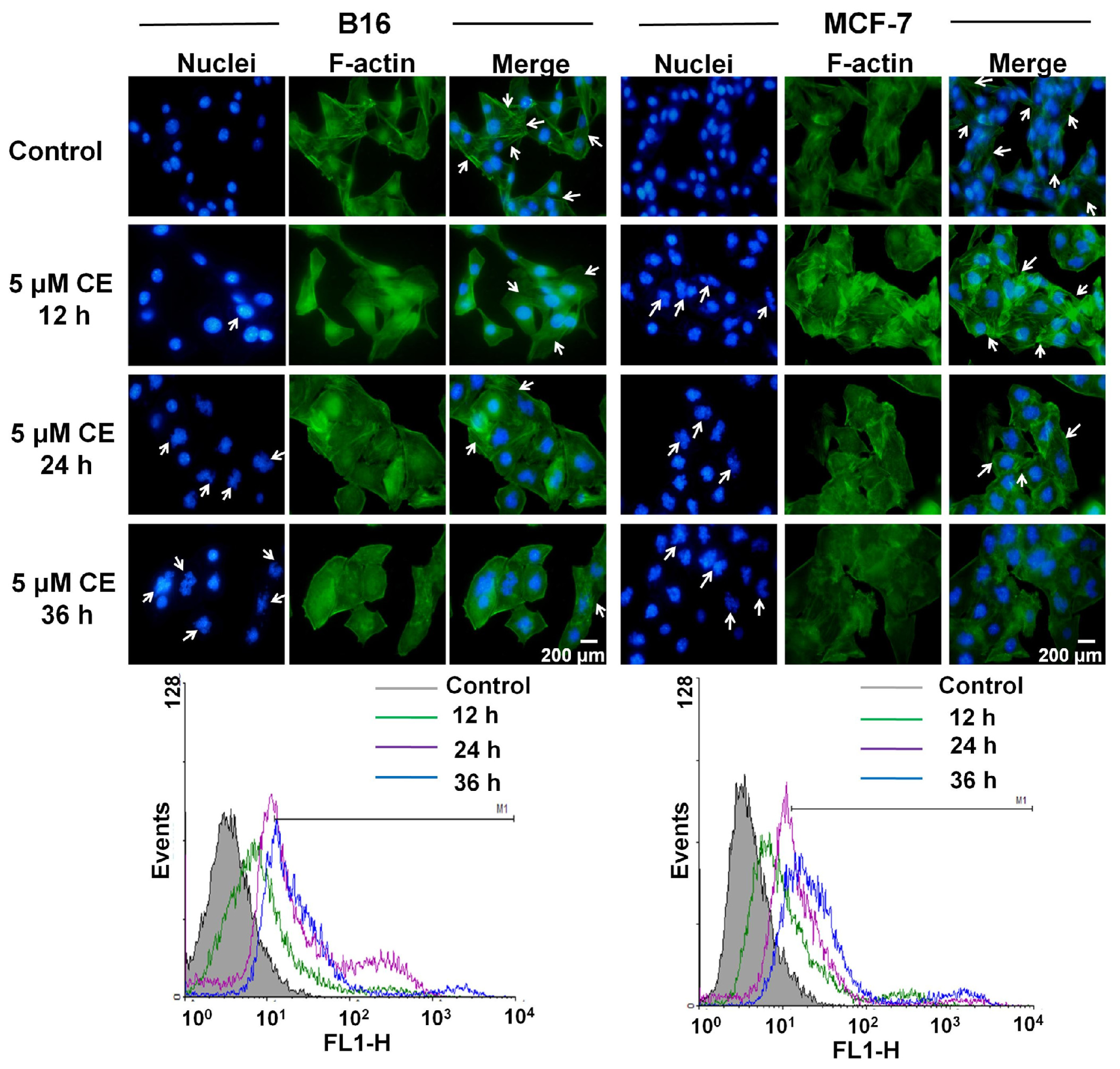
2.6. Cytoskeleton Destruction Effect of CE + DTX
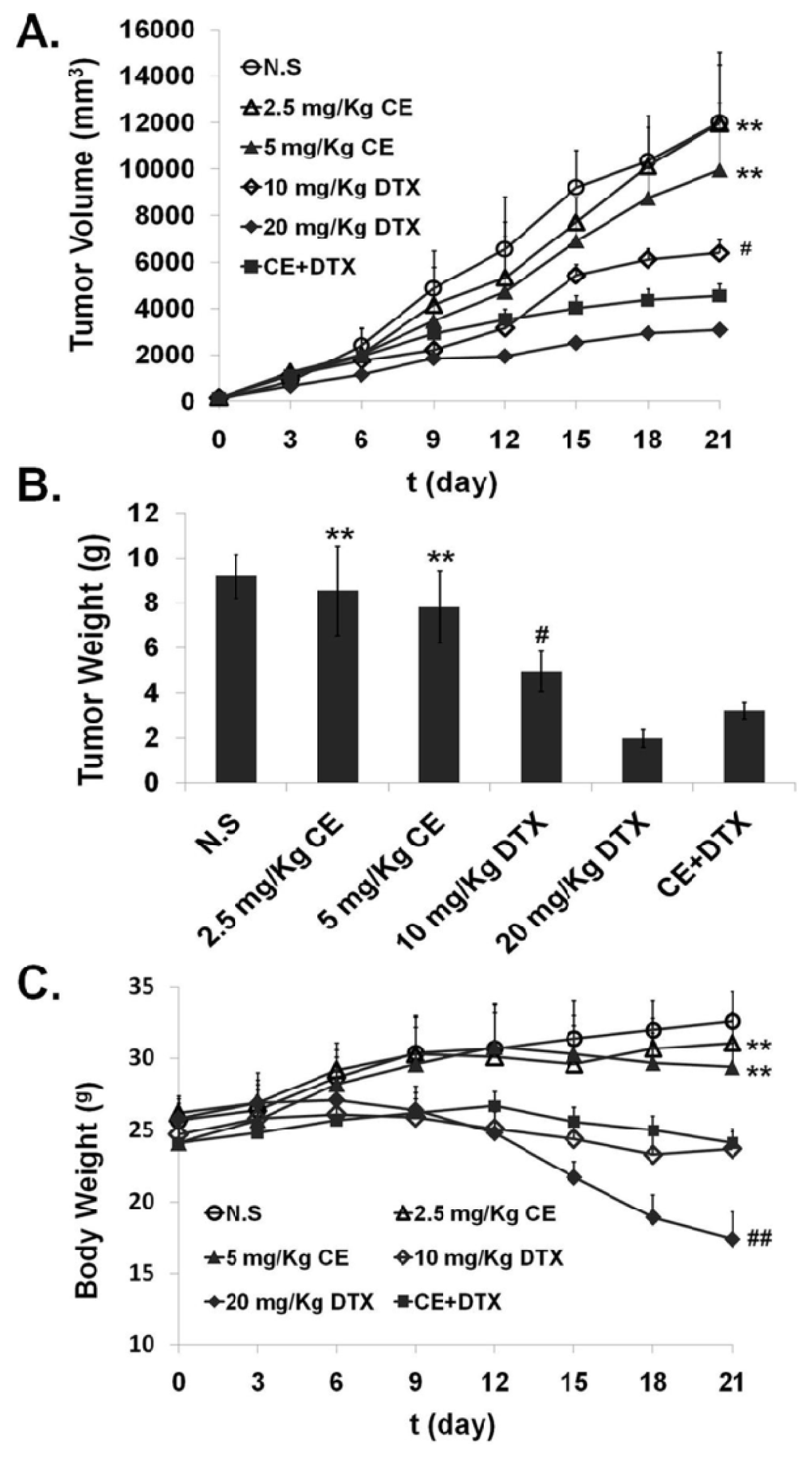
2.7. In Vivo Anti-Tumor Effect of CE + DTX
3. Discussion
4. Experimental Section
4.1. Materials
4.2. Cell Lines and Cell Culture
4.3. Animals
4.4. Anti-Proliferation Test in Vitro
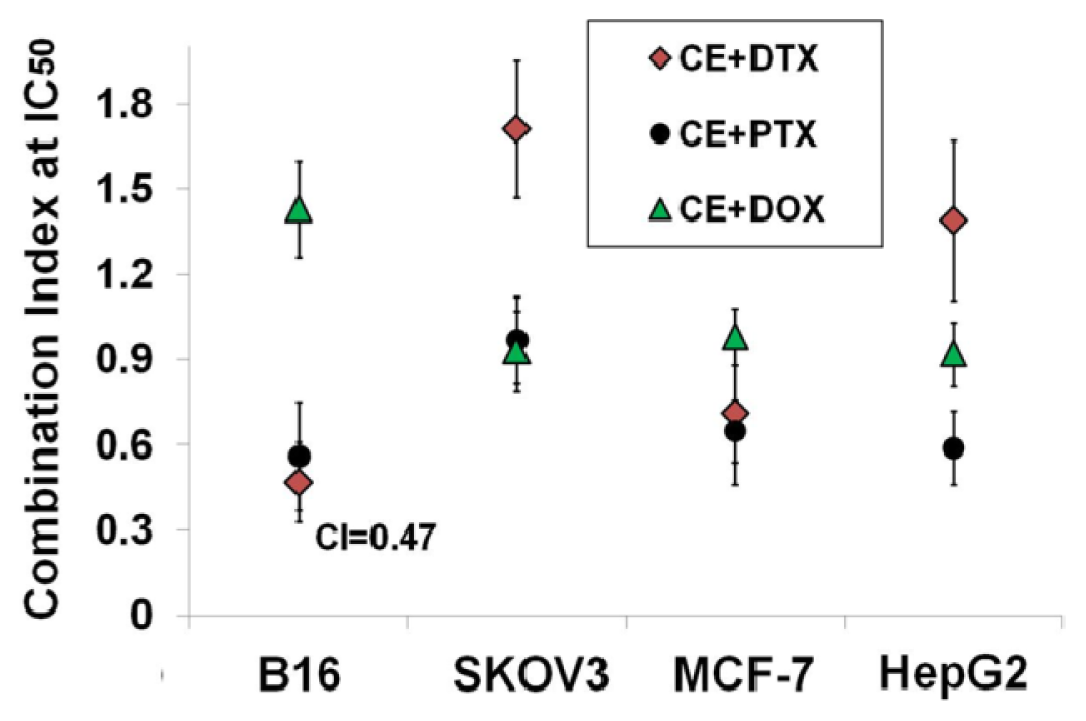
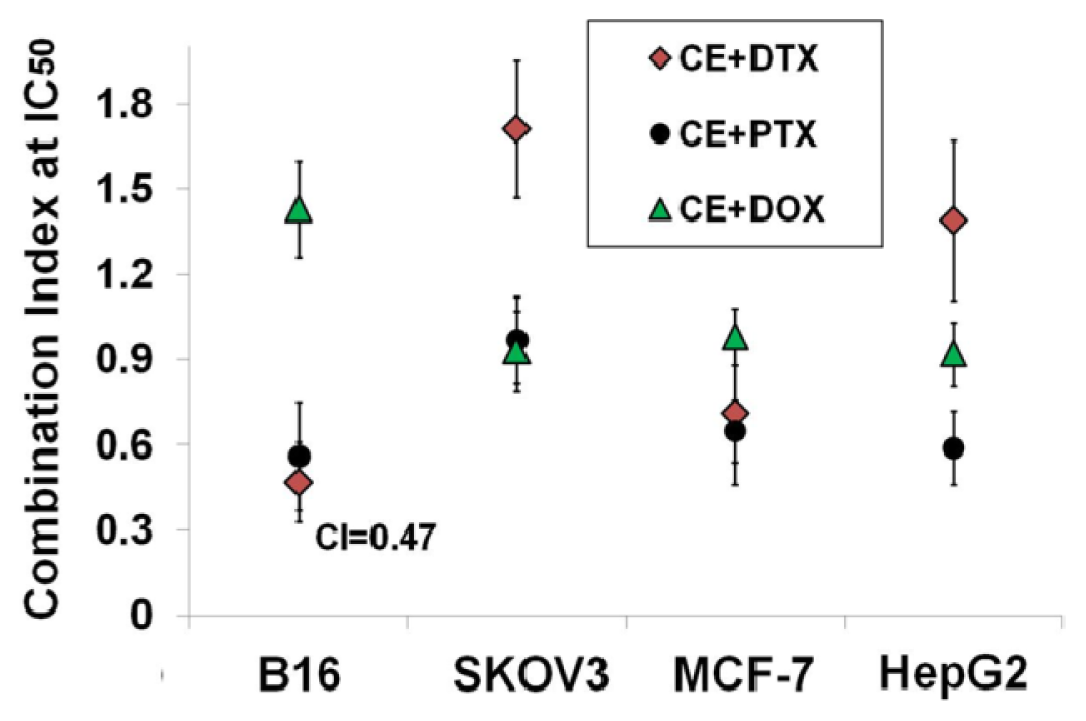
4.5. Calculation of Combination Index (CI)
4.6. Induction of Apoptosis on B16 and MCF-7 Cells
4.7. Evaluation of Caspase-3 Activity
4.8. Cell Cycle Analysis of B16 and MCF-7 Cells
4.9. Cytoskeleton Destruction and Actin-Polymerization Study
4.10. In Vivo Antitumor Efficacy Evaluation
4.11. Statistical Analysis
5. Conclusions
Supplementary Information
ijms-15-04201-s001.pdfAcknowledgments
Conflicts of Interest
References
- Jemal, A.; Siegel, R.; Ward, E.; Hao, Y.; Xu, J.; Murray, T.; Thun, M.J. Cancer statistics 2008. CA Cancer J. Clin. 2008, 58, 71–96. [Google Scholar]
- Parkin, D.M.; Bray, F.; Ferlay, J.; Pisani, P. Global cancer statistics 2002. CA Cancer. J. Clin. 2005, 55, 74–108. [Google Scholar]
- Schally, A.V.; Engel, J.B.; Emons, G.; Block, N.L.; Pinski, J. Use of analogs of peptide hormones conjugated to cytotoxic radicals for chemotherapy targeted to receptors on tumors. Curr. Drug Deliv. 2011, 8, 11–25. [Google Scholar]
- DeVita, V.T.; Chu, E. A history of cancer chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar]
- Bevis, K.S.; Erwin, J.L.; Barnes, M.N.; Straughn, J.M., Jr. The efficacy and toxicity of bevacizumab in combination with gemcitabine in patients with recurrent ovarian Cancer. Clin. Ovarian Cancer 2011, 4, 34–37. [Google Scholar]
