Galectin-9 Induced Myeloid Suppressor Cells Expand Regulatory T Cells in an IL-10-Dependent Manner in CVB3-Induced Acute Myocarditis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
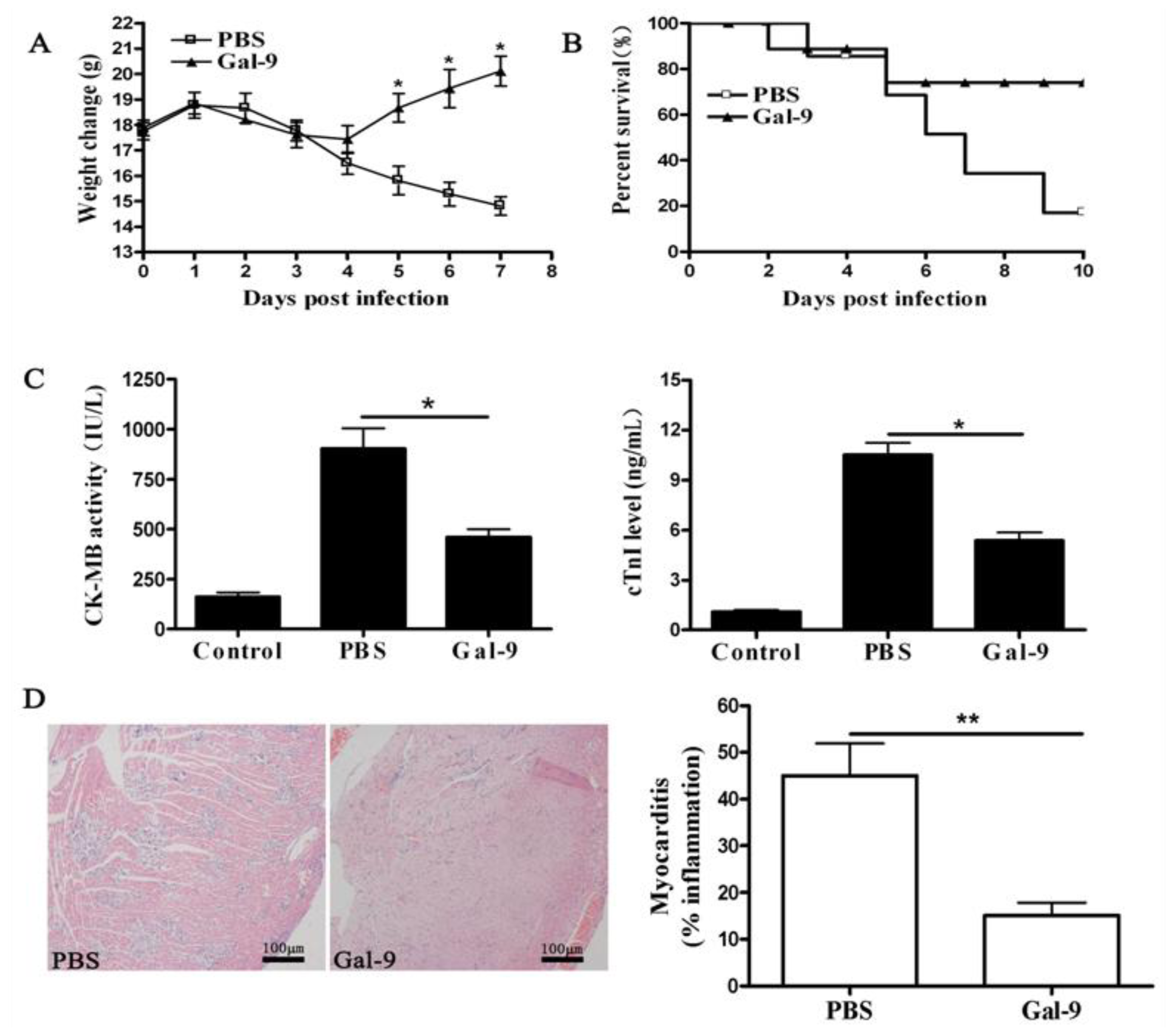
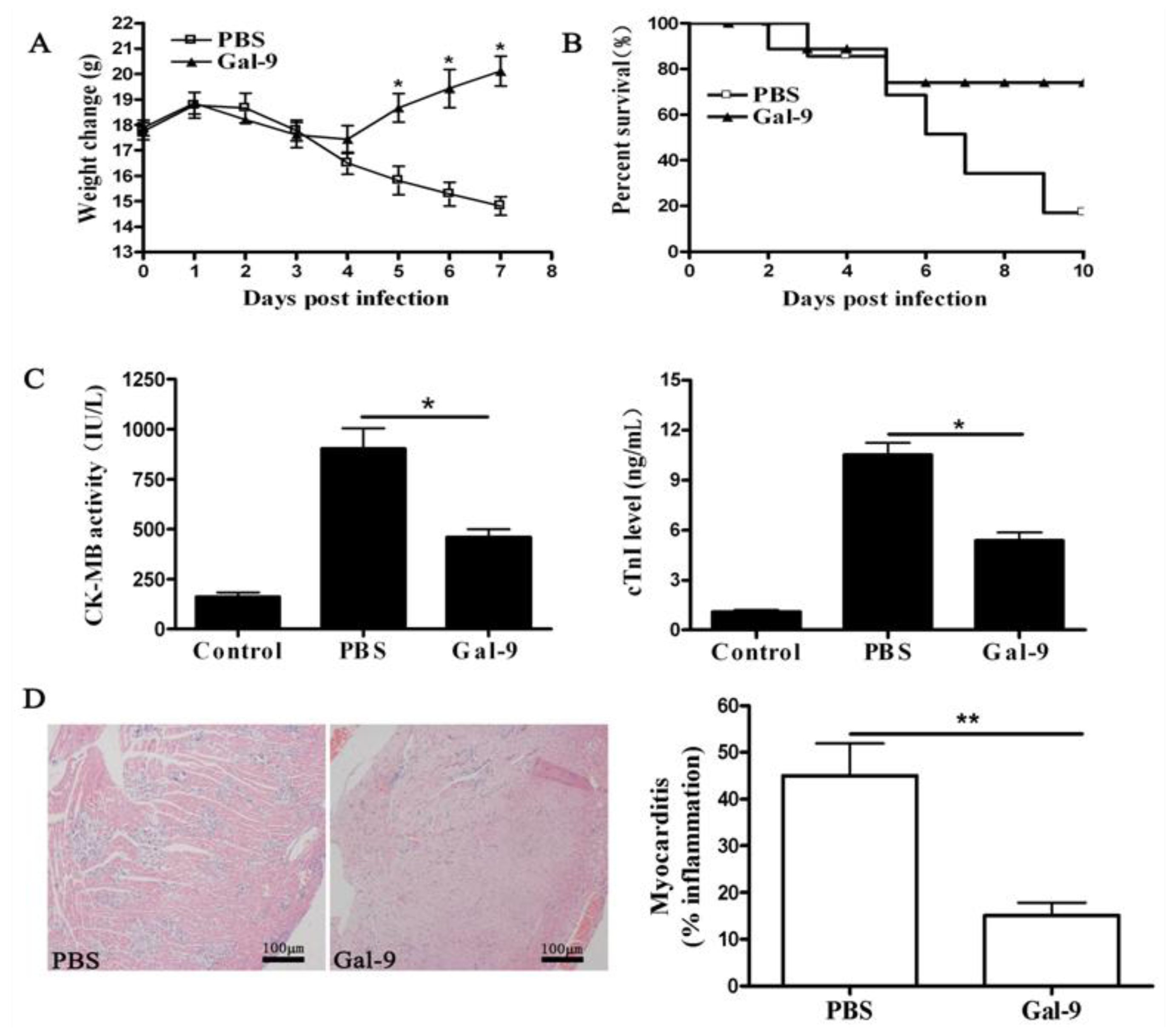
2.1.1. Remission of CVB3-Induced Myocarditis by Galectin-9 Administration
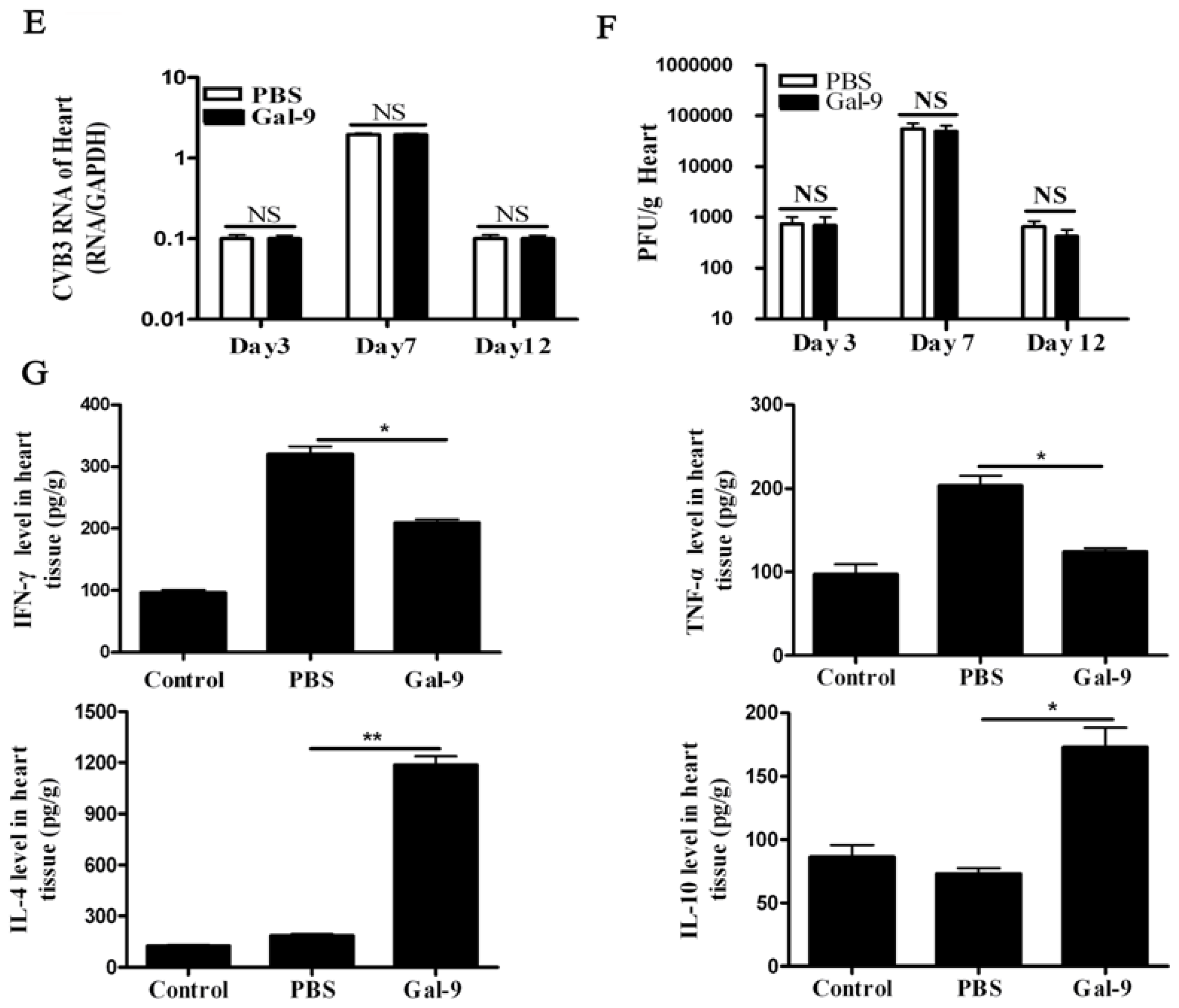
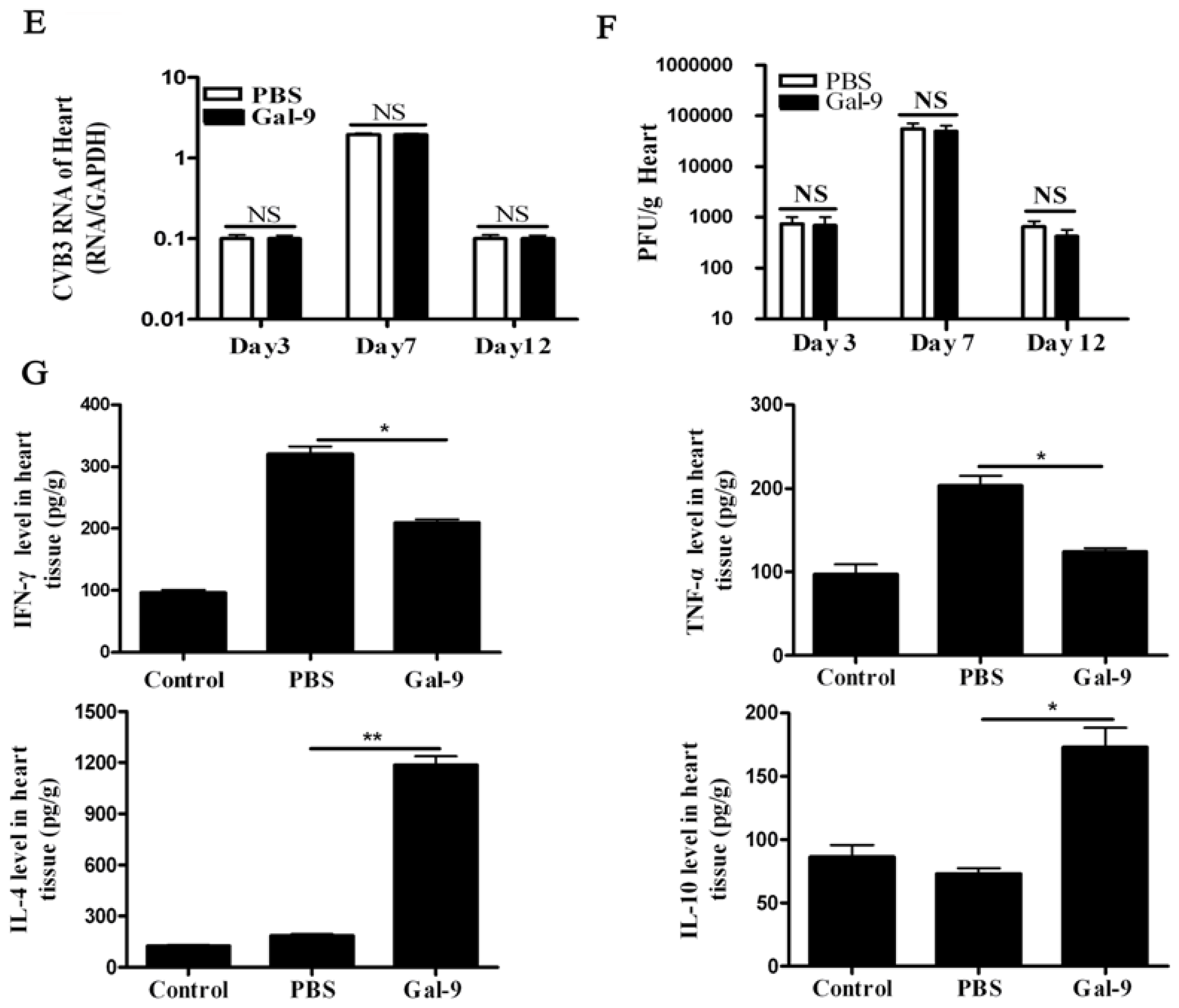
2.1.2. The Systemic and Local Immune Responses after Galectin-9 Treatment
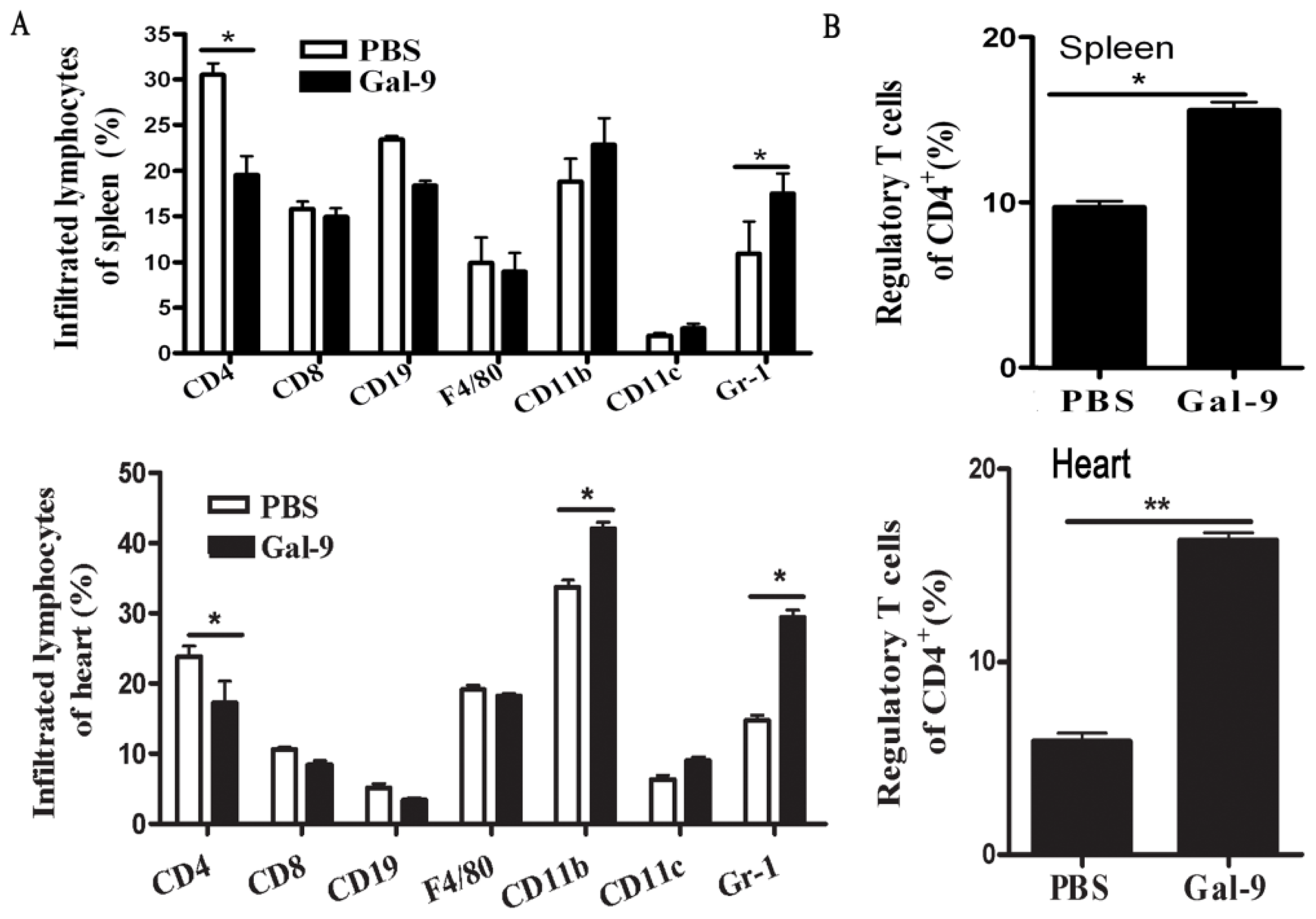
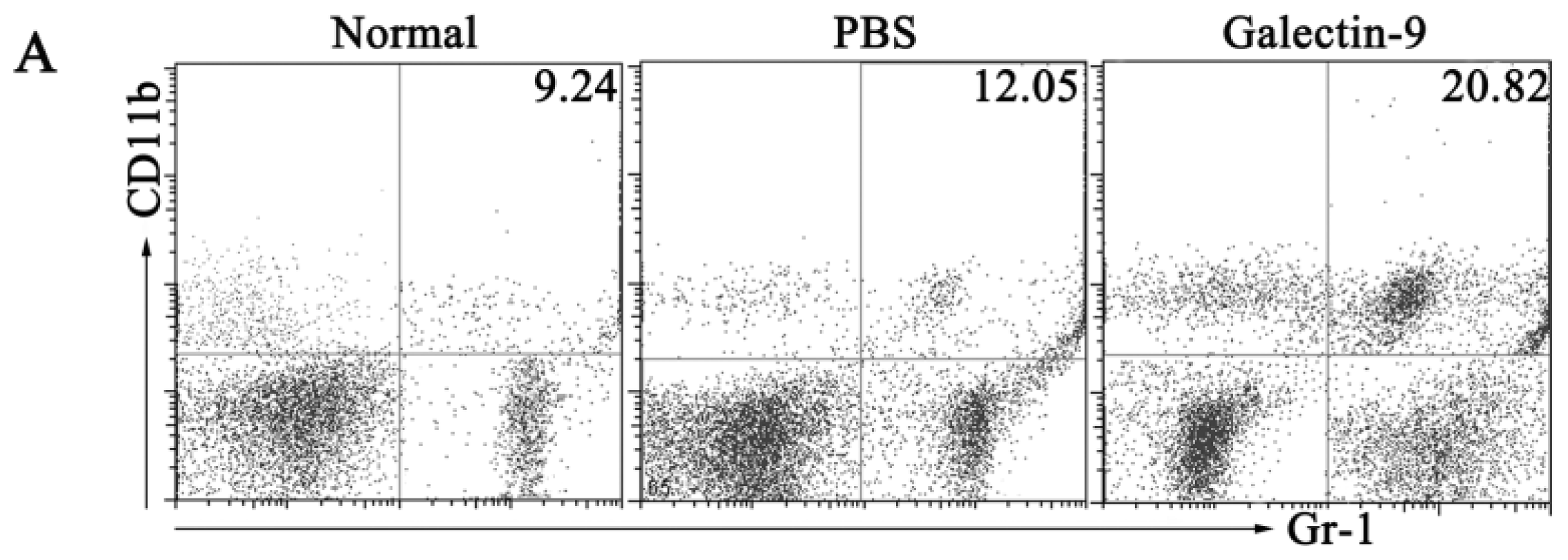
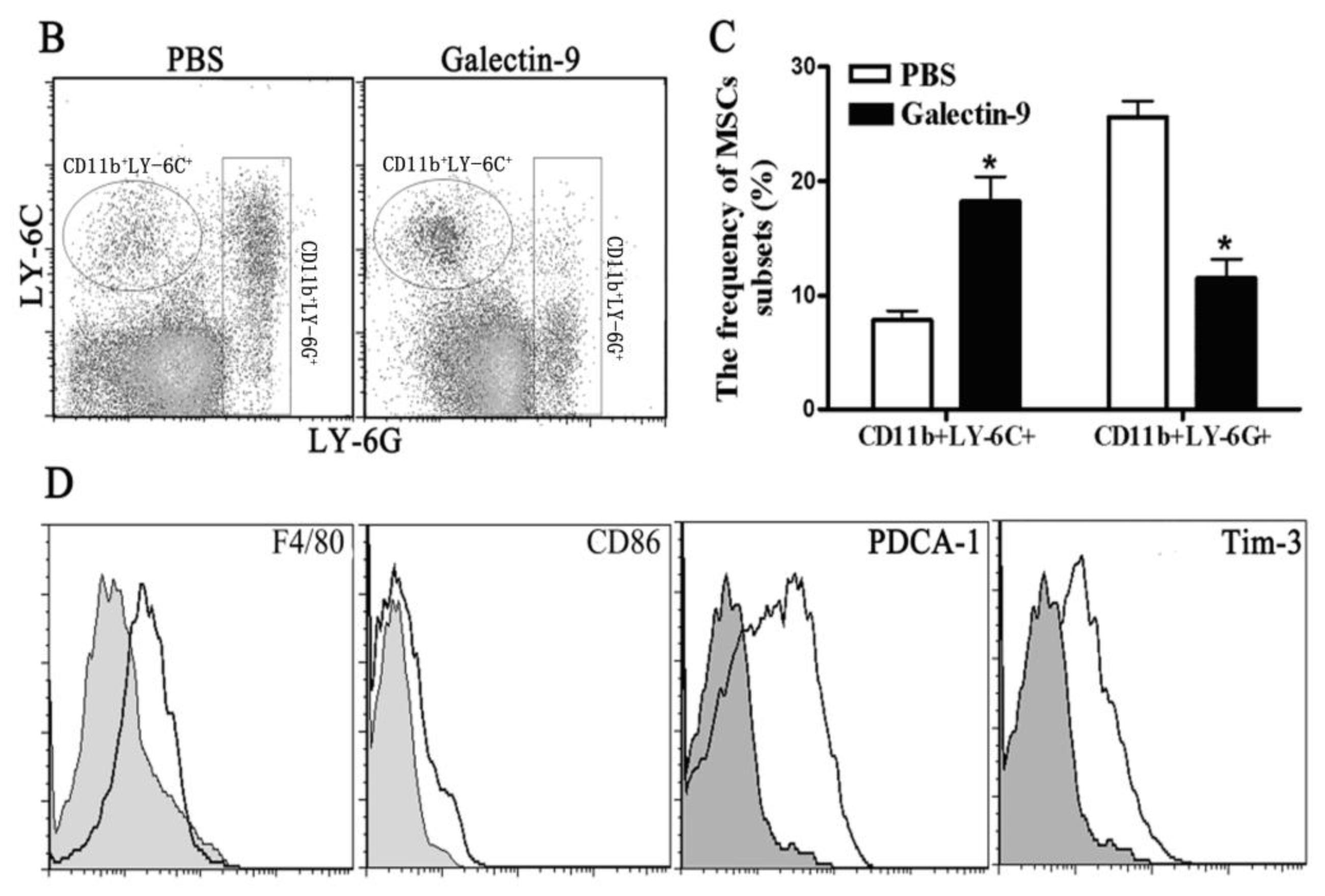
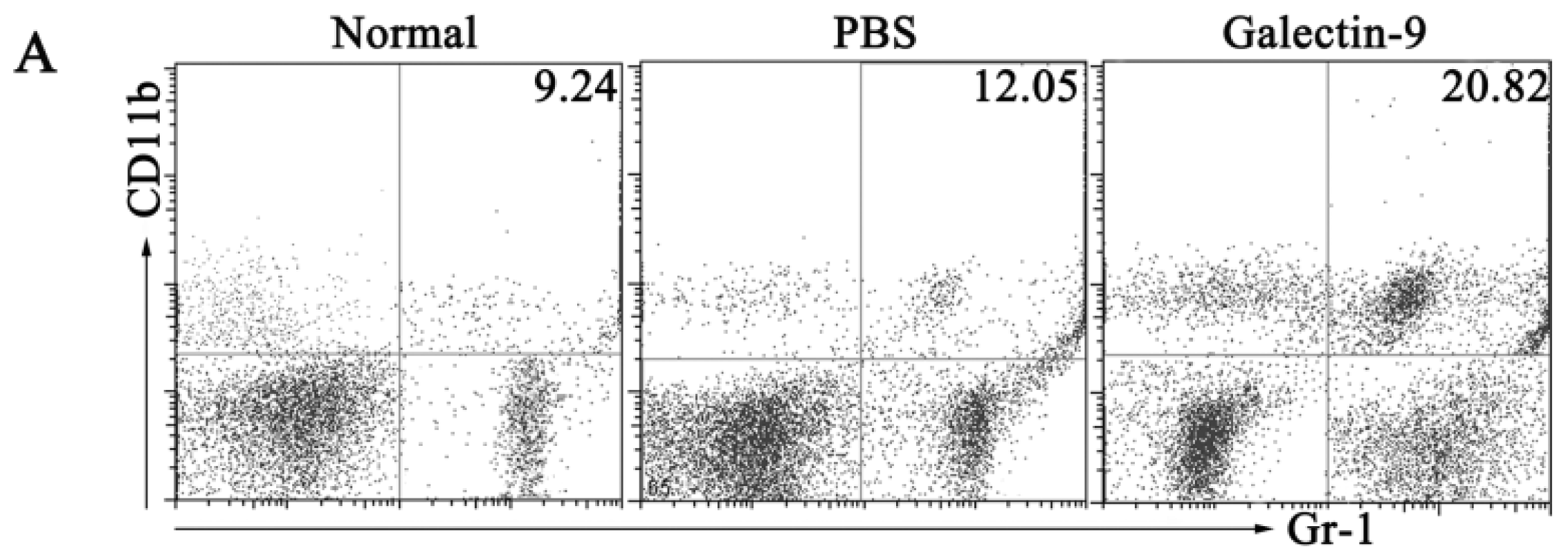
2.1.3. Frequency and Phenotypes of CD11b+Gr-1+ Myeloid Suppressor Cells in Galectin-9-Treated Mice
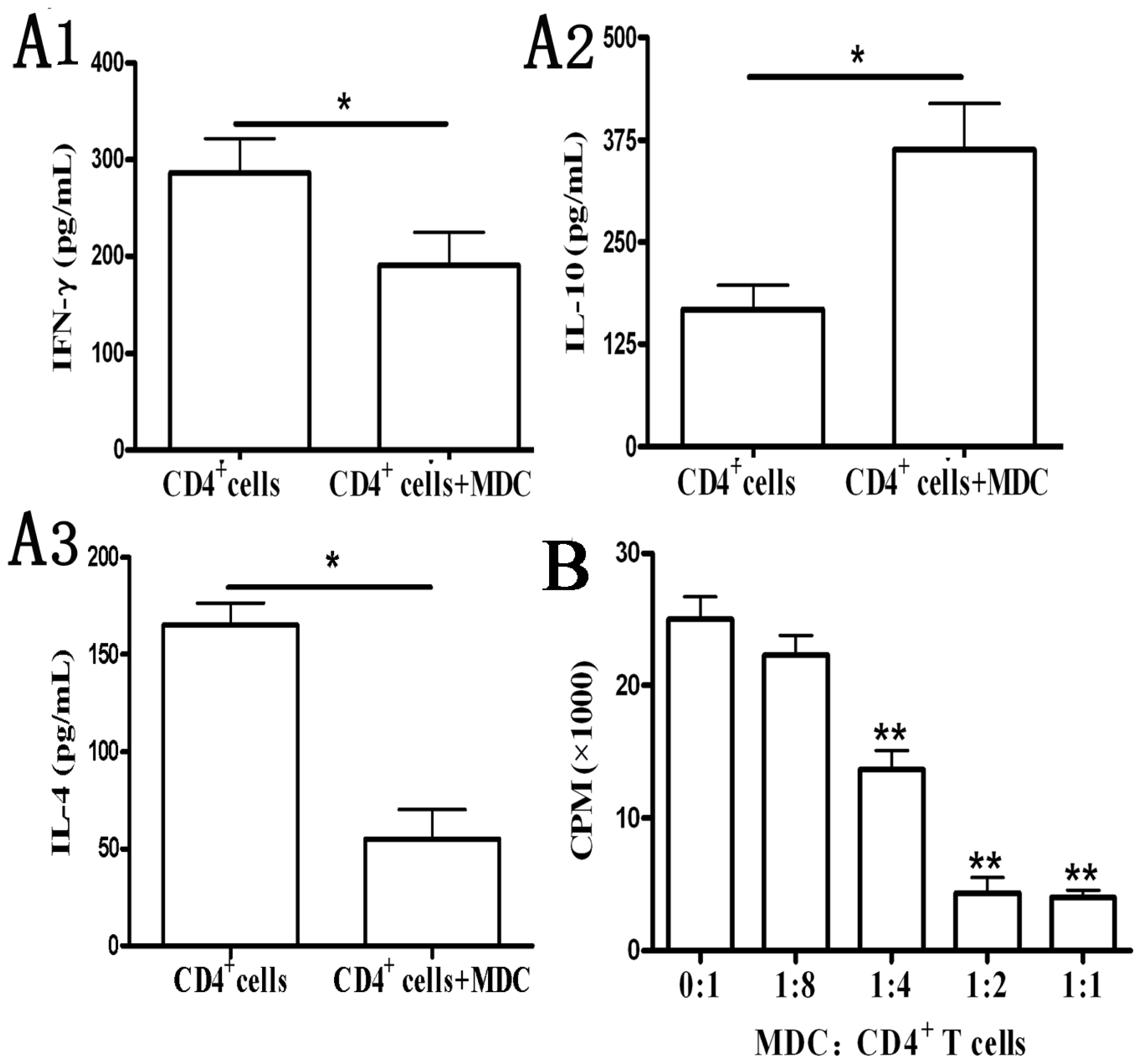
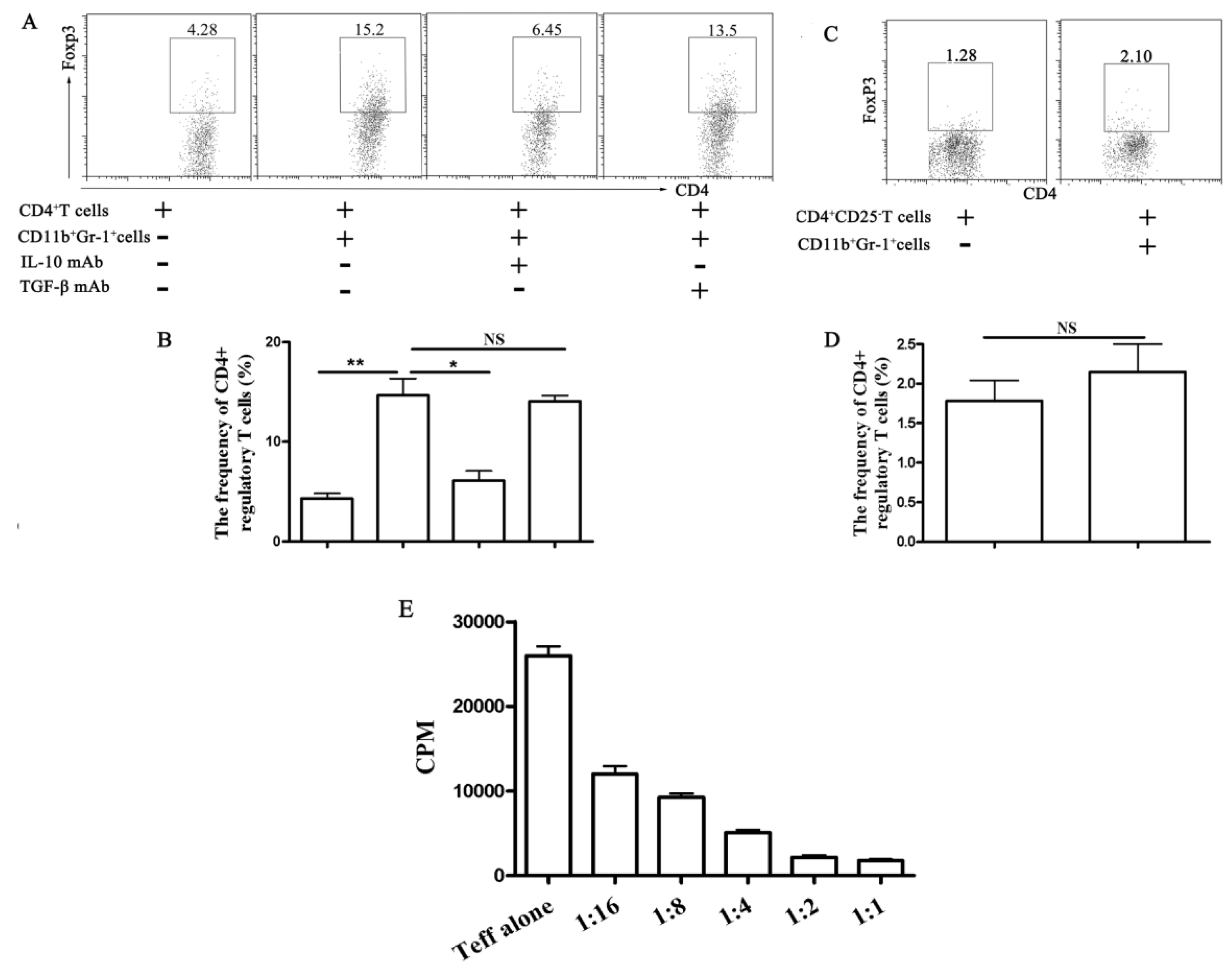
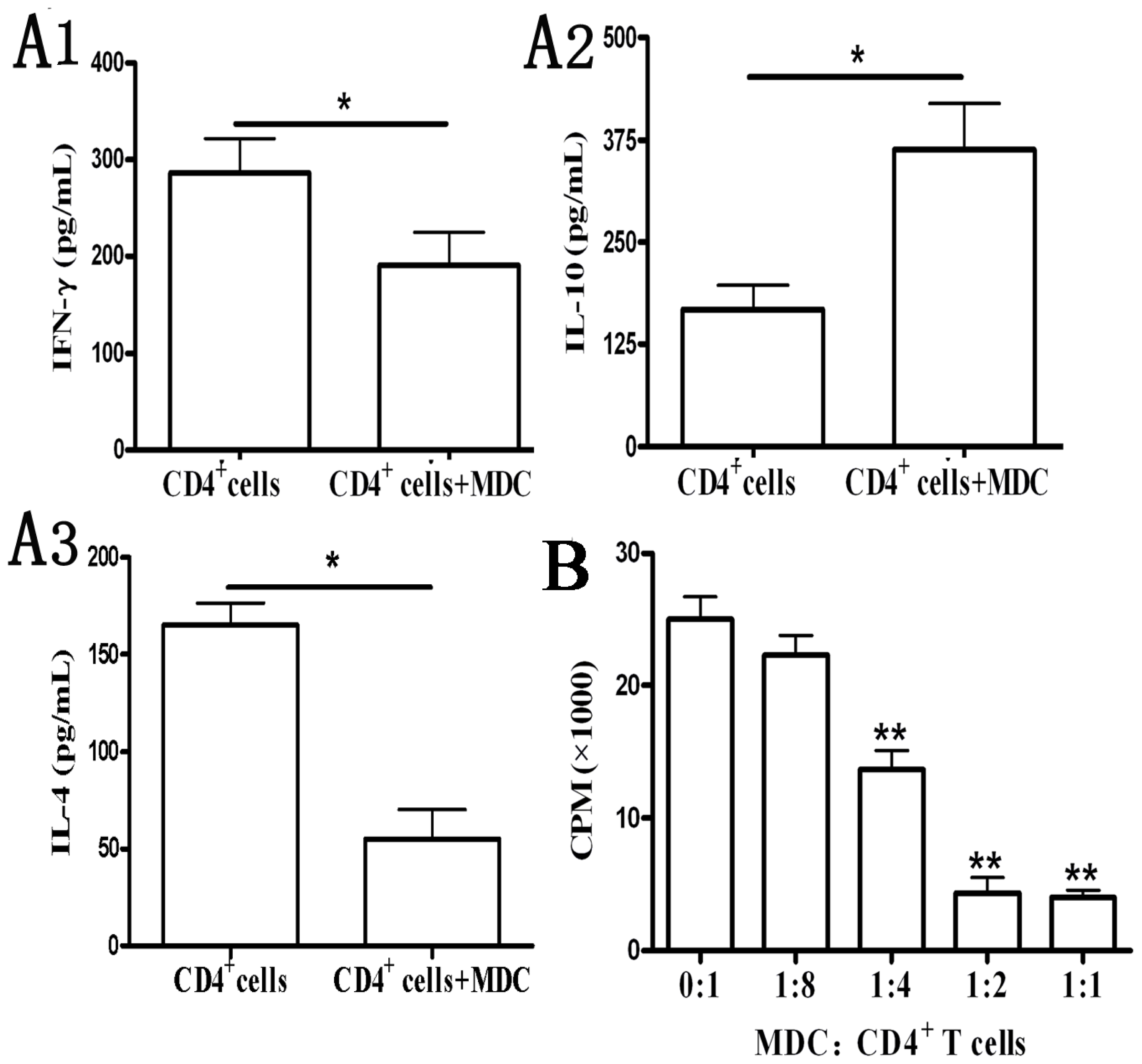
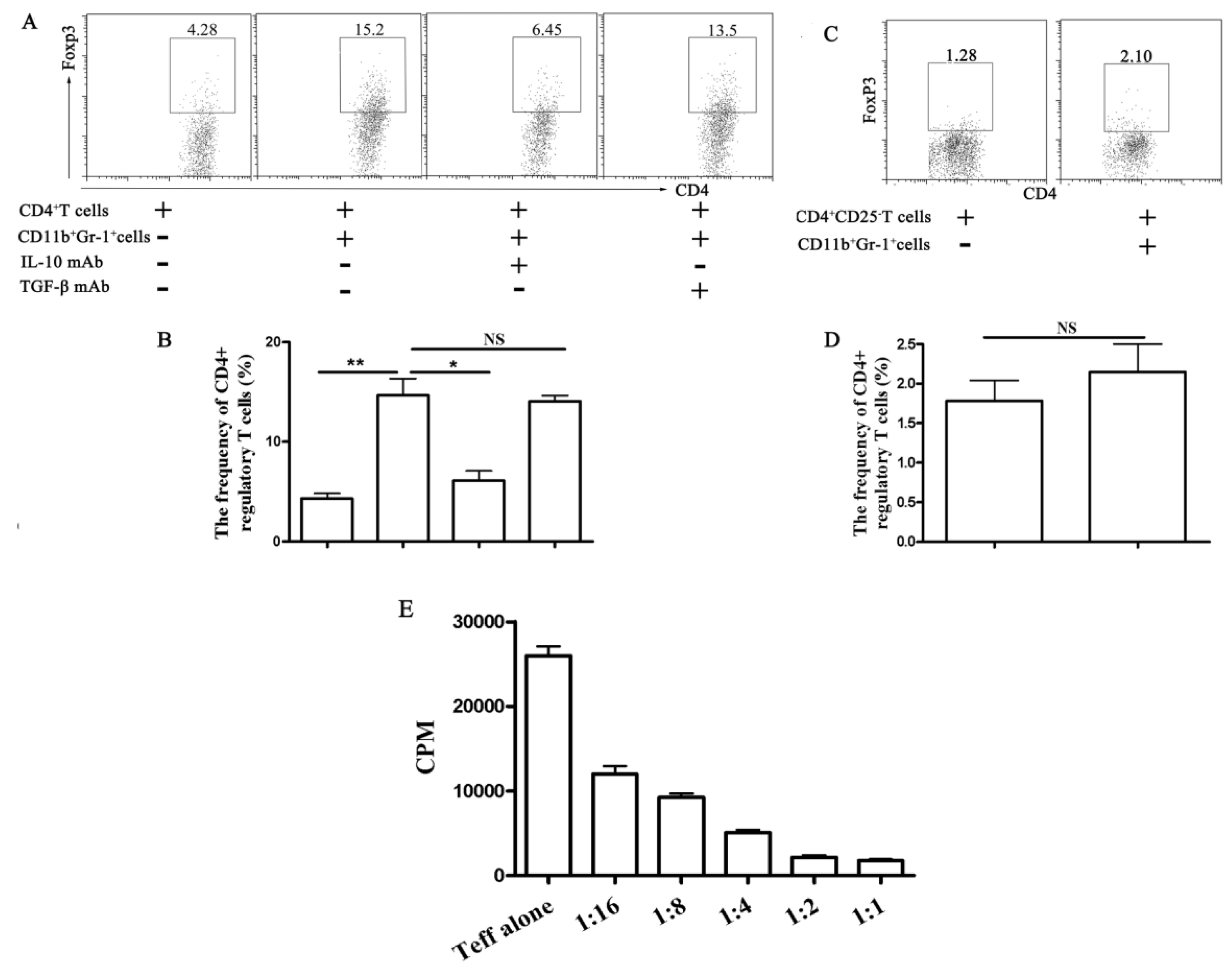
2.1.4. Functional Analyses of CD11b+Gr-1+ Myeloid Suppressor Cells in Galectin-9-Treated Mice
2.2. Discussion
3. Experimental Section
3.1. Animals
3.2. Virus
3.3. Myocarditis
3.4. Real-Time Polymerase Chain Reaction (RT-PCR)
3.5. Serological Index of Myocarditis
3.6. Cytokines Enzyme-Linked Immunosorbent Assay (ELISA)
3.7. Myeloid Cell Isolation and Culture
3.8. FACS Analysis
3.9. T Cell Proliferation and Cytokine Assays
3.10. Induction of Tregs in Vitro
3.11. Suppression Assays
3.12. Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Calabrese, F.; Thiene, G. Myocarditis and inflammatory cardiomyopathy: Microbiological and molecular biological aspects. Cardiovasc. Res 2003, 60, 11–25. [Google Scholar]
