C2-Ceramide Induces Cell Death and Protective Autophagy in Head and Neck Squamous Cell Carcinoma Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
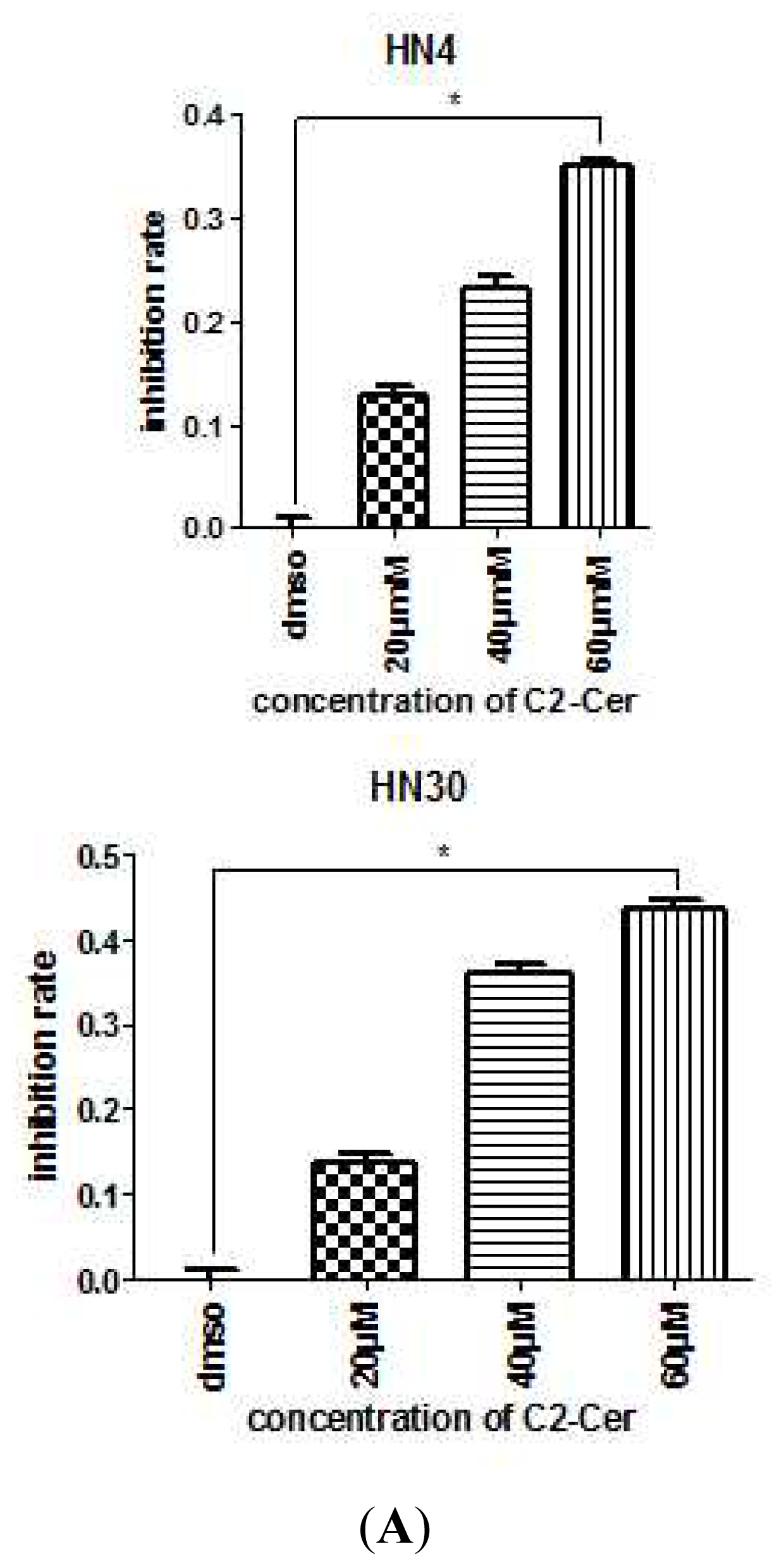
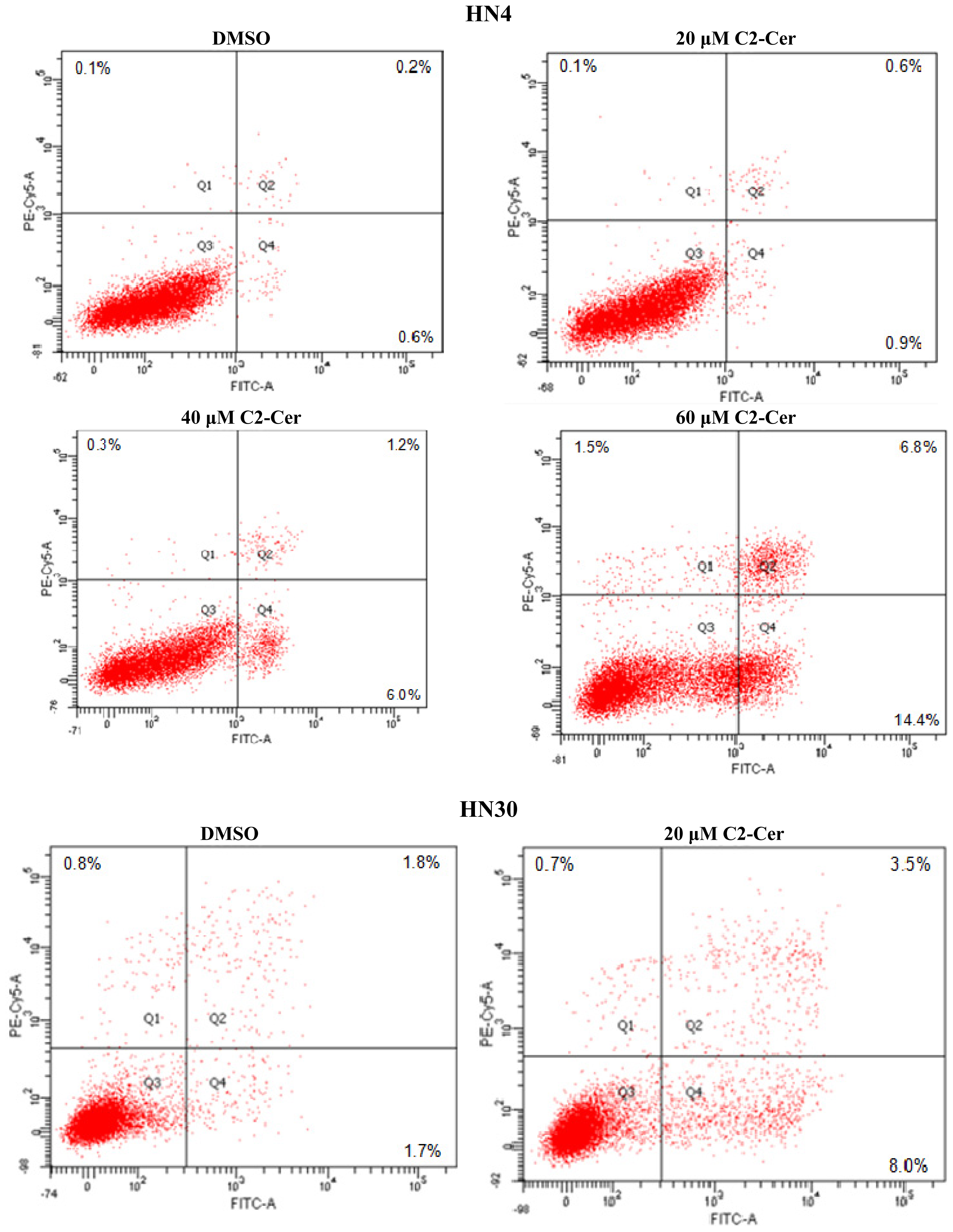
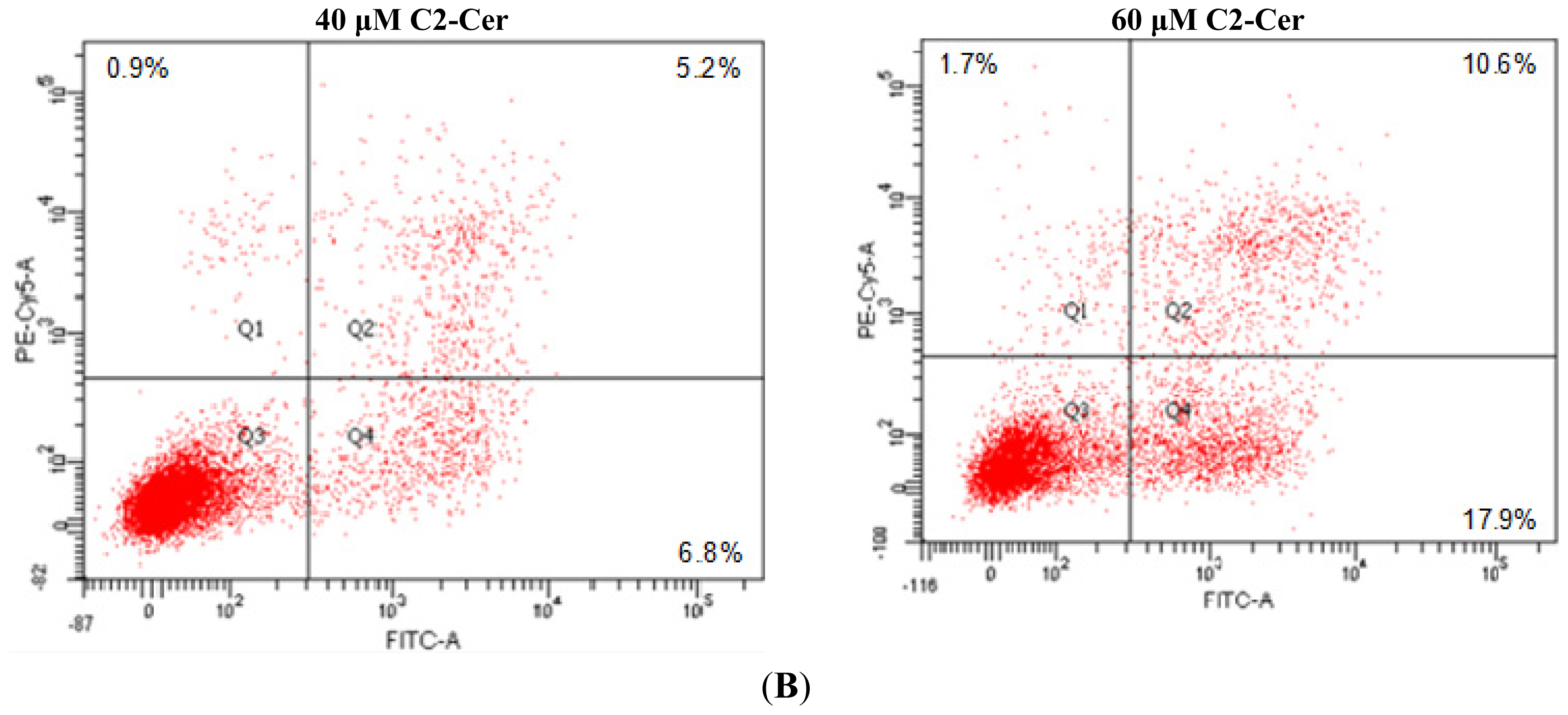
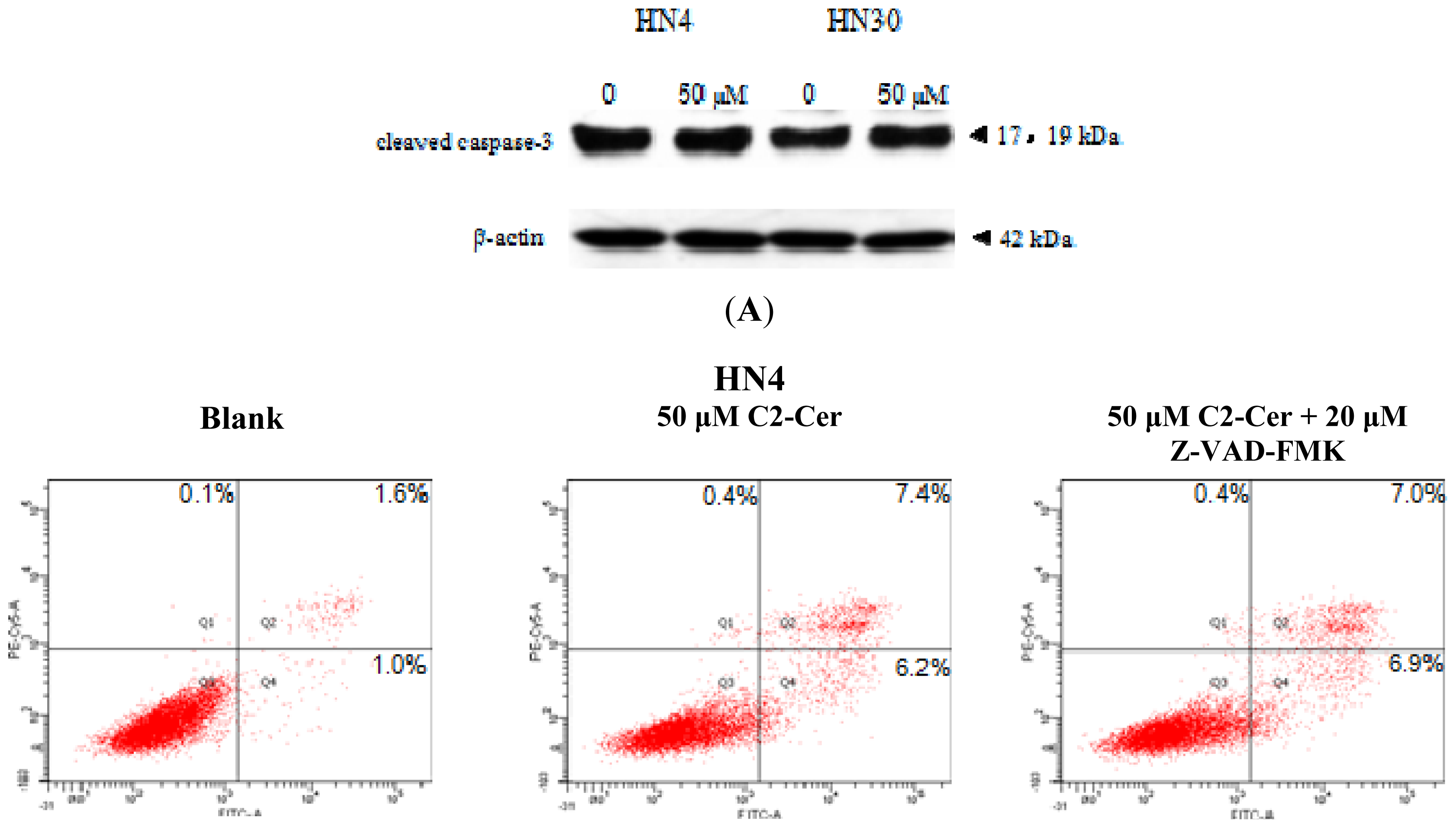
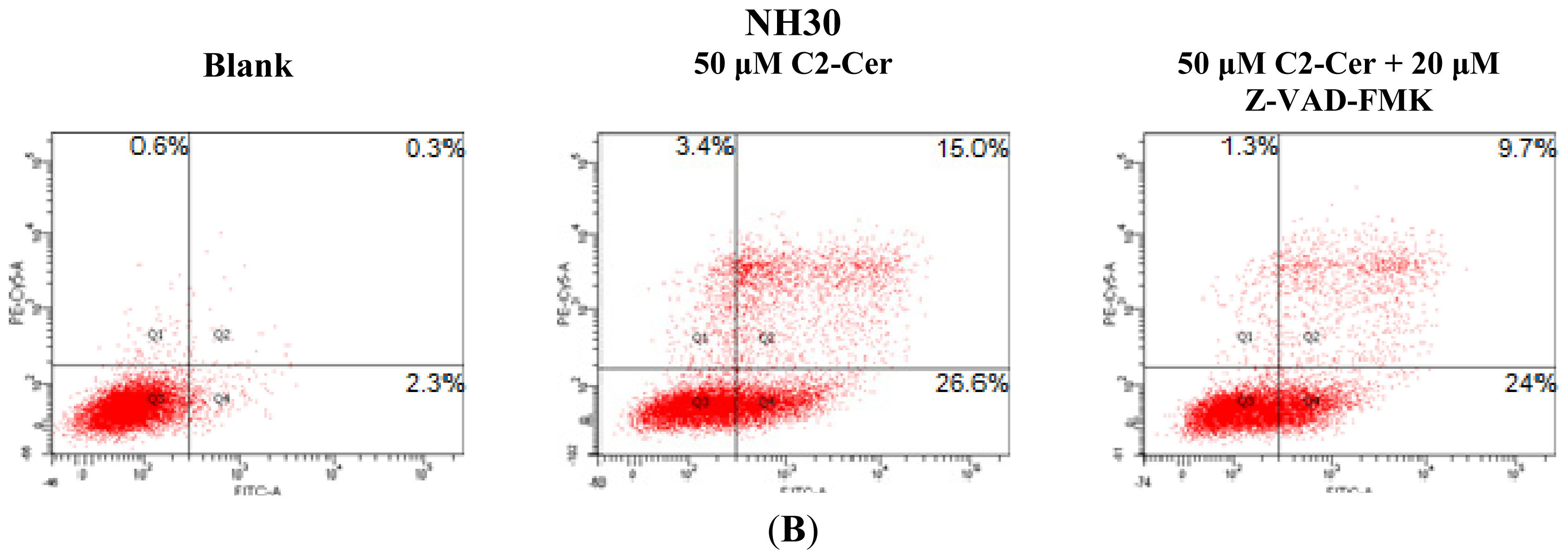
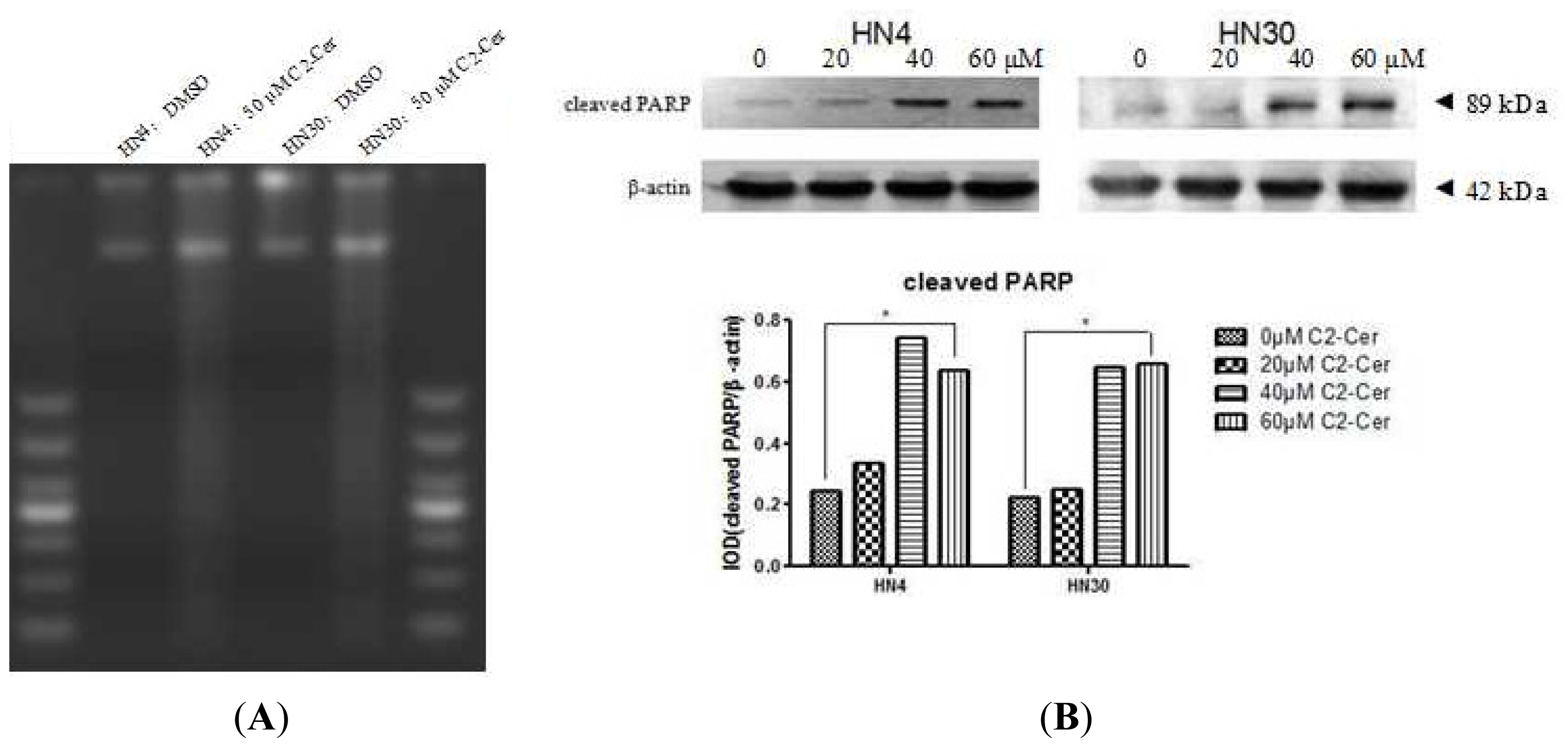
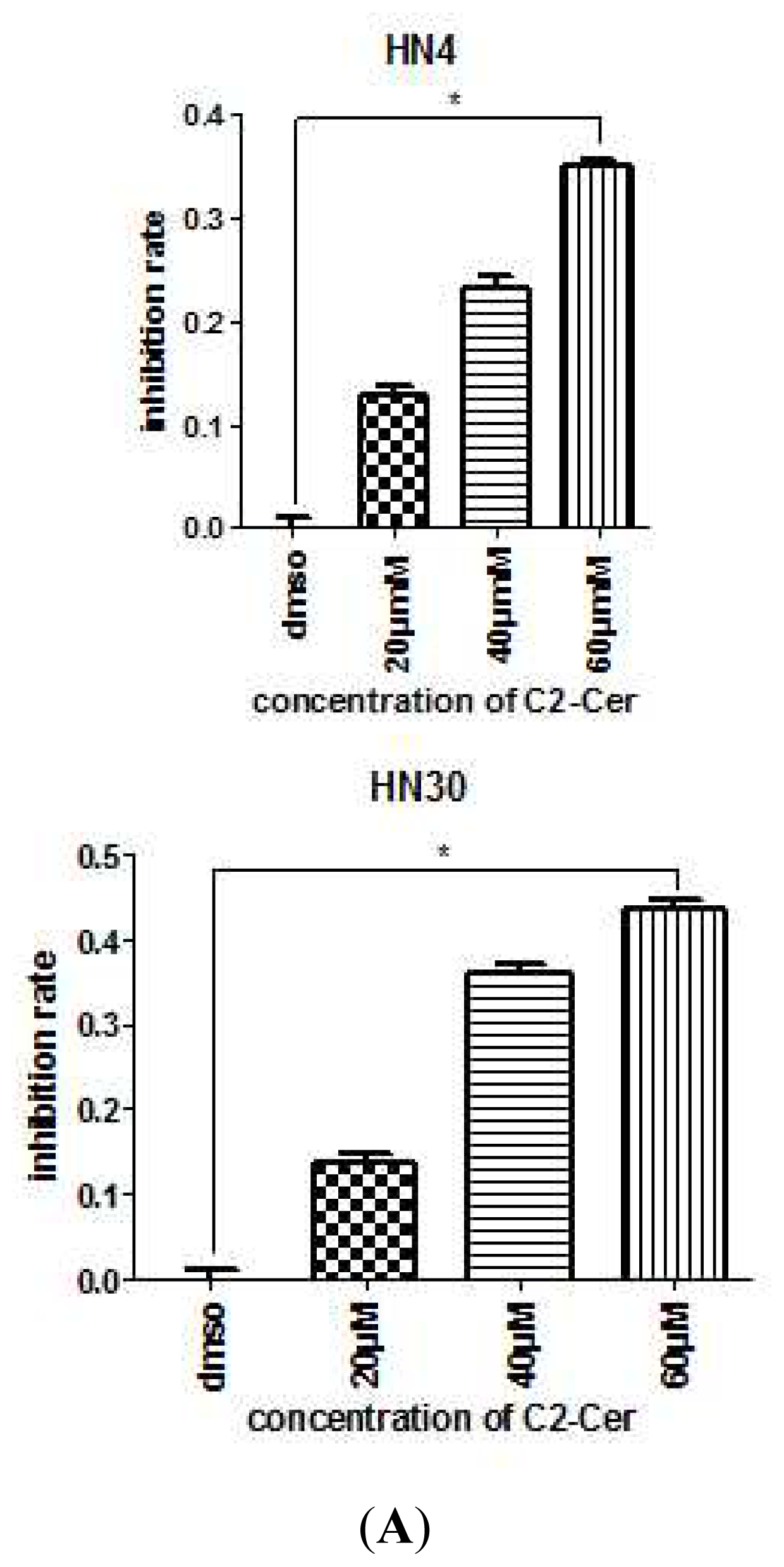
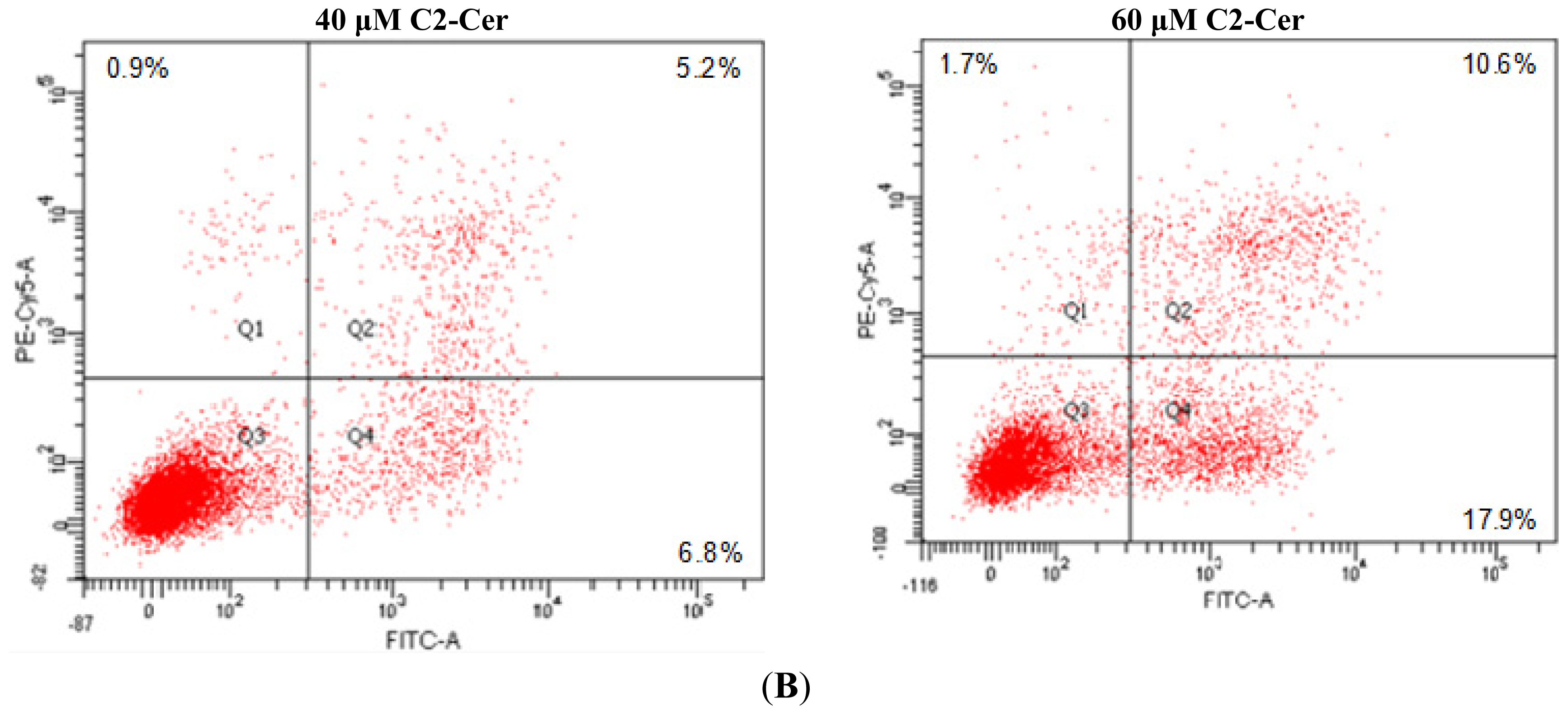
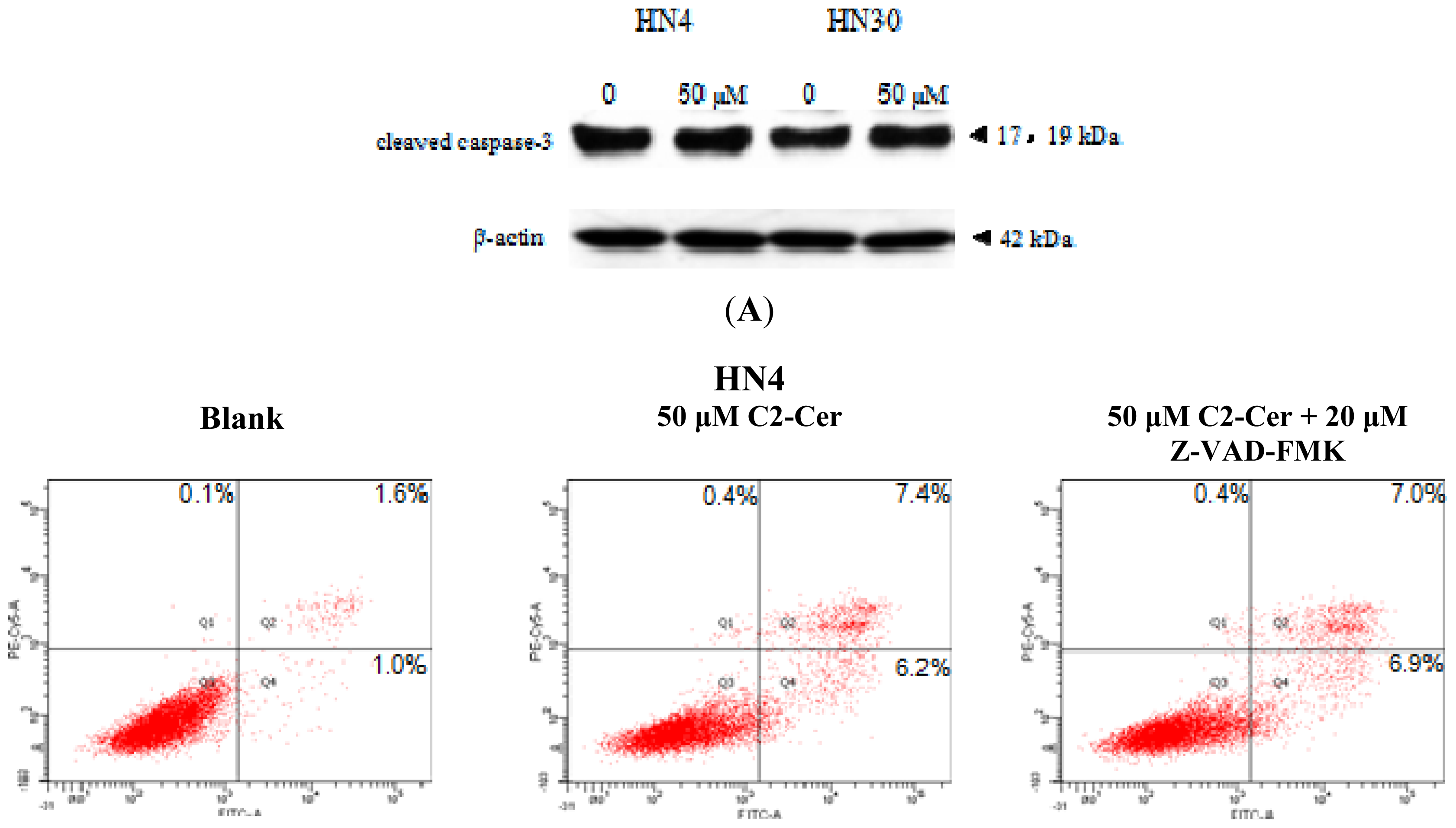
2.1.1. C2-Cer Induced Apoptosis in HNSCC
2.1.2. C2-Cer Induced DNA Fragmentation of HNSCC
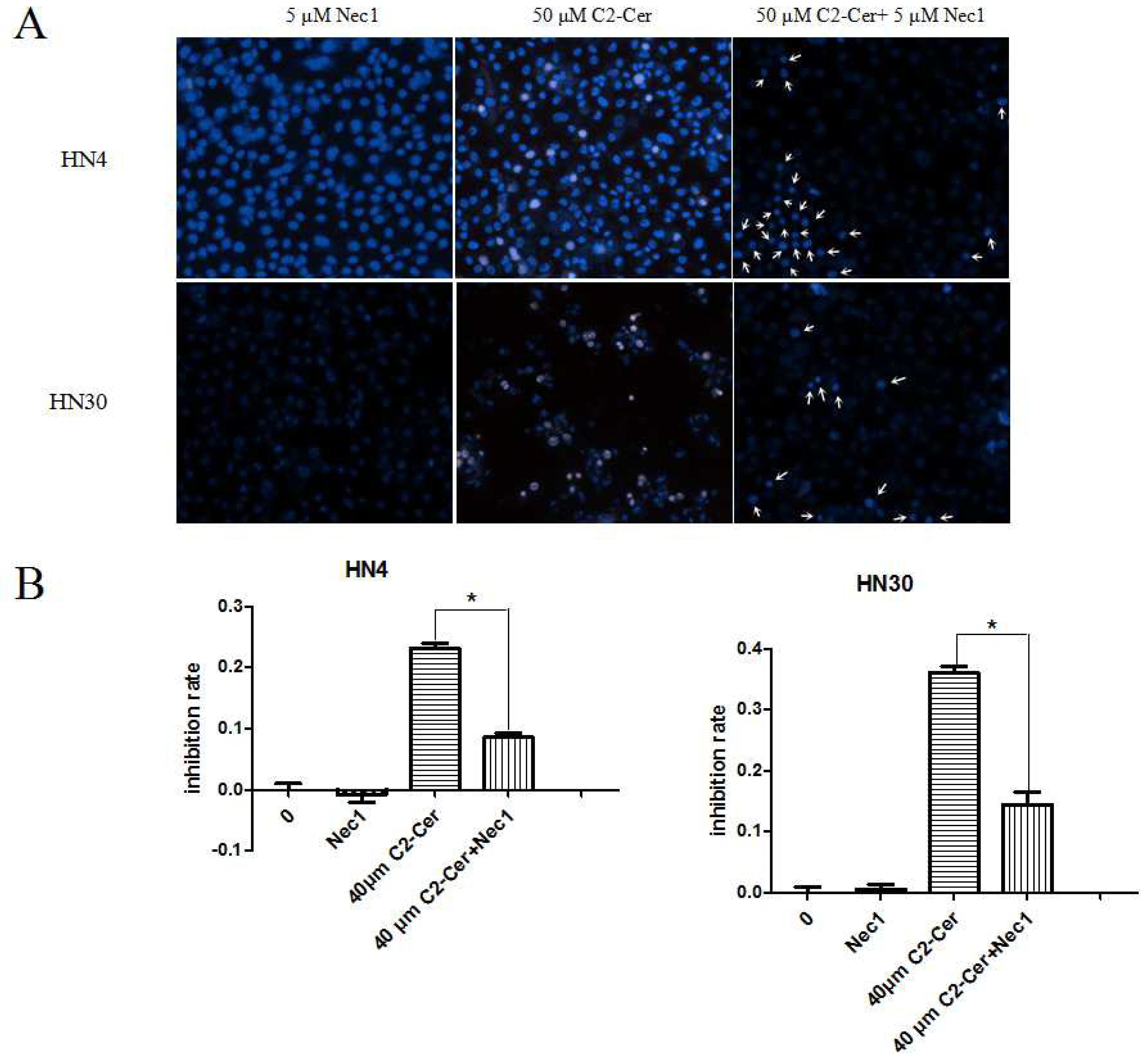
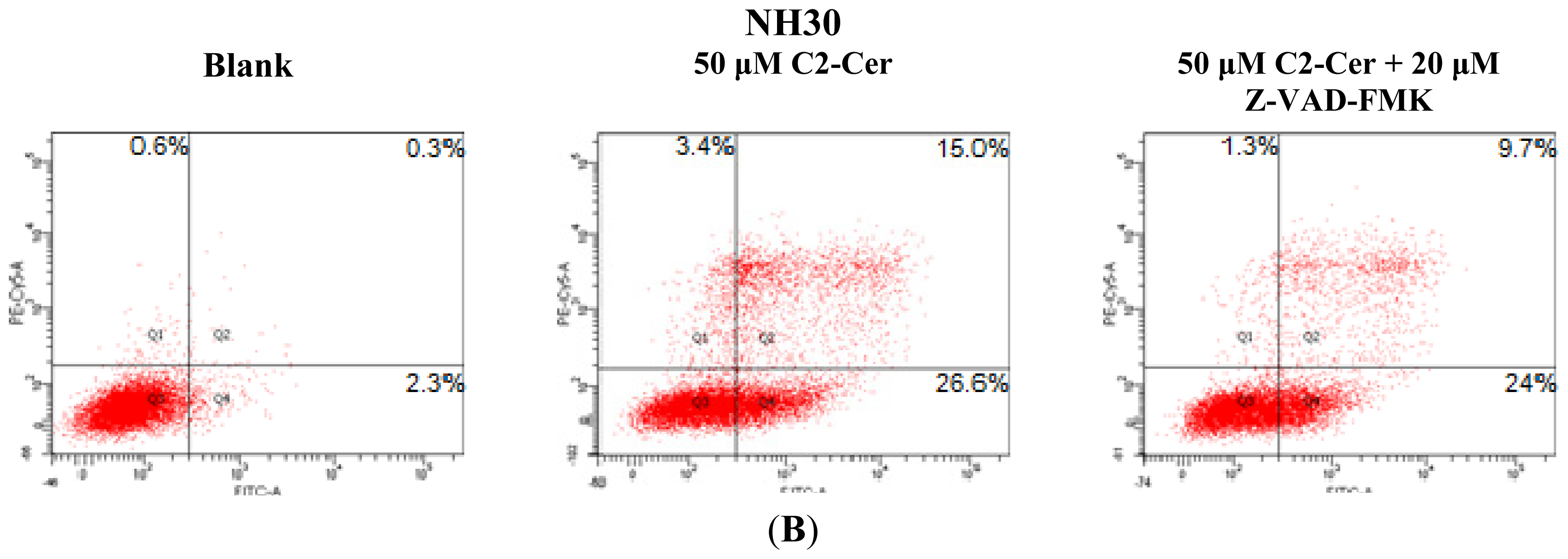
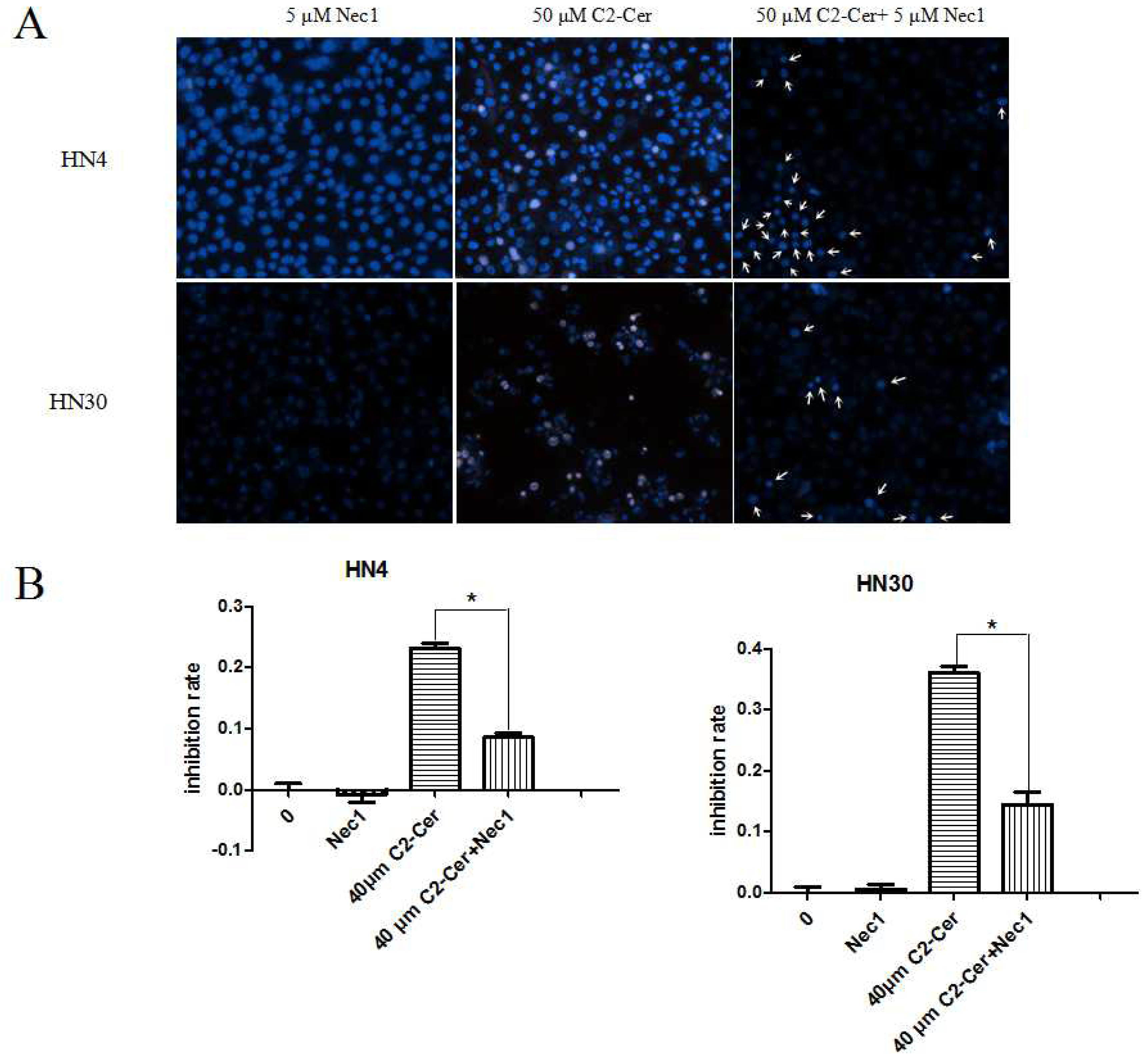
2.1.3. C2-Cer Induced Programmed Necrosis in HNSCC Cells
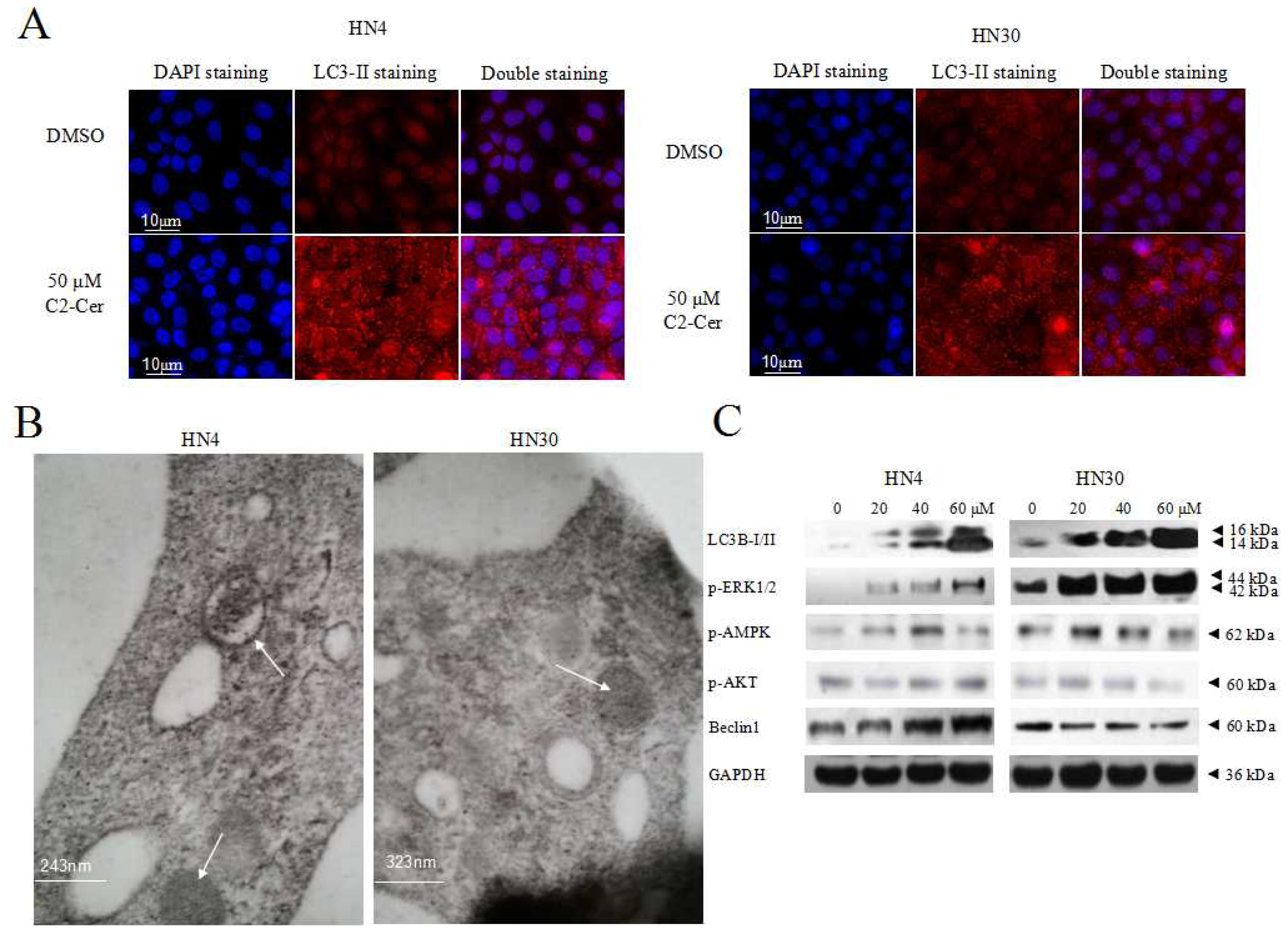
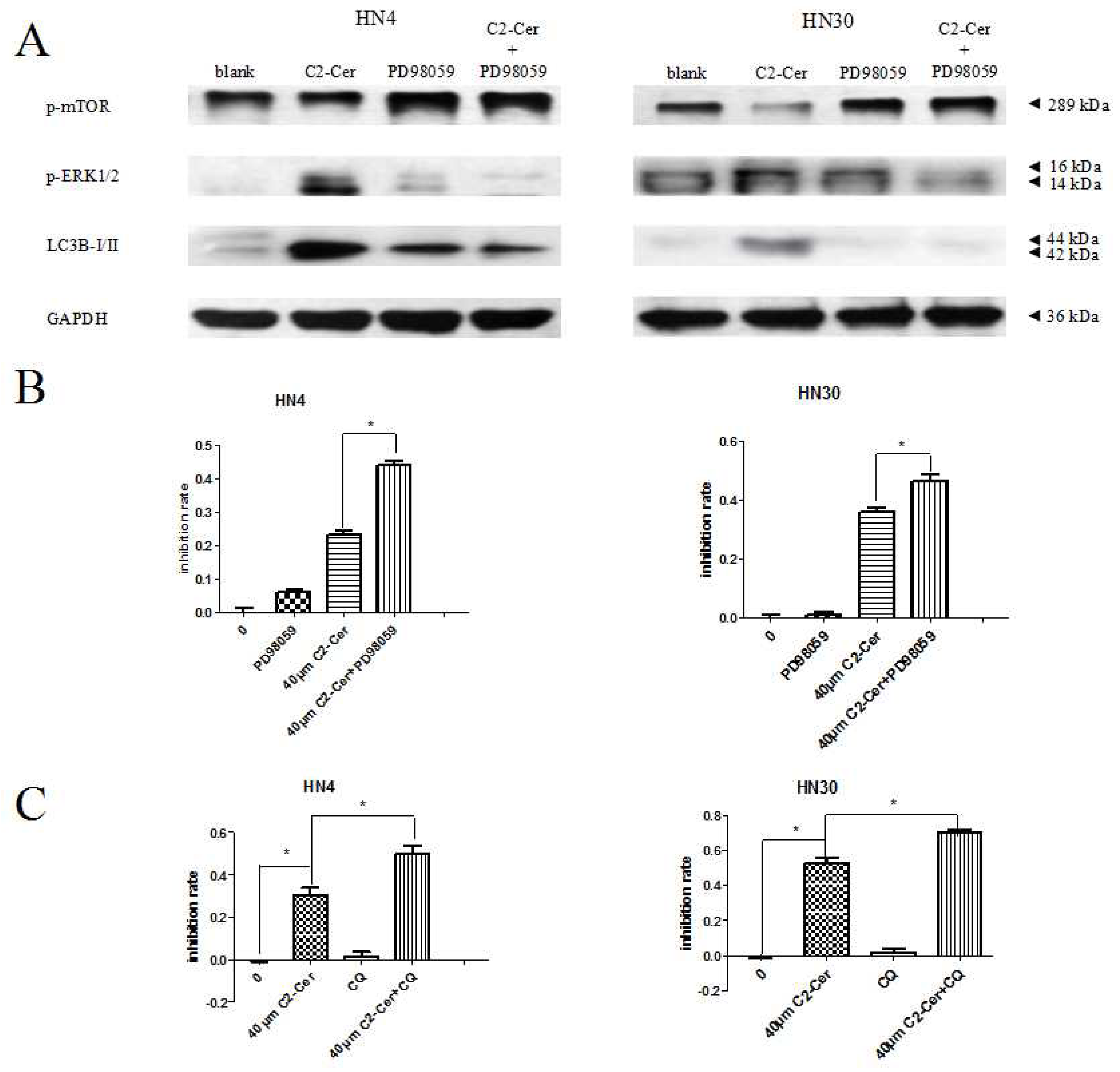
2.1.4. C2-Cer Induced Autophagy
2.1.5. Autophagy Inhibition Sensitized Cells to C2-Cer-Mediated Cytotoxicity
2.2. Discussion
3. Experimental Section
3.1. Chemicals and Cell Culture
3.2. CCK-8 Assay
3.3. Assay for DNA Fragmentation
3.4. Flow Cytometry Analysis
3.5. Immunofluorescence
3.6. Electron Microscopy
3.7. Western Blot Analysis
3.8. Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Chidzonga, M.M.; Mahomva, L. Squamous cell carcinoma of the oral cavity, maxillary antrum andlip in a Zimbabwean population: A descriptive epidemiological study. Oral Oncol 2006, 42, 184–189. [Google Scholar]
- Jemal, A.; Murray, T.; Samuels, A.; Ghafoor, A.; Ward, E.; Thun, M.J. Cancer statistics, 2003. CA: Cancer J. Clin 2003, 53, 5–26. [Google Scholar]
- Pierce, J.P.; Messer, K.; White, M.M.; Cowling, D.W.; Thomas, D.P. Prevalence of heavy smoking in California and the United States, 1965–2007. JAMA 2011, 305, 1106–1112. [Google Scholar]