- Colomer, R. Review of gemcitabine plus taxane combination therapy in the first-line treatment of metastatic breast cancer. Eur. J. Cancer Suppl. 2008, 6, 9–12. [Google Scholar]
- Colomer, R. Gemcitabine plus taxane combinations in metastatic breast cancer: A comprehensive review. Eur. J. Cancer Suppl. 2005, 3, 9–16. [Google Scholar]
- Makrantonakis, P.; Ziotopoulos, P.; Agelidou, A.; Polyzos, A.; Ziras, A.; Chandrinos, V.; Vossos, A.; Kalykaki, A.; Androulakis, N.; Geroyianni, A. Vinorelbine and cisplatin combination in pretreated patients with advanced non-small cell lung cancer pretreated with a taxane-based regimen: A multicenter phase II study. Lung Cancer 2006, 53, 85–90. [Google Scholar]
- Pereira, J.R.; Fein, L.; del Giglio, A.; Blajman, C.R.; Richardet, E.; Schwartsmann, G.; Orlando, M.; Hall, B.J.; West, T.M.; van Kooten, M. Gemcitabine administered as a short infusion versus a fixed dose rate in combination with cisplatin for the treatment of patients with advanced non-small cell lung cancer. Lung Cancer 2007, 58, 80–87. [Google Scholar]
- Scripture, C.D.; Figg, W.D. Drug interactions in cancer therapy. Nat. Rev. Cancer 2006, 6, 546–558. [Google Scholar]
- Aird, R.; Cummings, J.; Ritchie, A.; Muir, M.; Morris, R.; Chen, H.; Sadler, P.; Jodrell, D. In vitro and in vivo activity and cross resistance profiles of novel ruthenium (II) organometallic arene complexes in human ovarian cancer. Br. J. Cancer 2002, 86, 1652–1657. [Google Scholar]
- Tkaczuk, K.H.R. Review of the contemporary cytotoxic and biologic combinations available for the treatment of metastatic breast cancer. Clin. Ther. 2009, 31, 2273–2289. [Google Scholar]