- Maisch, B.; Ristic, A.D.; Hufnagel, G.; Pankuweit, S. Pathophysiology of viral myocarditis: The role of humoral immune response. Cardiovasc. Pathol 2002, 11, 112–122. [Google Scholar]
- Luo, H.; Wong, J.; Wong, B. Protein degradation systems in viral myocarditis leading to dilated cardiomyopathy. Cardiovasc. Res 2010, 85, 347–356. [Google Scholar]
- Jiang, Z.; Xu, W.; Li, K.; Yue, Y.; Xu, L.; Ye, F.; Xiong, S. Remission of CVB3-induced viral myocarditis by in vivo Th2 polarization via hydrodynamics-based interleukin-4 gene transfer. J. Gene Med 2008, 10, 918–929. [Google Scholar]
- Huber, S.A.; Feldman, A.M.; Sartini, D. Coxsackievirus B3 induces T regulatory cells which inhibit cardiomyopathy in tumor necrosis factor-alpha transgenic mice. Circ. Res 2006, 99, 1109–1116. [Google Scholar]
- Seko, Y.; Takahashi, N.; Azuma, M.; Yagita, H.; Okumura, K.; Yazaki, Y. Effects of in vivo administration of anti-B7-1/B7-2 monoclonal antibodies on murine acute myocarditis caused by coxsackievirus B3. Circ. Res 1998, 82, 613–618. [Google Scholar]
- Pinkert, S.; Westermann, D.; Wang, X.; Poller, W.; Henry, F. Prevention of cardiac dysfunction in acute coxsackievirus B3 cardiomyopathy by inducible expression of a soluble coxsackievirus-adenovirus receptor. Circulation 2009, 120, 2358–2366. [Google Scholar]
- Opavsky, M.A.; Penninger, J.; Aitken, K.; Wen, W.H.; Dawood, F.; Mak, T.; Liu, P. Susceptibility to myocarditis is dependent on the response of alphabeta T lymphocytes to coxsackieviral infection. Circ. Res 1999, 85, 551–558. [Google Scholar]