- Cowling, D.W.; Yang, J. Smoking-attributable cancer mortality in California, 1979–2005. Tob. Control 2010, 19, i62–i67. [Google Scholar]
- Rodu, B.; Cole, P. Declining mortality from smoking in the United States. Nicotine Tob. Res 2007, 9, 781–784. [Google Scholar]
- Boyle, P. Cancer, cigarette smoking and premature death in Europe: A review including the Recommendations of European Cancer Experts Consensus Meeting, Helsinki, October 1996. Lung Cancer 1997, 17, 1–60. [Google Scholar]
- Wagner, S.; Mayer, C.; Wittekindt, C.; Klussmann, J.P. Human papillomavirus (HPV) and head and neck cancer. Der Hautarzt; Zeitschrift fur Dermatologie, Venerologie, und verwandte Gebiete 2012, 63, 24–29. [Google Scholar]
- Smith, E.M.; Rubenstein, L.M.; Haugen, T.H.; Pawlita, M.; Turek, L.P. Complex etiology underlies risk and survival in head and neck cancer human papillomavirus, tobacco, and alcohol: A case for multifactor disease. J. Oncol 2012, 2012, 571862. [Google Scholar]
- Rikiishi, H. Autophagic action of new targeting agents in head and neck oncology. Cancer Biol. Ther 2012, 13, 978–991. [Google Scholar]
- Beckham, T.H.; Elojeimy, S.; Cheng, J.C.; Turner, L.S.; Hoffman, S.R.; Norris, J.S.; Liu, X. Targeting sphingolipid metabolism in head and neck cancer: Rational therapeutic potentials. Expert Opin. Ther. Targets 2010, 14, 529–539. [Google Scholar]
- Kimura, T.; Takabatake, Y.; Takahashi, A.; Isaka, Y. Chloroquine in cancer therapy: A double-edged sword of autophagy. Cancer Res. 2013, 73, 3–7. [Google Scholar]
- White, E.; DiPaola, R.S. The double-edged sword of autophagy modulation in cancer. Clin. Cancer Res 2009, 15, 5308–5316. [Google Scholar]