- Friedberg, J.W.; Neuberg, D.; Gribben, J.G.; Fisher, D.C.; Koval, M.; Poor, C.M.; Green, L.M.; Daley, J.; Soiffer, R.; Ritz, J. Combination immunotherapy with rituximab and interleukin 2 in patients with relapsed or refractory follicular non-Hodgkin’s lymphoma. Br. J. Haematol. 2002, 117, 828–834. [Google Scholar]
- Pandey, S.; Murphy, R.F.; Agrawal, D.K. Recent advances in the immunobiology of ceramide. Exp. Mol. Pathol. 2007, 82, 298–309. [Google Scholar]
- Vlerken, L.E.; Duan, Z.; Seiden, M.V.; Amiji, M.M. Modulation of intracellular ceramide using polymeric nanoparticles to overcome multidrug resistance in cancer. Cancer Res. 2007, 67, 4843–4850. [Google Scholar]
- Taniguchi, M.; Okazaki, T. The role of sphingomyelin and sphingomyelin synthases in cell death proliferation and migration-from cell and animal models to human disorders. Biochim. Biophys. Acta 2013. [Google Scholar] [CrossRef]
- Henry, B.; Möller, C.; Dimanche-Boitrel, M.T.; Gulbins, E.; Becker, K.A. Targeting the ceramide system in cancer. Cancer Lett. 2011, 332, 286–294. [Google Scholar]
- Stover, T.; Kester, M. Liposomal delivery enhances short-chain ceramide-induced apoptosis of breast cancer cells. J. Pharmacol. Exp. Ther. 2003, 307, 468–475. [Google Scholar]
- Haynes, T.A.S.; Filippov, V.; Filippova, M.; Yang, J.; Zhang, K.; Duerksen-Hughes, P.J. DNA damage induces down-regulation of UDP-glucose ceramide glucosyltransferase increases ceramide levels and triggers apoptosis in p53-deficient cancer cells. Biochim. Biophys. Acta 2012, 1821, 943–953. [Google Scholar]