- Huber, S.A.; Sartini, D.; Exley, M. Vγ4+ T cells promote autoimmune CD8+ cytolytic T-lymphocyte activation in coxsackievirus B3-induced myocarditis in mice: Role for CD4+ Th1 cells. J. Virol 2002, 76, 10785–10790. [Google Scholar]
- Huber, S.A.; Shi, C.; Budd, R.C. Gammadelta T cells promote a Th1 response during coxsackievirus B3 infection in vivo: Role of Fas and Fas ligand. J. Virol 2002, 76, 6487–6494. [Google Scholar]
- Afanasyeva, M.; Wang, Y.; Kaya, Z.; Stafford, E.A.; Dohmen, K.M.; Sadighi Akha, A.A.; Rose, N.R. Interleukin-12 receptor/STAT4 signaling is required for the development of autoimmune myocarditis in mice by an interferon-γ-independent pathway. Circulation 2001, 104, 3145–3151. [Google Scholar]
- Tang, H.; Sharp, G.C.; Peterson, K.P.; Braley-Mullen, H. IFN-γ-deficient mice develop severe granulomatous experimental autoimmune thyroiditis with eosinophil infiltration of the thyroids. J. Immunol. 1998, 160, 5105–5112. [Google Scholar]
- Tarrant, T.K.; Silver, P.B.; Wahlsten, J.L.; Rizzo, L.V.; Chan, C.C.; Wiggert, B.; Caspi, R.R. Interleukin 12 protects from a T helper type 1-mediated autoimmune disease experimental autoimmune uveitis through mechanisms involving interferon-γ nitric oxide and apoptosis. J. Exp. Med. 1999, 189, 219–230. [Google Scholar]
- Eriksson, U.; Kurrer, M.O.; Sebald, W.; Brombacher, F.; Kopf, M. Dual role of the IL-12/IFN-γ axis in the development of autoimmune myocarditis: Induction by IL-12 and protection by IFN-γ. J. Immunol. 2001, 167, 5464–5469. [Google Scholar]
