Mitochondria in the Center of Human Eosinophil Apoptosis and Survival

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Mitochondria and Bcl-2 Family Members
2.1. Bcl-2 Members and Pore-Forming Activity of Bax and Bid
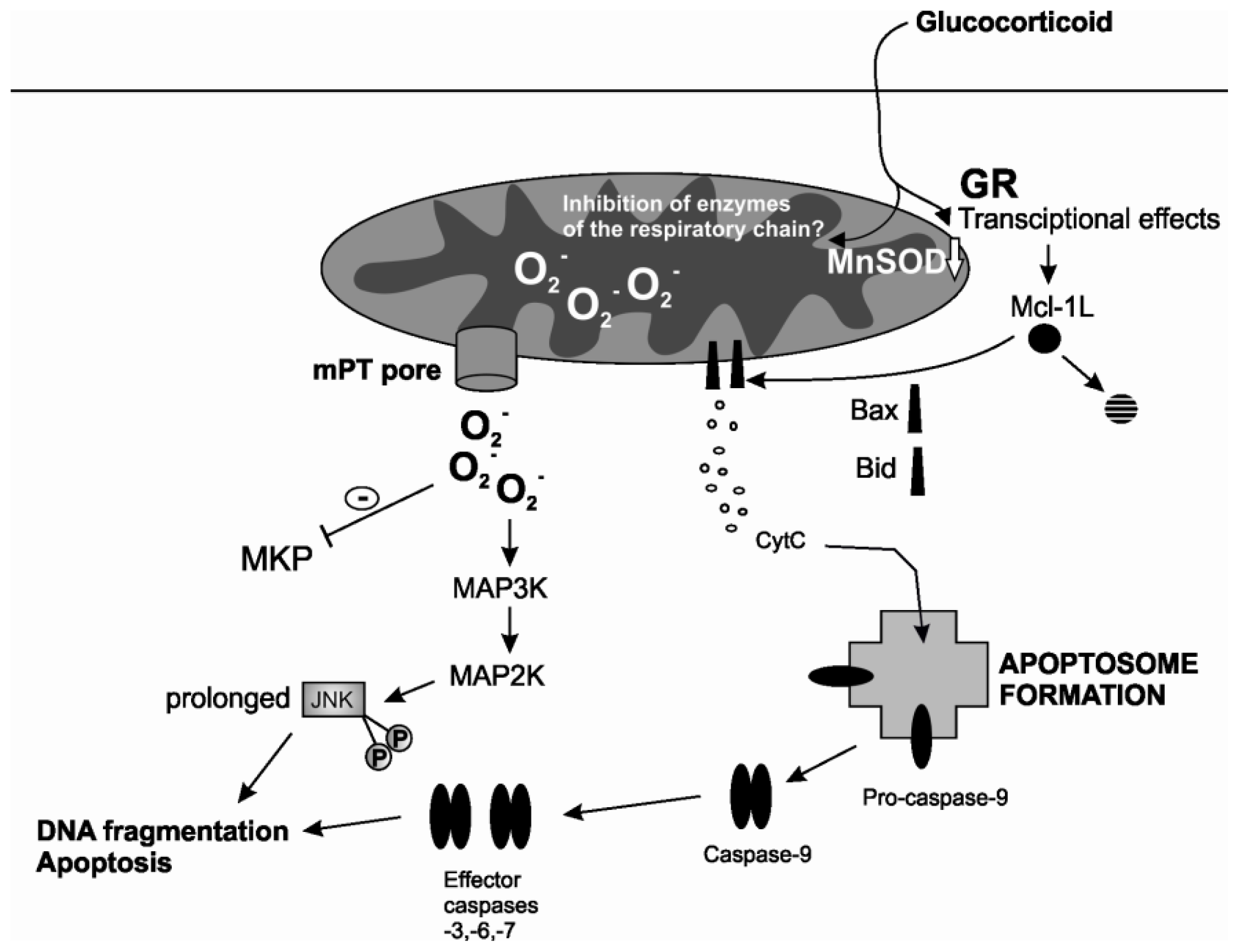
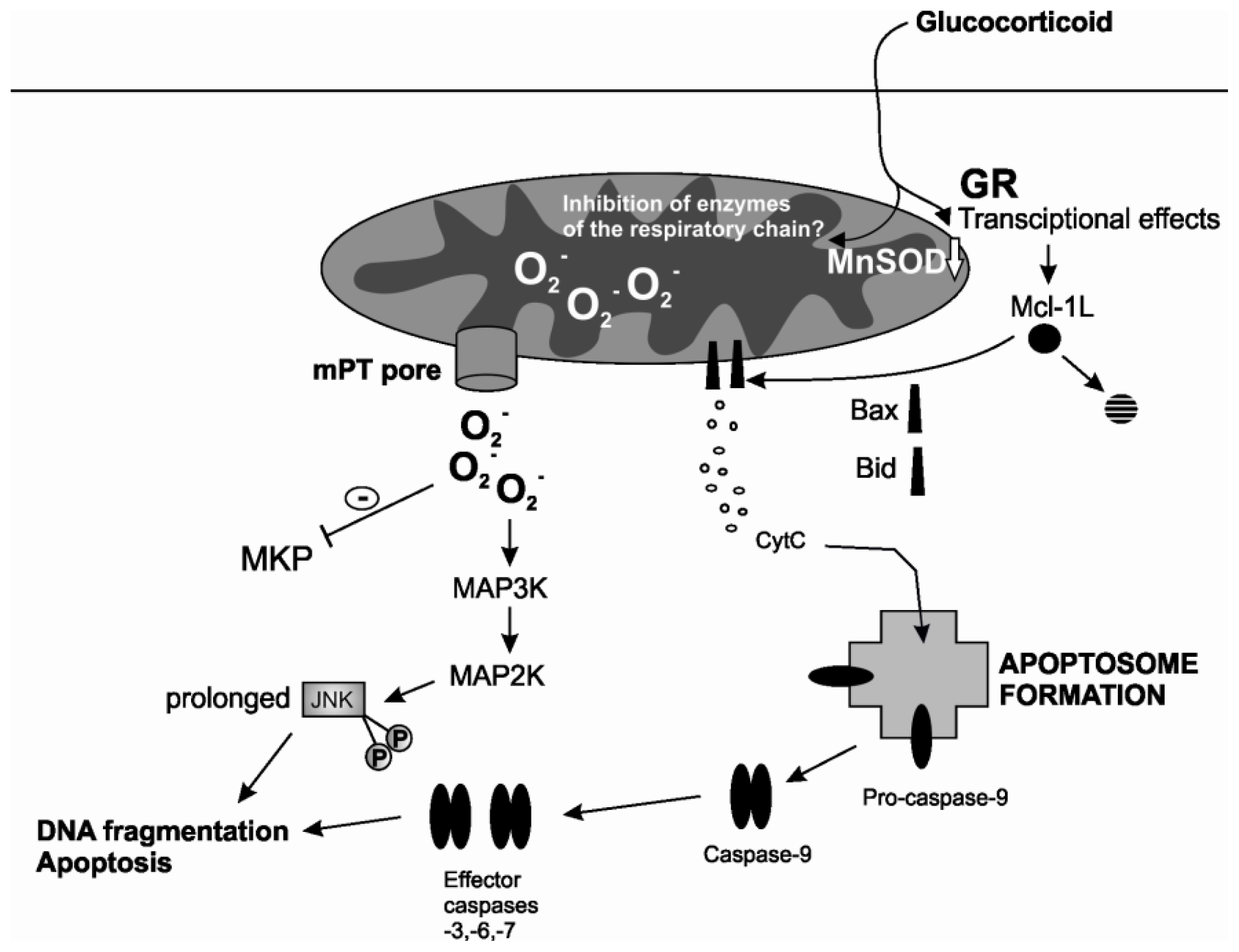
2.2. Mitochondrial Permeability Transition
3. Reactive Oxygen Species (ROS) and Pro-Apoptotic Signalling Pathways
3.1. ROS
3.2. Kinases Activated by ROS
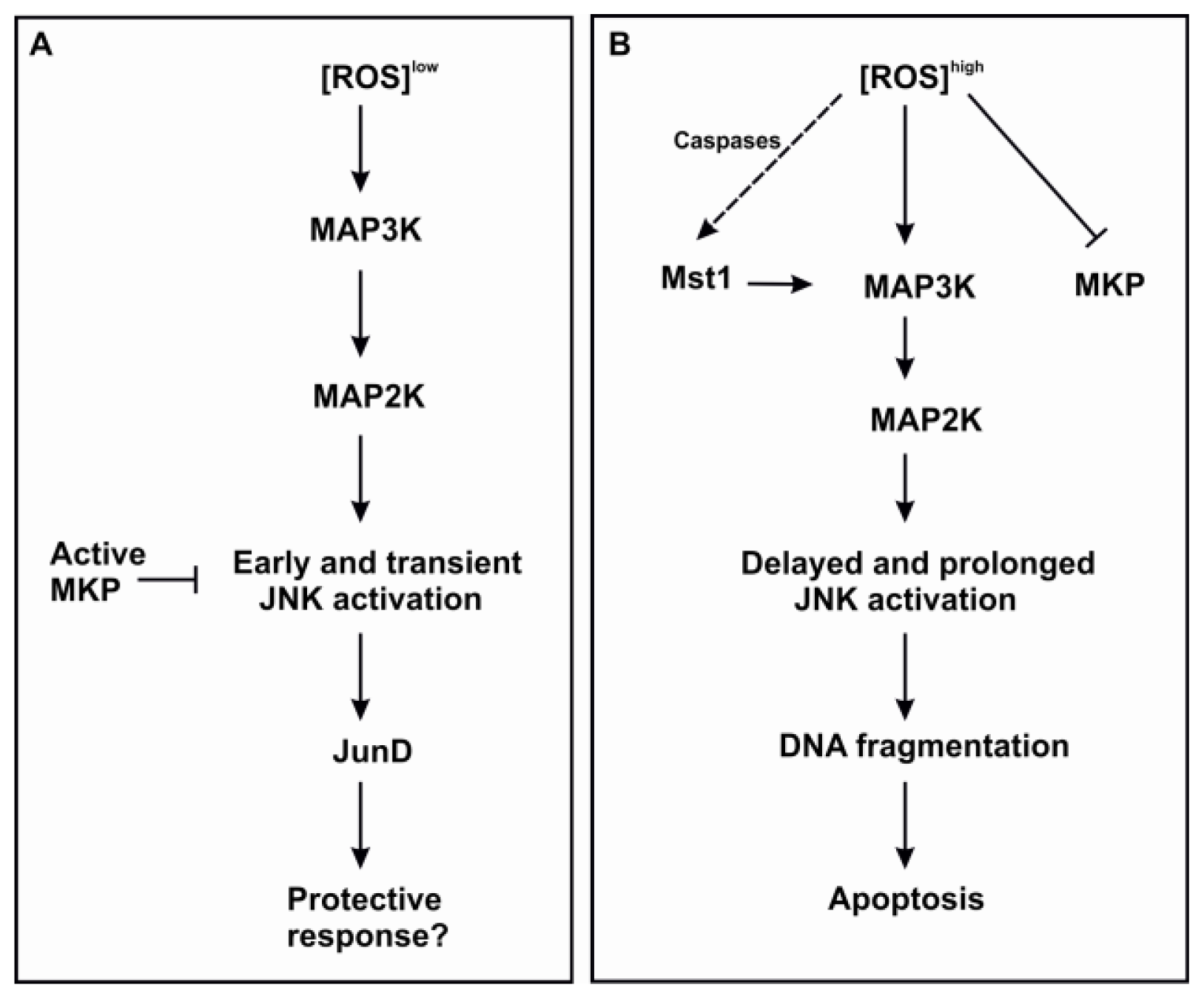
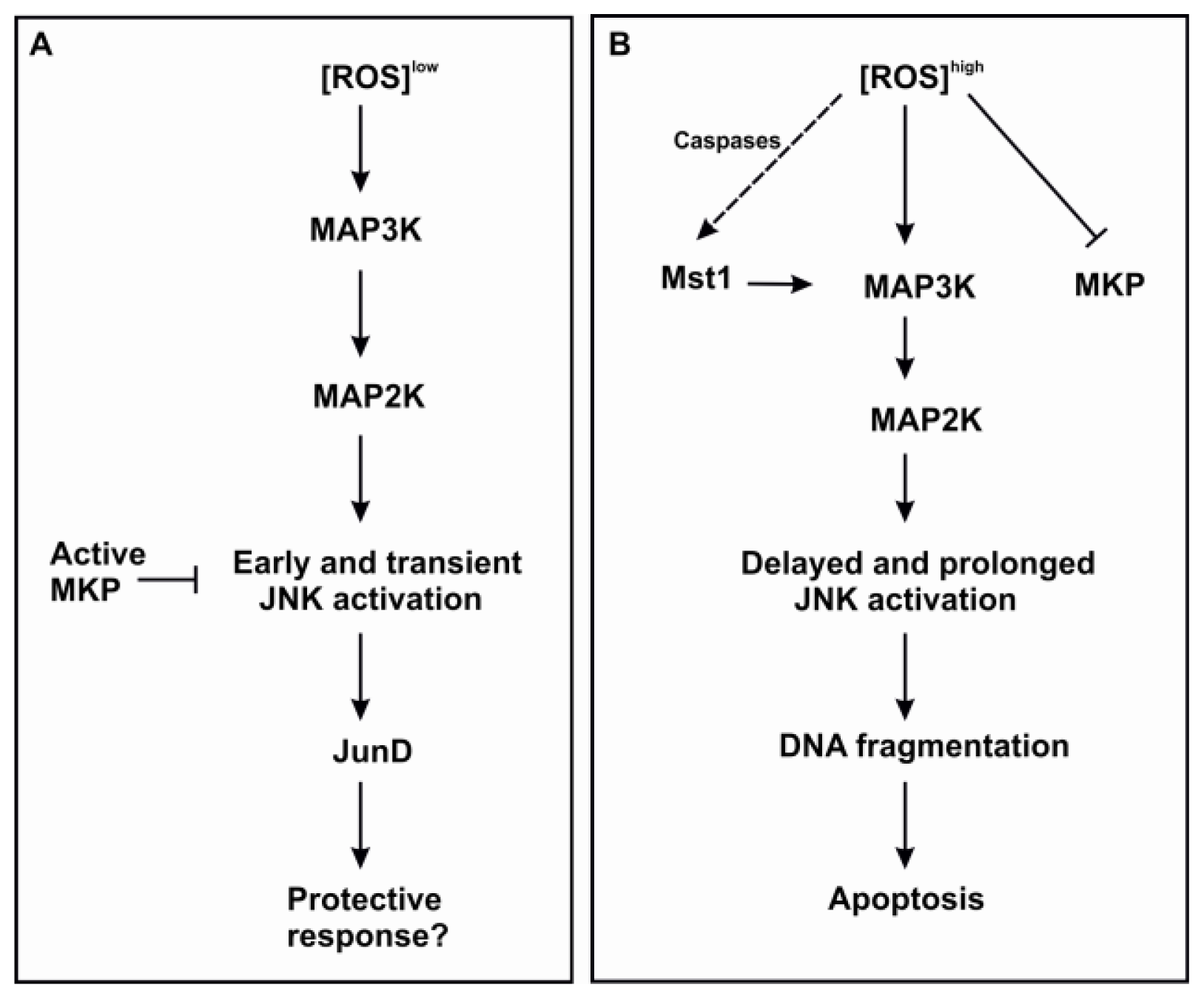
3.2.1. JNK
3.2.2. ERK
3.2.3. p38
3.2.4. Mst 1/2
4. Summary and Conclusions
Acknowledgments
Conflicts of Interest
References
- Trivedi, S.G.; Lloyd, C.M. Eosinophils in the pathogenesis of allergic airways disease. Cell. Mol. Life Sci. 2007, 64, 1269–1289. [Google Scholar]
- Zuo, L.; Rothenberg, M.E. Gastrointestinal eosinophilia. Immunol. Allergy Clin. N. Am. 2007, 27, 443–455. [Google Scholar]
- Gleich, G.J.; Leiferman, K.M. The hypereosinophilic syndromes: Current concepts and treatments. Br. J. Haematol. 2009, 145, 271–285. [Google Scholar]
- Ellyard, J.I.; Simson, L.; Parish, C.R. Th2-mediated anti-tumour immunity: Friend or foe? Tissue Antigens 2007, 70, 1–11. [Google Scholar]
- Klion, A.D.; Nutman, T.B. The role of eosinophils in host defense against helminth parasites. J. Allergy Clin. Immunol. 2004, 113, 30–37. [Google Scholar]
- Hogan, S.P.; Rosenberg, H.F.; Moqbel, R.; Phipps, S.; Foster, P.S.; Lacy, P.; Kay, A.B.; Rothenberg, M.E. Eosinophils: Biological properties and role in health and disease. Clin. Exp. Allergy 2008, 38, 709–750. [Google Scholar]
- Jacobsen, E.A.; Ochkur, S.I.; Pero, R.S.; Taranova, A.G.; Protheroe, C.A.; Colbert, D.C.; Lee, N.A.; Lee, J.J. Allergic pulmonary inflammation in mice is dependent on eosinophil-induced recruitment of effector T cells. J. Exp. Med. 2008, 205, 699–710. [Google Scholar]
- Humbles, A.A.; Lloyd, C.M.; McMillan, S.J.; Friend, D.S.; Xanthou, G.; McKenna, E.E.; Ghiran, S.; Gerard, N.P.; Yu, C.; Orkin, S.H.; et al. A critical role for eosinophils in allergic airways remodeling. Science 2004, 305, 1776–1779. [Google Scholar]
- Nair, P.; Pizzichini, M.M.; Kjarsgaard, M.; Inman, M.D.; Efthimiadis, A.; Pizzichini, E.; Hargreave, F.E.; O’Byrne, P.M. Mepolizumab for prednisone-dependent asthma with sputum eosinophilia. N. Engl. J. Med. 2009, 360, 985–993. [Google Scholar]
- Haldar, P.; Brightling, C.E.; Hargadon, B.; Gupta, S.; Monteiro, W.; Sousa, A.; Marshall, R.P.; Bradding, P.; Green, R.H.; Wardlaw, A.J.; et al. Mepolizumab and exacerbations of refractory eosinophilic asthma. N. Engl. J. Med. 2009, 360, 973–984. [Google Scholar]
- Wenzel, S.E. Eosinophils in asthma—Closing the loop or opening the door? N. Engl. J. Med. 2009, 360, 1026–1028. [Google Scholar]
- Lukawska, J.J.; Livieratos, L.; Sawyer, B.M.; Lee, T.; O’Doherty, M.; Blower, P.J.; Kofi, M.; Ballinger, J.R.; Corrigan, C.J.; Gnanasegaran, G.; et al. Real-time differential tracking of human neutrophil and eosinophil migration in vivo. J. Allergy Clin. Immunol. 2014, 133, 233–239. [Google Scholar]
- Farahi, N.; Singh, N.R.; Heard, S.; Loutsios, C.; Summers, C.; Solanki, C.K.; Solanki, K.; Balan, K.K.; Ruparelia, P.; Peters, A.M.; et al. Use of 111-Indium-labeled autologous eosinophils to establish the in vivo kinetics of human eosinophils in healthy subjects. Blood 2012, 120, 4068–4070. [Google Scholar]