- Li, C.; Iida, M.; Dunn, E.F.; Wheeler, D.L. Dasatinib blocks cetuximab- and radiation-induced nuclear translocation of the epidermal growth factor receptor in head and neck squamous cell carcinoma. Radiother. Oncol 2010, 97, 330–337. [Google Scholar]
- Hiratsuka, T.; Inomata, M.; Kono, Y.; Yokoyama, S.; Shiraishi, N.; Kitano, S. DHLTauZnNa, a newly synthesized alpha-lipoic acid derivative, induces autophagy in human colorectal cancer cells. Oncol. Rep 2013, 29, 2140–2146. [Google Scholar]
- Ren, S.X.; Shen, J.; Cheng, A.S.; Lu, L.; Chan, R.L.; Li, Z.J.; Wang, X.J.; Wong, C.C.; Zhang, L.; Ng, S.S.; et al. FK-16 derived from the anticancer peptide LL-37 induces caspase-independent apoptosis and autophagic cell death in colon cancer cells. PLoS One 2013, 8, e63641. [Google Scholar]
- Ponnusamy, S.; Meyers-Needham, M.; Senkal, C.E.; Saddoughi, S.A.; Sentelle, D.; Selvam, S.P.; Salas, A.; Ogretmen, B. Sphingolipids and cancer: Ceramide and sphingosine-1-phosphate in the regulation of cell death and drug resistance. Future Oncol 2010, 6, 1603–1624. [Google Scholar]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol 2008, 9, 139–150. [Google Scholar]
- Parra, V.; Moraga, F.; Kuzmicic, J.; Lopez-Crisosto, C.; Troncoso, R.; Torrealba, N.; Criollo, A.; Diaz-Elizondo, J.; Rothermel, B.A.; Quest, A.F.; et al. Calcium and mitochondrial metabolism in ceramide-induced cardiomyocyte death. Biochim. Biophys. Acta 2013, 1832, 1334–1344. [Google Scholar]
- Obeid, L.M.; Linardic, C.M.; Karolak, L.A.; Hannun, Y.A. Programmed cell death induced by ceramide. Science 1993, 259, 1769–1771. [Google Scholar]
- Zhang, X.F.; Li, B.X.; Dong, C.Y.; Ren, R. Apoptosis of human colon carcinoma HT-29 cells induced by ceramide. World J. Gastroenterol 2006, 12, 3581–3584. [Google Scholar]
- Wang, J.; Lv, X.W.; Shi, J.P.; Hu, X.S. Mechanisms involved in ceramide-induced cell cycle arrest in human hepatocarcinoma cells. World J. Gastroenterol 2007, 13, 1129–1134. [Google Scholar]