- Ono, M.; Shimizu, J.; Miyachi, Y. Control of autoimmune myocarditis and multiorgan inflammation by glucocorticoid-induced TNF receptor family related protein (high) Foxp3-expressing CD25+ and CD25− regulatory T cells. J. Immunol 2006, 176, 4748–4756. [Google Scholar]
- Shevach, E.M.; Stephens, G.L. The GITR–GITRL interaction: Co-stimulation or contra-suppression of regulatory activity? Nat. Rev. Immunol 2006, 6, 613–618. [Google Scholar]
- Fairweather, D.; Yusung, S.; Frisancho, S.; Barrett, M.; Gatewood, S.; Steele, R.; Rose, N.R. IL-12 receptor β 1 and Toll-like receptor 4 increase IL-1 β and IL-18 associated myocarditis and coxsackievirus replication. J. Immunol 2003, 170, 4731–4737. [Google Scholar]
- Lane, J.R.; Neumann, D.A.; Lafond-Walker, A.; Herskowitz, A.; Rose, N.R. Interleukin 1 or tumor necrosis factor can promote coxsackie B3-induced myocarditis in resistant B1 mice. J. Exp. Med 1992, 175, 1123–1129. [Google Scholar]
- Kuchroo, V.K.; Meyers, J.H.; Umetsu, D.T.; DeKruyff, R.H. Tim family of genes in immunity and tolerance. Adv. Immunol 2006, 91, 227–249. [Google Scholar]
- Zhu, C.; Anderson, A.C.; Schubart, A.; Xiong, H.; Imitola, J.; Khoury, S.J.; Zheng, X.X.; Strom, T.B.; Kuchroo, V.K. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 2005, 6, 1245–1252. [Google Scholar]
- Kuchroo, V.K.; Dardalhon, V.; Xiao, S.; Anderson, A.C. New roles for Tim family members in immune regulation. Nat. Rev. Immunol 2008, 8, 577–580. [Google Scholar]