- Walsh, G. Eosinophil apoptosis and clearance in asthma. J. Cell Death 2013, 6, 17–25. [Google Scholar]
- Kankaanranta, H.; Moilanen, E.; Zhang, X. Pharmacological regulation of human eosinophil apoptosis. Curr. Drug Targets Inflamm. Allergy 2005, 4, 433–445. [Google Scholar]
- Rothenberg, M.E.; Owen, W.F., Jr; Silberstein, D.S.; Soberman, R.J.; Austen, K.F.; Stevens, R.L. Eosinophils cocultured with endothelial cells have increased survival and functional properties. Science 1987, 237, 645–647. [Google Scholar]
- Kankaanranta, H.; Lindsay, M.A.; Giembycz, M.A.; Zhang, X.; Moilanen, E.; Barnes, P.J. Delayed eosinophil apoptosis in asthma. J. Allergy Clin. Immunol. 2000, 106, 77–83. [Google Scholar]
- Simon, H.U.; Yousefi, S.; Schranz, C.; Schapowal, A.; Bachert, C.; Blaser, K. Direct demonstration of delayed eosinophil apoptosis as a mechanism causing tissue eosinophilia. J. Immunol. 1997, 158, 3902–3908. [Google Scholar]
- Duffin, R.; Leitch, A.E.; Fox, S.; Haslett, C.; Rossi, A.G. Targeting granulocyte apoptosis: Mechanisms models and therapies. Immunol. Rev. 2010, 236, 28–40. [Google Scholar]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the nomenclature committee on cell death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar]
- Rasola, A.; Bernardi, P. Mitochondrial permeability transition in Ca2+-dependent apoptosis and necrosis. Cell Calcium 2011, 50, 222–233. [Google Scholar]
- Letuve, S.; Druilhe, A.; Grandsaigne, M.; Aubier, M.; Pretolani, M. Involvement of caspases and of mitochondria in Fas ligation-induced eosinophil apoptosis: Modulation by interleukin-5 and interferon-gamma. J. Leukoc. Biol. 2001, 70, 767–775. [Google Scholar]
- Sharma, S.K.; Almeida, F.A.; Kierstein, S.; Hortobagyi, L.; Lin, T.; Larkin, A.; Peterson, J.; Yagita, H.; Zangrilli, J.G.; Haczku, A. Systemic FasL neutralization increases eosinophilic inflammation in a mouse model of asthma. Allergy 2012, 67, 328–335. [Google Scholar]
- Beauvais, F.; Joly, F. Effects of nitric oxide on the eosinophil survival in vitro A role for nitrosyl-heme. FEBS Lett. 1999, 443, 37–40. [Google Scholar]
- Zhang, X.; Msc Moilanen, E.; Lahti, A.; Hamalainen, M.; Giembycz, M.A.; Barnes, P.J.; Lindsay, M.A.; Kankaanranta, H. Regulation of eosinophil apoptosis by nitric oxide: Role of c-Jun-N-terminal kinase and signal transducer and activator of transcription 5. J. Allergy Clin. Immunol. 2003, 112, 93–101. [Google Scholar]
- Hebestreit, H.; Dibbert, B.; Balatti, I.; Braun, D.; Schapowal, A.; Blaser, K.; Simon, H.U. Disruption of fas receptor signaling by nitric oxide in eosinophils. J. Exp. Med. 1998, 187, 415–425. [Google Scholar]
- Oliveira, M.S.; de O Barreto, E.; Zamuner, S.; Pires, A.L.; Ferreira, T.P.; Cordeiro, R.S.; Lagente, V.; Martins, M.A.; Wallace, J.L.; e Silva, P.M. Suppressive effects of nitric oxide-releasing prednisolone NCX-1015 on the allergic pleural eosinophil recruitment in rats. Clin. Exp. Allergy 2008, 38, 1830–1837. [Google Scholar]
- Feder, L.S.; Stelts, D.; Chapman, R.W.; Manfra, D.; Crawley, Y.; Jones, H.; Minnicozzi, M.; Fernandez, X.; Paster, T.; Egan, R.W.; et al. Role of nitric oxide on eosinophilic lung inflammation in allergic mice. Am. J. Respir. Cell Mol. Biol. 1997, 17, 436–442. [Google Scholar]