- Dam, A.D.; Mitchell, A.S.; Quadrilatero, J. Induction of mitochondrial biogenesis protects against caspase-dependent and caspase-independent apoptosis in L6 myoblasts. Biochim. Biophys. Acta 2013, 1833, 3426–3435. [Google Scholar]
- Karahatay, S.; Thomas, K.; Koybasi, S.; Senkal, C.E.; Elojeimy, S.; Liu, X.; Bielawski, J.; Day, T.A.; Gillespie, M.B.; Sinha, D.; et al. Clinical relevance of ceramide metabolism in the pathogenesis of human head and neck squamous cell carcinoma (HNSCC): Attenuation of C(18)-ceramide in HNSCC tumors correlates with lymphovascular invasion and nodal metastasis. Cancer Lett 2007, 256, 101–111. [Google Scholar]
- Saddoughi, S.A.; Garrett-Mayer, E.; Chaudhary, U.; O’Brien, P.E.; Afrin, L.B.; Day, T.A.; Gillespie, M.B.; Sharma, A.K.; Wilhoit, C.S.; Bostick, R.; et al. Results of a phase II trial of gemcitabine plus doxorubicin in patients with recurrent head and neck cancers: Serum C(1)(8)-ceramide as a novel biomarker for monitoring response. Clin. Cancer Res 2011, 17, 6097–6105. [Google Scholar]
- Yang, Y.L.; Ji, C.; Bi, Z.G.; Lu, C.C.; Wang, R.; Gu, B.; Cheng, L. Deguelin induces both apoptosis and autophagy in cultured head and neck squamous cell carcinoma cells. PLoS One 2013, 8, e54736. [Google Scholar]
- Young, M.R.; Neville, B.W.; Chi, A.C.; Lathers, D.M.; Gillespie, M.B.; Day, T.A. Autocrine motility-stimulatory pathways of oral premalignant lesion cells. Clin. Exp. Metastasis 2007, 24, 131–139. [Google Scholar]
- Mizushima, N.; Koike, R.; Kohsaka, H.; Kushi, Y.; Handa, S.; Yagita, H.; Miyasaka, N. Ceramide induces apoptosis via CPP32 activation. FEBS Lett 1996, 395, 267–271. [Google Scholar]