- Anderson, D.E. Tim-3 as a therapeutic target in human inflammatory diseases. Expert. Opin. Ther. Targets 2007, 11, 1005–1009. [Google Scholar]
- Sanchez-Fueyo, A.; Tian, J.; Picarella, D.; Kuchroo, V.K.; Gutierrez-Ramos, J.C.; Coyle, A.J.; Strom, T.B. Tim-3 inhibits T helper type 1-mediated auto- and allo-immune responses and promotes immunological tolerance. Nat. Immunol 2003, 4, 1093–1101. [Google Scholar]
- Saita, N.; Goto, E.; Yamamoto, T.; Maruo, K.; Ono, T.; Takeya, M.; Kashio, Y.; Nakamura, K.; Hirashima, M. Association of galectin-9 with eosinophil apoptosis. Int. Arch. Allergy Immunol 2002, 128, 42–50. [Google Scholar]
- Kageshita, T.; Kashio, Y.; Yamauchi, A.; Nishi, N.; Shoji, H.; Nakamura, T.; Ono, T.; Hirashima, M. Possible role of galectin-9 in cell aggregation and apoptosis of human melanoma cell lines and its clinical significance. Int. J. Cancer 2002, 99, 809–816. [Google Scholar]
- Wada, J.; Ota, K.; Kumar, A.; Wallner, E.I.; Kanwar, Y.S. Developmental regulation expression and apoptotic potential of galectin-9 a β-galactoside binding lectin. J. Clin. Investig 1997, 99, 2452–2461. [Google Scholar]
- Matsumoto, R.; Matsumoto, H.; Seki, M.; Kanegasaki, S.; Stevens, R.L.; Hirashima, M. A variant of human galectin-9 is a novel eosinophil chemoattractant produced by T lymphocytes. J. Biol. Chem 1998, 273, 16976–16984. [Google Scholar]
- Kashio, Y.; Nakamura, K.; Abedin, M.J.; Yoshida, N.; Nakamura, T.; Hirashima, M. Galectin-9 induces apoptosis through the calcium-calpain-caspase-1 pathway. J. Immunol 2003, 170, 3631–3636. [Google Scholar]