- Lehtimaki, L.; Kankaanranta, H.; Saarelainen, S.; Hahtola, P.; Jarvenpaa, R.; Koivula, T.; Turjanmaa, V.; Moilanen, E. Extended exhaled NO measurement differentiates between alveolar and bronchial inflammation. Am. J. Respir. Crit. Care Med. 2001, 163, 1557–1561. [Google Scholar]
- Zhang, X.; Moilanen, E.; Kankaanranta, H. Enhancement of human eosinophil apoptosis by fluticasone propionate budesonide and beclomethasone. Eur. J. Pharmacol. 2000, 406, 325–332. [Google Scholar]
- Lamas, A.M.; Leon, O.G.; Schleimer, R.P. Glucocorticoids inhibit eosinophil responses to granulocyte-macrophage colony-stimulating factor. J. Immunol. 1991, 147, 254–259. [Google Scholar]
- Wallen, N.; Kita, H.; Weiler, D.; Gleich, G.J. Glucocorticoids inhibit cytokine-mediated eosinophil survival. J. Immunol. 1991, 147, 3490–3495. [Google Scholar]
- Lee, E.; Robertson, T.; Smith, J.; Kilfeather, S. Leukotriene receptor antagonists and synthesis inhibitors reverse survival in eosinophils of asthmatic individuals. Am. J. Respir. Crit. Care Med. 2000, 161, 1881–1886. [Google Scholar]
- Yasui, K.; Hu, B.; Nakazawa, T.; Agematsu, K.; Komiyama, A. Theophylline accelerates human granulocyte apoptosis not via phosphodiesterase inhibition. J. Clin. Investig. 1997, 100, 1677–1684. [Google Scholar]
- Ilmarinen, P.; Kankaanranta, H. Eosinophil apoptosis as a therapeutic target in allergic asthma. Basic Clin. Pharmacol. Toxicol. 2013, 114, 109–117. [Google Scholar]
- Woolley, K.L.; Gibson, P.G.; Carty, K.; Wilson, A.J.; Twaddell, S.H.; Woolley, M.J. Eosinophil apoptosis and the resolution of airway inflammation in asthma. Am. J. Respir. Crit. Care Med. 1996, 154, 237–243. [Google Scholar]
- Druilhe, A.; Wallaert, B.; Tsicopoulos, A.; Lapa e Silva, J.R.; Tillie-Leblond, I.; Tonnel, A.B.; Pretolani, M. Apoptosis proliferation and expression of Bcl-2 Fas and Fas ligand in bronchial biopsies from asthmatics. Am. J. Respir. Cell Mol. Biol. 1998, 19, 747–757. [Google Scholar]
- Vignola, A.M.; Chanez, P.; Chiappara, G.; Siena, L.; Merendino, A.; Reina, C.; Gagliardo, R.; Profita, M.; Bousquet, J.; Bonsignore, G. Evaluation of apoptosis of eosinophils macrophages and T lymphocytes in mucosal biopsy specimens of patients with asthma and chronic bronchitis. J. Allergy Clin. Immunol. 1999, 103, 563–573. [Google Scholar]
- Duncan, C.J.; Lawrie, A.; Blaylock, M.G.; Douglas, J.G.; Walsh, G.M. Reduced eosinophil apoptosis in induced sputum correlates with asthma severity. Eur. Respir. J. 2003, 22, 484–490. [Google Scholar]
- Montuschi, P.; Peters-Golden, M.L. Leukotriene modifiers for asthma treatment. Clin. Exp. Allergy 2010, 40, 1732–1741. [Google Scholar]
- Lim, S.; Tomita, K.; Caramori, G.; Jatakanon, A.; Oliver, B.; Keller, A.; Adcock, I.; Chung, K.F.; Barnes, P.J. Low-dose theophylline reduces eosinophilic inflammation but not exhaled nitric oxide in mild asthma. Am. J. Respir. Crit. Care Med. 2001, 164, 273–276. [Google Scholar]
- Jetzek-Zader, M.; Gudowius, S.; Feyen, O.; Stevens, M.; Lipfert, P.; Niehues, T. A single intravenous dose of prednisolone induces phosphatidylserine externalization loss of surface marker expression and a 24-h net increase in human peripheral blood lymphocytes ex vivo. Rheumatol. Int. 2007, 27, 667–673. [Google Scholar]