- Tavarini, S.; Colombaioni, L.; Garcia-Gil, M. Sphingomyelinase metabolites control survival and apoptotic death in SH-SY5Y neuroblastoma cells. Neurosci. Lett 2000, 285, 185–188. [Google Scholar]
- Ji, L.; Zhang, G.; Uematsu, S.; Akahori, Y.; Hirabayashi, Y. Induction of apoptotic DNA fragmentation and cell death by natural ceramide. FEBS Lett 1995, 358, 211–214. [Google Scholar]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol 2005, 1, 112–119. [Google Scholar]
- Christofferson, D.E.; Yuan, J. Necroptosis as an alternative form of programmed cell death. Curr. Opin. Cell Biol 2010, 22, 263–268. [Google Scholar]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch’en, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol 2008, 4, 313–321. [Google Scholar]
- Jiang, W.; Ogretmen, B. Autophagy paradox and ceramide. Biochim. Biophys. Acta 2013. [Google Scholar] [CrossRef]
- Zhang, G.; Park, M.A.; Mitchell, C.; Walker, T.; Hamed, H.; Studer, E.; Graf, M.; Rahmani, M.; Gupta, S.; Hylemon, P.B.; et al. Multiple cyclin kinase inhibitors promote bile acid-induced apoptosis and autophagy in primary hepatocytes via p53-CD95-dependent signaling. J. Biol. Chem 2008, 283, 24343–24358. [Google Scholar]
- Pattingre, S.; Bauvy, C.; Levade, T.; Levine, B.; Codogno, P. Ceramide-induced autophagy: To junk or to protect cells? Autophagy 2009, 5, 558–560. [Google Scholar]
- Samaddar, J.S.; Gaddy, V.T.; Duplantier, J.; Thandavan, S.P.; Shah, M.; Smith, M.J.; Browning, D.; Rawson, J.; Smith, S.B.; Barrett, J.T.; et al. A role for macroautophagy in protection against 4-hydroxytamoxifen-induced cell death and the development of antiestrogen resistance. Mol. Cancer Ther 2008, 7, 2977–2987. [Google Scholar]
- Degtyarev, M.; De Maziere, A.; Orr, C.; Lin, J.; Lee, B.B.; Tien, J.Y.; Prior, W.W.; van Dijk, S.; Wu, H.; Gray, D.C.; et al. Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents. J. Cell Biol 2008, 183, 101–116. [Google Scholar]