- Tsuboi, Y.; Abe, H.; Nakagawa, R.; Nishi, N.; Nakamura, T.; Yamauchi, A.; Hirashima, M. Galectin-9 protects mice from the Shwartzman reaction by attracting prostaglandin E2-producing polymorphonuclear leukocytes. Clin. Immunol 2007, 124, 221–233. [Google Scholar]
- Fukushima, A.; Tamaki, S.; Ken, F. Roles of galectin-9 in the development of experimental allergic conjunctivitis in mice. Int. Arch. Allergy Immunol 2008, 146, 36–43. [Google Scholar]
- Seki, M.; Oomizu, S.; Sakata, K.; Nishi, N.; Yamauchi, A.; Katoh, S.; Matsukawa, A.; Kuchroo, V.; Hirashima, M. Galectin-9 suppresses the generation of Th17 promotes the induction of regulatory T cells and regulates experimental autoimmune arthritis. Clin. Immunol 2008, 127, 78–88. [Google Scholar]
- Sharvan, S.; Amol, S.; Hirashima, M.; Rouse, B.T. Role of Tim-3/galectin-9 inhibitory interaction in viral-induced immunopathology: Shifting the balance toward regulators. J. Immunol 2009, 182, 3191–3201. [Google Scholar]
- Lv, K.; Xu, W.; Wang, C.N.; Niki, T.; Hirashima, M.; Xiong, S.D. Galectin-9 administration ameliorates CVB3 induced myocarditis by promoting the proliferation of regulatory T cells and alternatively activated Th2 cells. Clin. Immunol. 2011, 140, 92–101. [Google Scholar]
- Tam, P.E. Coxsackievirus myocarditis: Interplay between virus and host in the pathogenesis of heart disease. Viral Immunol 2006, 19, 133–146. [Google Scholar]