- Wiegers, G.J.; Knoflach, M.; Bock, G.; Niederegger, H.; Dietrich, H.; Falus, A.; Boyd, R.; Wick, G. CD4(+)CD8(+)TCR(low) thymocytes express low levels of glucocorticoid receptors while being sensitive to glucocorticoid-induced apoptosis. Eur. J. Immunol. 2001, 31, 2293–2301. [Google Scholar]
- Leussink, V.I.; Jung, S.; Merschdorf, U.; Toyka, K.V.; Gold, R. High-dose methylprednisolone therapy in multiple sclerosis induces apoptosis in peripheral blood leukocytes. Arch. Neurol. 2001, 58, 91–97. [Google Scholar]
- Meagher, L.C.; Cousin, J.M.; Seckl, J.R.; Haslett, C. Opposing effects of glucocorticoids on the rate of apoptosis in neutrophilic and eosinophilic granulocytes. J. Immunol. 1996, 156, 4422–4428. [Google Scholar]
- Zhang, X.; Moilanen, E.; Kankaanranta, H. Beclomethasone budesonide and fluticasone propionate inhibit human neutrophil apoptosis. Eur. J. Pharmacol. 2001, 431, 365–371. [Google Scholar]
- Zhang, X.; Moilanen, E.; Adcock, I.M.; Lindsay, M.A.; Kankaanranta, H. Divergent effect of mometasone on human eosinophil and neutrophil apoptosis. Life Sci. 2002, 71, 1523–1534. [Google Scholar]
- Marwick, J.A.; Dorward, D.A.; Lucas, C.D.; Jones, K.O.; Sheldrake, T.A.; Fox, S.; Ward, C.; Murray, J.; Brittan, M.; Hirani, N.; et al. Oxygen levels determine the ability of glucocorticoids to influence neutrophil survival in inflammatory environments. J. Leukoc. Biol. 2013, 94, 1285–1292. [Google Scholar]
- Peachman, K.K.; Lyles, D.S.; Bass, D.A. Mitochondria in eosinophils: Functional role in apoptosis but not respiration. Proc. Natl. Acad. Sci. USA 2001, 98, 1717–1722. [Google Scholar]
- Dewson, G.; Cohen, G.M.; Wardlaw, A.J. Interleukin-5 inhibits translocation of Bax to the mitochondria cytochrome c release and activation of caspases in human eosinophils. Blood 2001, 98, 2239–2247. [Google Scholar]
- Gardai, S.J.; Hoontrakoon, R.; Goddard, C.D.; Day, B.J.; Chang, L.Y.; Henson, P.M.; Bratton, D.L. Oxidant-mediated mitochondrial injury in eosinophil apoptosis: Enhancement by glucocorticoids and inhibition by granulocyte-macrophage colony-stimulating factor. J. Immunol. 2003, 170, 556–566. [Google Scholar]
- Segal, M.; Niazi, S.; Simons, M.P.; Galati, S.A.; Zangrilli, J.G. Bid activation during induction of extrinsic and intrinsic apoptosis in eosinophils. Immunol. Cell Biol. 2007, 85, 518–524. [Google Scholar]
- Maret, M.; Ruffie, C.; Letuve, S.; Phelep, A.; Thibaudeau, O.; Marchal, J.; Pretolani, M.; Druilhe, A. A role for Bid in eosinophil apoptosis and in allergic airway reaction. J. Immunol. 2009, 182, 5740–5747. [Google Scholar]
- Lee, Y.A.; Shin, M.H. Mitochondrial respiration is required for activation of ERK1/2 and caspase-3 in human eosinophils stimulated with hydrogen peroxide. J. Investig. Allergol. Clin. Immunol. 2009, 19, 188–194. [Google Scholar]
- Van Der Vliet, H.J.; Wever, P.C.; van Diepen, F.N.; Yong, S.L.; Ten Berge, I.J. Quantification of Bax/Bcl-2 ratios in peripheral blood lymphocytes monocytes and granulocytes and their relation to susceptibility to anti-Fas (anti-CD95)-induced apoptosis. Clin. Exp. Immunol. 1997, 110, 324–328. [Google Scholar]
- Druilhe, A.; Arock, M.; le Goff, L.; Pretolani, M. Human eosinophils express bcl-2 family proteins: Modulation of Mcl-1 expression by IFN-gamma. Am. J. Respir. Cell Mol. Biol. 1998, 18, 315–322. [Google Scholar]
- Shen, Z.J.; Esnault, S.; Schinzel, A.; Borner, C.; Malter, J.S. The peptidyl-prolyl isomerase Pin1 facilitates cytokine-induced survival of eosinophils by suppressing Bax activation. Nat. Immunol. 2009, 10, 257–265. [Google Scholar]