- Itoh, M.; Kitano, T.; Watanabe, M.; Kondo, T.; Yabu, T.; Taguchi, Y.; Iwai, K.; Tashima, M.; Uchiyama, T.; Okazaki, T. Possible role of ceramide as an indicator of chemoresistance decrease of the ceramide content via activation of glucosylceramide synthase and sphingomyelin synthase in chemoresistant leukemia. Clin. Cancer Res. 2003, 9, 415–423. [Google Scholar]
- Vlerken, L.E.; Duan, Z.; Little, S.R.; Seiden, M.V.; Amiji, M.M. Augmentation of therapeutic efficacy in drug-resistant tumor models using ceramide coadministration in temporal-controlled polymer-blend nanoparticle delivery systems. AAPS J. 2010, 12, 171–180. [Google Scholar]
- Tallarida, R.J. Drug synergism: Its detection and applications. J. Pharmacol. Exp. Ther. 2001, 298, 865–872. [Google Scholar]
- Pinato, D.J.; Graham, J.; Gabra, H.; Sharma, R. Evolving concepts in the management of drug resistant ovarian cancer: Dose dense chemotherapy and the reversal of clinical platinum resistance. Cancer Treat. Rev. 2013, 39, 153–160. [Google Scholar]
- Kono, K.; Kojima, C.; Hayashi, N.; Nishisaka, E.; Kiura, K.; Watarai, S.; Harada, A. Preparation and cytotoxic activity of poly (ethylene glycol)-modified poly (amidoamine) dendrimers bearing adriamycin. Biomaterials 2008, 29, 1664–1675. [Google Scholar]
- Liu, S.; Guo, Y.; Huang, R.; Li, J.; Huang, S.; Kuang, Y.; Han, L.; Jiang, C. Gene and doxorubicin co-delivery system for targeting therapy of glioma. Biomaterials 2012, 33, 4907–4916. [Google Scholar]
- Chougule, M.; Patel, A.R.; Sachdeva, P.; Jackson, T.; Singh, M. Anticancer activity of Noscapine an opioid alkaloid in combination with Cisplatin in human non-small cell lung cancer. Lung Cancer 2011, 71, 271–282. [Google Scholar]