- Shi, Y.; Chen, C.; Lisewski, U.; Radke, M.; Westermann, D.; Sauter, M.; Tschope, C.; Poller, W.; Klingel, K.; Gotthardt, M. Cardiac deletion of the coxsackievirus-adenovirus receptor abolishes Coxsackievirus B3 infection and prevents myocarditis in vivo. J. Am. Coll. Cardiol 2009, 53, 1219–1226. [Google Scholar]
- Maekawa, Y.; Ouzounian, M.; Opavsky, M.A.; Liu, P. Connecting the missing link between dilated cardiomyopathy and viral myocarditis: Virus cytoskeleton and innate immunity. Circulation 2007, 115, 5–8. [Google Scholar]
- Maier, R.; Krebs, P.; Ludewig, B. Immunopathological basis of virus-induced myocarditis. Clin. Dev. Immunol 2004, 11, 1–5. [Google Scholar]
- Esfandiarei, M.; McManus, B.M. Molecular biology and pathogenesis of viral myocarditis. Annu. Rev. Pathol 2008, 3, 127–155. [Google Scholar]
- Frisancho-Kiss, S.; Coronado, M.J.; Frisancho, J.A.; Lau, V.M.; Rose, N.R.; Klein, S.L.; Fairweather, D. Gonadectomy of male BALB/c mice increases Tim-3(+) alternatively activated M2 macrophages Tim-3(+) T cells Th2 cells and Treg in the heart during acute oxsackievirus-induced myocarditis. Brain Behav. Immun 2009, 23, 649–657. [Google Scholar]
- Frisancho-Kiss, S.; Sarah, E.D.; Jennifer, F.N.; Frisancho, J.A.; Cihakova, D.; Barrett, M.A.; Rose, N.R.; Fairweather, D. Cutting edge: Cross-regulation by TLR4 and T cell Ig mucin-3 determines sex differences in inflammatory heart disease. J. Immunol 2007, 178, 6710–6714. [Google Scholar]
- Mouzaki, A.; Deraos, S.; Chatzantoni, K. Advances in the treatment of autoimmune diseases cellular activity type-1/type-2 cytokine secretion patterns and their modulation by therapeutic peptides. Curr. Med. Chem 2005, 12, 1537–1550. [Google Scholar]