- Oh, J.; Malter, J.S. Pin1-FADD interactions regulate Fas-mediated apoptosis in activated eosinophils. J. Immunol. 2013, 190, 4937–4945. [Google Scholar]
- Zangrilli, J.; Robertson, N.; Shetty, A.; Wu, J.; Hastie, A.; Fish, J.E.; Litwack, G.; Peters, S.P. Effect of IL-5 glucocorticoid and Fas ligation on Bcl-2 homologue expression and caspase activation in circulating human eosinophils. Clin. Exp. Immunol. 2000, 120, 12–21. [Google Scholar]
- Duffin, R.; Leitch, A.E.; Sheldrake, T.A.; Hallett, J.M.; Meyer, C.; Fox, S.; Alessandri, A.L.; Martin, M.C.; Brady, H.J.; Teixeira, M.M.; et al. The CDK inhibitor R-roscovitine promotes eosinophil apoptosis by down-regulation of Mcl-1. FEBS Lett. 2009, 583, 2540–2546. [Google Scholar]
- Farahi, N.; Uller, L.; Juss, J.K.; Langton, A.J.; Cowburn, A.S.; Gibson, A.; Foster, M.R.; Farrow, S.N.; Marco-Casanova, P.; Sobolewski, A.; et al. Effects of the cyclin-dependent kinase inhibitor R-roscovitine on eosinophil survival and clearance. Clin. Exp. Allergy 2011, 41, 673–687. [Google Scholar]
- Sivertson, K.L.; Seeds, M.C.; Long, D.L.; Peachman, K.K.; Bass, D.A. The differential effect of dexamethasone on granulocyte apoptosis involves stabilization of Mcl-1L in neutrophils but not in eosinophils. Cell. Immunol. 2007, 246, 34–45. [Google Scholar]
- Germain, M.; Milburn, J.; Duronio, V. MCL-1 inhibits BAX in the absence of MCL-1/BAX interaction. J. Biol. Chem. 2008, 283, 6384–6392. [Google Scholar]
- Saffar, A.S.; Dragon, S.; Ezzati, P.; Shan, L.; Gounni, A.S. Phosphatidylinositol 3-kinase and p38 mitogen-activated protein kinase regulate induction of Mcl-1 and survival in glucocorticoid-treated human neutrophils. J. Allergy Clin. Immunol. 2008, 121, 492–498. [Google Scholar]
- Dewson, G.; Walsh, G.M.; Wardlaw, A.J. Expression of Bcl-2 and its homologues in human eosinophils Modulation by interleukin-5. Am. J. Respir. Cell Mol. Biol. 1999, 20, 720–728. [Google Scholar]
- Kaasik, A.; Safiulina, D.; Zharkovsky, A.; Veksler, V. Regulation of mitochondrial matrix volume. Am. J. Physiol. Cell Physiol. 2007, 292, C157–C163. [Google Scholar]
- Ilmarinen-Salo, P.; Moilanen, E.; Kinnula, V.L.; Kankaanranta, H. Nitric oxide-induced eosinophil apoptosis is dependent on mitochondrial permeability transition (mPT), JNK and oxidative stress: Apoptosis is preceded but not mediated by early mPT-dependent JNK activation. Respir. Res. 2012, 13. [Google Scholar] [CrossRef]
- Letuve, S.; Druilhe, A.; Grandsaigne, M.; Aubier, M.; Pretolani, M. Critical role of mitochondria but not caspases during glucocorticosteroid-induced human eosinophil apoptosis. Am. J. Respir. Cell Mol. Biol. 2002, 26, 565–571. [Google Scholar]
- Garcia-Perez, C.; Roy, S.S.; Naghdi, S.; Lin, X.; Davies, E.; Hajnoczky, G. Bid-induced mitochondrial membrane permeabilization waves propagated by local reactive oxygen species (ROS) signaling. Proc. Natl. Acad. Sci. USA 2012, 109, 4497–4502. [Google Scholar]
- Petronilli, V.; Penzo, D.; Scorrano, L.; Bernardi, P.; di Lisa, F. The mitochondrial permeability transition release of cytochrome c and cell death Correlation with the duration of pore openings in situ. J. Biol. Chem. 2001, 276, 12030–12034. [Google Scholar]