- Chou, T.C. Theoretical basis experimental design and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar]
- Lakhani, S.A.; Masud, A.; Kuida, K.; Porter, G.A.; Booth, C.J.; Mehal, W.Z.; Inayat, I.; Flavell, R.A. Caspases 3 and 7: Key mediators of mitochondrial events of apoptosis. Science 2006, 311, 847–851. [Google Scholar]
- Nehme, A.; Varadarajan, P.; Sellakumar, G.; Gerhold, M.; Niedner, H.; Zhang, Q.; Lin, X.; Christen, R. Modulation of docetaxel-induced apoptosis and cell cycle arrest by all-trans retinoic acid in prostate cancer cells. Br. J. Cancer 2001, 84, 1571–1576. [Google Scholar]
- Louzao, M.C.; Ares, I.R.; Cagide, E.; Espiña, B.; Vilariño, N.; Alfonso, A.; Vieytes, M.R.; Botana, L.M. Palytoxins and cytoskeleton: An overview. Toxicon 2011, 57, 460–469. [Google Scholar]
- Riedl, S.J.; Shi, Y. Molecular mechanisms of caspase regulation during apoptosis. Nat. Rev. Mol. Cell Biol. 2004, 5, 897–907. [Google Scholar]
- Yang, Y.; Wang, J.; Zhang, X.; Lu, W.; Zhang, Q. A novel mixed micelle gel with thermo-sensitive property for the local delivery of docetaxel. J. Control. Release 2009, 135, 175–182. [Google Scholar]
- Oh, K.T.; Lee, E.S.; Kim, D.; Bae, Y.H. l-histidine-based pH-sensitive anticancer drug carrier micelle: Reconstitution and brief evaluation of its systemic toxicity. Int. J. Pharm. 2008, 358, 177–183. [Google Scholar]
- Chapman, J.V.; Gouazé-Andersson, V.; Messner, M.C.; Flowers, M.; Karimi, R.; Kester, M.; Barth, B.M.; Liu, X.; Liu, Y.Y.; Giuliano, A.E. Metabolism of short-chain ceramide by human cancer cells-Implications for therapeutic approaches. Biochem. Pharmacol. 2010, 80, 308–315. [Google Scholar]