- Guenther, G.G.; Peralta, E.R.; Rosales, K.R.; Wong, S.Y.; Siskind, L.J.; Edinger, A.L. Ceramide starves cells to death by downregulating nutrient transporter proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 17402–17407. [Google Scholar]
- Park, M.A.; Zhang, G.; Martin, A.P.; Hamed, H.; Mitchell, C.; Hylemon, P.B.; Graf, M.; Rahmani, M.; Ryan, K.; Liu, X.; et al. Vorinostat and sorafenib increase ER stress, autophagy and apoptosis via ceramide-dependent CD95 and PERK activation. Cancer Biol. Ther 2008, 7, 1648–1662. [Google Scholar]
- Scarlatti, F.; Bauvy, C.; Ventruti, A.; Sala, G.; Cluzeaud, F.; Vandewalle, A.; Ghidoni, R.; Codogno, P. Ceramide-mediated macroautophagy involves inhibition of protein kinase B and up-regulation of beclin 1. J. Biol. Chem 2004, 279, 18384–18391. [Google Scholar]
- Nowak, G. Protein kinase C-alpha and ERK1/2 mediate mitochondrial dysfunction, decreases in active Na+ transport, and cisplatin-induced apoptosis in renal cells. J. Biol. Chem. 2002, 277, 43377–43388. [Google Scholar]
- Brozovic, A.; Osmak, M. Activation of mitogen-activated protein kinases by cisplatin and their role in cisplatin-resistance. Cancer Lett 2007, 251, 1–16. [Google Scholar]
- Rieber, M.; Rieber, M.S. Signalling responses linked to betulinic acid-induced apoptosis are antagonized by MEK inhibitor U0126 in adherent or 3D spheroid melanoma irrespective of p53 status. Int. J. Cancer 2006, 118, 1135–1143. [Google Scholar]
- Fluhr, H.; Spratte, J.; Bredow, M.; Heidrich, S.; Zygmunt, M. Constitutive activity of Erk1/2 and NF-kappaB protects human endometrial stromal cells from death receptor-mediated apoptosis. Reprod. Biol 2013, 13, 113–121. [Google Scholar]