- Ichas, F.; Jouaville, L.S.; Mazat, J.P. Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell 1997, 89, 1145–1153. [Google Scholar]
- Bernardi, P.; Petronilli, V. The permeability transition pore as a mitochondrial calcium release channel: A critical appraisal. J. Bioenerg. Biomembr. 1996, 28, 131–138. [Google Scholar]
- Barsukova, A.; Komarov, A.; Hajnoczky, G.; Bernardi, P.; Bourdette, D.; Forte, M. Activation of the mitochondrial permeability transition pore modulates Ca2+ responses to physiological stimuli in adult neurons. Eur. J. Neurosci. 2011, 33, 831–842. [Google Scholar]
- Bernardi, P.; von Stockum, S. The permeability transition pore as a Ca2+ release channel: New answers to an old question. Cell Calcium 2012, 52, 22–27. [Google Scholar]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial ROS-induced ROS release: An update and review. Biochim. Biophys. Acta 2006, 1757, 509–517. [Google Scholar]
- Ma, Q.; Fang, H.; Shang, W.; Liu, L.; Xu, Z.; Ye, T.; Wang, X.; Zheng, M.; Chen, Q.; Cheng, H. Superoxide flashes: Early mitochondrial signals for oxidative stress-induced apoptosis. J. Biol. Chem. 2011, 286, 27573–27581. [Google Scholar]
- Wedi, B.; Straede, J.; Wieland, B.; Kapp, A. Eosinophil apoptosis is mediated by stimulators of cellular oxidative metabolisms and inhibited by antioxidants: Involvement of a thiol-sensitive redox regulation in eosinophil cell death. Blood 1999, 94, 2365–2373. [Google Scholar]
- Martinez-Losa, M.; Cortijo, J.; Juan, G.; Ramon, M.; Sanz, M.J.; Morcillo, E.J. Modulatory effects of N-acetyl-l-cysteine on human eosinophil apoptosis. Eur. Respir. J. 2007, 30, 436–442. [Google Scholar]
- Nissim Ben Efraim, A.H.; Eliashar, R.; Levi-Schaffer, F. Hypoxia modulates human eosinophil function. Clin. Mol. Allergy 2010, 8. [Google Scholar] [CrossRef]
- Kano, G.; Almanan, M.; Bochner, B.S.; Zimmermann, N. Mechanism of siglec-8-mediated cell death in IL-5-activated eosinophils: Role for reactive oxygen species-enhanced MEK/ERK activation. J. Allergy Clin. Immunol. 2013, 132, 437–445. [Google Scholar]
- Orrenius, S.; Gogvadze, V.; Zhivotovsky, B. Mitochondrial oxidative stress: Implications for cell death. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 143–183. [Google Scholar]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation oxidative stress and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar]
- Gough, D.R.; Cotter, T.G. Hydrogen peroxide: A Jekyll and Hyde signalling molecule. Cell Death Dis. 2011, 2, e213. [Google Scholar]
- Kankaanranta, H.; Giembycz, M.A.; Barnes, P.J.; Haddad, E.l.-B.; Saarelainen, S.; Zhang, X.; Moilanen, E.; Lindsay, M.A. Hydrogen peroxide reverses IL-5 afforded eosinophil survival and promotes constitutive human eosinophil apoptosis. Int. Arch. Allergy Immunol. 2002, 127, 73–78. [Google Scholar]
- Serradell, M.C.; Guasconi, L.; Masih, D.T. Involvement of a mitochondrial pathway and key role of hydrogen peroxide during eosinophil apoptosis induced by excretory-secretory products from Fasciola hepatica. Mol. Biochem. Parasitol. 2009, 163, 95–106. [Google Scholar]
- Tonomura, N.; McLaughlin, K.; Grimm, L.; Goldsby, R.A.; Osborne, B.A. Glucocorticoid-induced apoptosis of thymocytes: requirement of proteasome-dependent mitochondrial activity. J. Immunol. 2003, 170, 2469–2478. [Google Scholar]