- Yang, X.; Li, Y.; Li, M.; Zhang, L.; Feng, L.; Zhang, N. Hyaluronic acid-coated nanostructured lipid carriers for targeting paclitaxel to cancer. Cancer Lett. 2013, 334, 338–345. [Google Scholar]
- Liu, D.; Liu, F.; Liu, Z.; Wang, L.; Zhang, N. Tumor specific delivery and therapy by double-targeted nanostructured lipid carriers with anti-VEGFR-2 antibody. Mol. Pharm. 2011, 8, 2291–2301. [Google Scholar]
- Montero, A.; Fossella, F.; Hortobagyi, G.; Valero, V. Docetaxel for treatment of solid tumours: A systematic review of clinical data. Lancet Oncol. 2005, 6, 229–239. [Google Scholar]
- Hari, M.; Loganzo, F.; Annable, T.; Tan, X.; Musto, S.; Morilla, D.B.; Nettles, J.H.; Snyder, J.P.; Greenberger, L.M. Paclitaxel-resistant cells have a mutation in the paclitaxel-binding region of β-tubulin (Asp26Glu) and less stable microtubules. Mol. Cancer Ther. 2006, 5, 270–278. [Google Scholar]
- Rao, J.; Li, N. Microfilament actin remodeling as a potential target for cancer drug development. Curr. Cancer Drug Targrts 2004, 4, 345–354. [Google Scholar]
- Lindberg, U.; Karlsson, R.; Lassing, I.; Schutt, C.E.; Höglund, A.S. The microfilament system and malignancy. Semin. Cancer Biol. 2008, 18, 2–11. [Google Scholar]
- Hayot, C.; Debeir, O.; van Ham, P.; van Damme, M.; Kiss, R.; Decaestecker, C. Characterization of the activities of actin-affecting drugs on tumor cell migration. Toxicol. Appl. Pharmacol. 2006, 211, 30–40. [Google Scholar]
- Wang, J.; Lv, X.W.; Du, Y.G. Potential mechanisms involved in ceramide-induced apoptosis in human colon cancer HT29 cells. Biomed. Environ. Sci. 2009, 22, 76–85. [Google Scholar]