- Cai, C.; Teng, L.; Vu, D.; He, J.Q.; Guo, Y.; Li, Q.; Tang, X.L.; Rokosh, G.; Bhatnagar, A.; Bolli, R. The heme oxygenase 1 inducer (CoPP) protects human cardiac stem cells against apoptosis through activation of the extracellular signal-regulated kinase (ERK)/NRF2 signaling pathway and cytokine release. J. Biol. Chem 2012, 287, 33720–33732. [Google Scholar]
- Taniguchi, M.; Kitatani, K.; Kondo, T.; Hashimoto-Nishimura, M.; Asano, S.; Hayashi, A.; Mitsutake, S.; Igarashi, Y.; Umehara, H.; Takeya, H.; et al. Regulation of autophagy and its associated cell death by “sphingolipid rheostat”: Reciprocal role of ceramide and sphingosine 1-phosphate in the mammalian target of rapamycin pathway. J. Biol. Chem. 2012, 287, 39898–39910. [Google Scholar]
- Daido, S.; Kanzawa, T.; Yamamoto, A.; Takeuchi, H.; Kondo, Y.; Kondo, S. Pivotal role of the cell death factor BNIP3 in ceramide-induced autophagic cell death in malignant glioma cells. Cancer Res 2004, 64, 4286–4293. [Google Scholar]
- Patel, V.; Ramesh, A.; Traicoff, J.L.; Baibakov, G.; Emmert-Buck, M.R.; Gutkind, J.S.; Knezevic, V. Profiling EGFR activity in head and neck squamous cell carcinoma by using a novel layered membrane Western blot technology. Oral Oncol 2005, 41, 503–508. [Google Scholar]
- Yu, C.; Xing, F.; Tang, Z.; Bronner, C.; Lu, X.; Di, J.; Zeng, S.; Liu, J. Anisomycin suppresses Jurkat T cell growth by the cell cycle-regulating proteins. Pharmacol. Rep 2013, 65, 435–444. [Google Scholar]
- Herman-Antosiewicz, A.; Johnson, D.E.; Singh, S.V. Sulforaphane causes autophagy to inhibit release of cytochrome C and apoptosis in human prostate cancer cells. Cancer Res 2006, 66, 5828–5835. [Google Scholar]
- Ahn, M.Y.; Ahn, S.G.; Yoon, J.H. Apicidin, a histone deaceylase inhibitor, induces both apoptosis and autophagy in human oral squamous carcinoma cells. Oral Oncol 2011, 47, 1032–1038. [Google Scholar]