- Tome, M.E.; Baker, A.F.; Powis, G.; Payne, C.M.; Briehl, M.M. Catalase-overexpressing thymocytes are resistant to glucocorticoid-induced apoptosis and exhibit increased net tumor growth. Cancer Res. 2001, 61, 2766–2773. [Google Scholar]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial metabolism of reactive oxygen species. Biochemistry 2005, 70, 200–214. [Google Scholar]
- Simon, N.; Jolliet, P.; Morin, C.; Zini, R.; Urien, S.; Tillement, J.P. Glucocorticoids decrease cytochrome c oxidase activity of isolated rat kidney mitochondria. FEBS Lett. 1998, 435, 25–28. [Google Scholar]
- Morin, C.; Zini, R.; Simon, N.; Charbonnier, P.; Tillement, J.P.; le Louet, H. Low glucocorticoid concentrations decrease oxidative phosphorylation of isolated rat brain mitochondria: An additional effect of dexamethasone. Fundam. Clin. Pharmacol. 2000, 14, 493–500. [Google Scholar]
- Mutsaers, H.A.; Tofighi, R. Dexamethasone enhances oxidative stress-induced cell death in murine neural stem cells. Neurotox. Res. 2012, 22, 127–137. [Google Scholar]
- Korhonen, R.; Moilanen, E. MAP kinase phosphatase-1 as an inflammatory factor and drug target. Basic Clin. Pharmacol. Toxicol. 2014, 114, 24–36. [Google Scholar]
- Raman, M.; Chen, W.; Cobb, M.H. Differential regulation and properties of MAPKs. Oncogene 2007, 26, 3100–3112. [Google Scholar]
- Wancket, L.M.; Frazier, W.J.; Liu, Y. Mitogen-activated protein kinase phosphatase (MKP)-1 in immunology physiology and disease. Life Sci. 2012, 90, 237–248. [Google Scholar]
- Yin, H.; Shi, Z.; Jiao, S.; Chen, C.; Wang, W.; Greene, M.I.; Zhou, Z. Germinal center kinases in immune regulation. Cell. Mol. Immunol. 2012, 9, 439–445. [Google Scholar]
- McCubrey, J.A.; Lahair, M.M.; Franklin, R.A. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxid. Redox Signal. 2006, 8, 1775–1789. [Google Scholar]
- Hasala, H.; Zhang, X.; Saarelainen, S.; Moilanen, E.; Kankaanranta, H. c-Jun N-terminal kinase mediates constitutive human eosinophil apoptosis. Pulm. Pharmacol. Ther. 2007, 20, 580–587. [Google Scholar]
- Kankaanranta, H.; Zhang, X.; Tumelius, R.; Ruotsalainen, M.; Haikala, H.; Nissinen, E.; Moilanen, E. Antieosinophilic activity of simendans. J. Pharmacol. Exp. Ther. 2007, 323, 31–38. [Google Scholar]
- Hasala, H.; Moilanen, E.; Janka-Junttila, M.; Giembycz, M.A.; Kankaanranta, H. First-generation antihistamines diphenhydramine and chlorpheniramine reverse cytokine-afforded eosinophil survival by enhancing apoptosis. Allergy Asthma Proc. 2007, 28, 79–86. [Google Scholar]
- Kankaanranta, H.; Ilmarinen, P.; Zhang, X.; Nissinen, E.; Moilanen, E. Antieosinophilic activity of orazipone. Mol. Pharmacol. 2006, 69, 1861–1870. [Google Scholar]
- Shrivastava, P.; Pantano, C.; Watkin, R.; McElhinney, B.; Guala, A.; Poynter, M.L.; Persinger, R.L.; Budd, R.; Janssen-Heininger, Y. Reactive nitrogen species-induced cell death requires Fas-dependent activation of c-Jun N-terminal kinase. Mol. Cell. Biol. 2004, 24, 6763–6772. [Google Scholar]
- Sanchez-Perez, I.; Murguia, J.R.; Perona, R. Cisplatin induces a persistent activation of JNK that is related to cell death. Oncogene 1998, 16, 533–540. [Google Scholar]
- Ventura, J.J.; Hubner, A.; Zhang, C.; Flavell, R.A.; Shokat, K.M.; Davis, R.J. Chemical genetic analysis of the time course of signal transduction by JNK. Mol. Cell 2006, 21, 701–710. [Google Scholar]