- Wang, L.; Li, M.; Zhang, N. Folate-targeted docetaxel-lipid-based-nanosuspensions for active-targeted cancer therapy. Int. J. Nanomed. 2012, 7, 3281–3294. [Google Scholar]
- Wang, L.; Liu, Z.; Liu, D.; Liu, C.; Juan, Z.; Zhang, N. Docetaxel-loaded-lipid-based-nanosuspensions (DTX-LNS): Preparation pharmacokinetics tissue distribution and antitumor activity. Int. J. Pharm. 2011, 413, 194–201. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CE + DTX | Combination molar ratio | |||||
|---|---|---|---|---|---|---|
| 0.25 | 0.5 | 1 | 2 | 4 | ||
| B16 | CI at IC50 | 0.49 ± 0.17 | 0.31 ± 0.13 | 0.47 ± 0.14 | 0.95 ± 0.26 | 1.08 ± 0.17 |
| Interpretation | synergism | strong synergism | synergism | additive effect | additive effect | |
| MCF-7 | CI at IC50 | 0.62 ± 0.13 | 0.48 ± 0.11 | 0.79 ± 0.25 | 1.17 ± 0.18 | 1.03 ± 0.19 |
| Interpretation | moderate synergism | synergism | moderate synergism | moderate antagonism | additive effect | |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Feng, L.-X.; Li, M.; Liu, Y.-J.; Yang, S.-M.; Zhang, N. Synergistic Enhancement of Cancer Therapy Using a Combination of Ceramide and Docetaxel. Int. J. Mol. Sci. 2014, 15, 4201-4220. https://doi.org/10.3390/ijms15034201
Feng L-X, Li M, Liu Y-J, Yang S-M, Zhang N. Synergistic Enhancement of Cancer Therapy Using a Combination of Ceramide and Docetaxel. International Journal of Molecular Sciences. 2014; 15(3):4201-4220. https://doi.org/10.3390/ijms15034201
Chicago/Turabian StyleFeng, Li-Xia, Min Li, Yong-Jun Liu, Shao-Mei Yang, and Na Zhang. 2014. "Synergistic Enhancement of Cancer Therapy Using a Combination of Ceramide and Docetaxel" International Journal of Molecular Sciences 15, no. 3: 4201-4220. https://doi.org/10.3390/ijms15034201