- Serafini, P.; Borrello, I.; Bronte, V. Myeloid suppressor cells in cancer: Recruitment phenotype properties and mechanisms of immune suppression. Semin. Cancer Biol 2006, 16, 53–65. [Google Scholar]
- Huang, B.; Pan, P.Y.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.; Divino, C.M.; Chen, S.H. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res 2006, 66, 1123–1131. [Google Scholar]
- Henke, A.; Huber, S.; Stelzner, A.; Whitton, J.L. Role of CD8+ T lymphocytes in coxsackievirus B3-induced myocarditis. J. Virol 1995, 69, 6720–6728. [Google Scholar]


© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, Y.; Jiang, L.; Zhang, M.; Lv, K. Galectin-9 Induced Myeloid Suppressor Cells Expand Regulatory T Cells in an IL-10-Dependent Manner in CVB3-Induced Acute Myocarditis. Int. J. Mol. Sci. 2014, 15, 3356-3372. https://doi.org/10.3390/ijms15033356
Zhang Y, Jiang L, Zhang M, Lv K. Galectin-9 Induced Myeloid Suppressor Cells Expand Regulatory T Cells in an IL-10-Dependent Manner in CVB3-Induced Acute Myocarditis. International Journal of Molecular Sciences. 2014; 15(3):3356-3372. https://doi.org/10.3390/ijms15033356
Chicago/Turabian StyleZhang, Yingying, Li Jiang, Mengying Zhang, and Kun Lv. 2014. "Galectin-9 Induced Myeloid Suppressor Cells Expand Regulatory T Cells in an IL-10-Dependent Manner in CVB3-Induced Acute Myocarditis" International Journal of Molecular Sciences 15, no. 3: 3356-3372. https://doi.org/10.3390/ijms15033356