- Lamb, J.A.; Ventura, J.J.; Hess, P.; Flavell, R.A.; Davis, R.J. JunD mediates survival signaling by the JNK signal transduction pathway. Mol. Cell 2003, 11, 1479–1489. [Google Scholar]
- Kamata, H.; Honda, S.; Maeda, S.; Chang, L.; Hirata, H.; Karin, M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 2005, 120, 649–661. [Google Scholar]
- Miike, S.; Nakao, A.; Hiraguri, M.; Kurasawa, K.; Saito, Y.; Iwamoto, I. Involvement of JAK2 but not PI 3-kinase/Akt and MAP kinase pathways in anti-apoptotic signals of GM-CSF in human eosinophils. J. Leukoc. Biol. 1999, 65, 700–706. [Google Scholar]
- Kankaanranta, H.; de Souza, P.M.; Barnes, P.J.; Salmon, M.; Giembycz, M.A.; Lindsay, M.A. SB 203580 an inhibitor of p38 mitogen-activated protein kinase enhances constitutive apoptosis of cytokine-deprived human eosinophils. J. Pharmacol. Exp. Ther. 1999, 290, 621–628. [Google Scholar]
- Zhang, J.P.; Wong, C.K.; Lam, C.W. Role of caspases in dexamethasone-induced apoptosis and activation of c-Jun NH2-terminal kinase and p38 mitogen-activated protein kinase in human eosinophils. Clin. Exp. Immunol. 2000, 122, 20–27. [Google Scholar]
- Baruch-Morgenstern, N.B.; Shik, D.; Moshkovits, I.; Itan, M.; Karo-Atar, D.; Bouffi, C.; Fulkerson, P.C.; Rashkovan, D.; Jung, S.; Rothenberg, M.E.; Munitz, A. Paired immunoglobulin-like receptor A is an intrinsic self-limiting suppressor of IL-5-induced eosinophil development. Nat. Immunol. 2013, 15. [Google Scholar] [CrossRef]
- Matsumoto, K.; Terakawa, M.; Miura, K.; Fukuda, S.; Nakajima, T.; Saito, H. Extremely rapid and intense induction of apoptosis in human eosinophils by anti-CD30 antibody treatment in vitro. J. Immunol. 2004, 172, 2186–2193. [Google Scholar]
- Tsukahara, K.; Nakao, A.; Hiraguri, M.; Miike, S.; Mamura, M.; Saito, Y.; Iwamoto, I. Tumor necrosis factor-alpha mediates antiapoptotic signals partially via p38 MAP kinase activation in human eosinophils. Int. Arch. Allergy Immunol. 1999, 120, 54–59. [Google Scholar]
- Wong, C.K.; Hu, S.; Cheung, P.F.; Lam, C.W. Thymic stromal lymphopoietin induces chemotactic and prosurvival effects in eosinophils: Implications in allergic inflammation. Am. J. Respir. Cell Mol. Biol. 2010, 43, 305–315. [Google Scholar]
- De Souza, P.M.; Kankaanranta, H.; Michael, A.; Barnes, P.J.; Giembycz, M.A.; Lindsay, M.A. Caspase-catalyzed cleavage and activation of Mst1 correlates with eosinophil but not neutrophil apoptosis. Blood 2002, 99, 3432–3438. [Google Scholar]
- Ura, S.; Nishina, H.; Gotoh, Y.; Katada, T. Activation of the c-Jun N-terminal kinase pathway by MST1 is essential and sufficient for the induction of chromatin condensation during apoptosis. Mol. Cell. Biol. 2007, 27, 5514–5522. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ilmarinen, P.; Moilanen, E.; Kankaanranta, H. Mitochondria in the Center of Human Eosinophil Apoptosis and Survival. Int. J. Mol. Sci. 2014, 15, 3952-3969. https://doi.org/10.3390/ijms15033952
Ilmarinen P, Moilanen E, Kankaanranta H. Mitochondria in the Center of Human Eosinophil Apoptosis and Survival. International Journal of Molecular Sciences. 2014; 15(3):3952-3969. https://doi.org/10.3390/ijms15033952
Chicago/Turabian StyleIlmarinen, Pinja, Eeva Moilanen, and Hannu Kankaanranta. 2014. "Mitochondria in the Center of Human Eosinophil Apoptosis and Survival" International Journal of Molecular Sciences 15, no. 3: 3952-3969. https://doi.org/10.3390/ijms15033952