- Shabbits, J.A.; Mayer, L.D. High ceramide content liposomes with in vivo antitumor activity. Anticancer Res 2003, 23, 3663–3669. [Google Scholar]
- Stover, T.C.; Sharma, A.; Robertson, G.P.; Kester, M. Systemic delivery of liposomal short-chain ceramide limits solid tumor growth in murine models of breast adenocarcinoma. Clin. Cancer Res 2005, 11, 3465–3474. [Google Scholar]
- Liu, X.; Ryland, L.; Yang, J.; Liao, A.; Aliaga, C.; Watts, R.; Tan, S.F.; Kaiser, J.; Shanmugavelandy, S.S.; Rogers, A.; et al. Targeting of survivin by nanoliposomal ceramide induces complete remission in a rat model of NK-LGL leukemia. Blood 2010, 116, 4192–4201. [Google Scholar]
- Tran, M.A.; Smith, C.D.; Kester, M.; Robertson, G.P. Combining nanoliposomal ceramide with sorafenib synergistically inhibits melanoma and breast cancer cell survival to decrease tumor development. Clin. Cancer Res 2008, 14, 3571–3581. [Google Scholar]





© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhu, W.; Wang, X.; Zhou, Y.; Wang, H. C2-Ceramide Induces Cell Death and Protective Autophagy in Head and Neck Squamous Cell Carcinoma Cells. Int. J. Mol. Sci. 2014, 15, 3336-3355. https://doi.org/10.3390/ijms15023336
Zhu W, Wang X, Zhou Y, Wang H. C2-Ceramide Induces Cell Death and Protective Autophagy in Head and Neck Squamous Cell Carcinoma Cells. International Journal of Molecular Sciences. 2014; 15(2):3336-3355. https://doi.org/10.3390/ijms15023336
Chicago/Turabian StyleZhu, Wenyuan, Xinhua Wang, Yi Zhou, and Huiming Wang. 2014. "C2-Ceramide Induces Cell Death and Protective Autophagy in Head and Neck Squamous Cell Carcinoma Cells" International Journal of Molecular Sciences 15, no. 2: 3336-3355. https://doi.org/10.3390/ijms15023336