Bacterial Cellular Engineering by Genome Editing and Gene Silencing

Abstract
:1. Introduction
2. Conventional Genome Editing
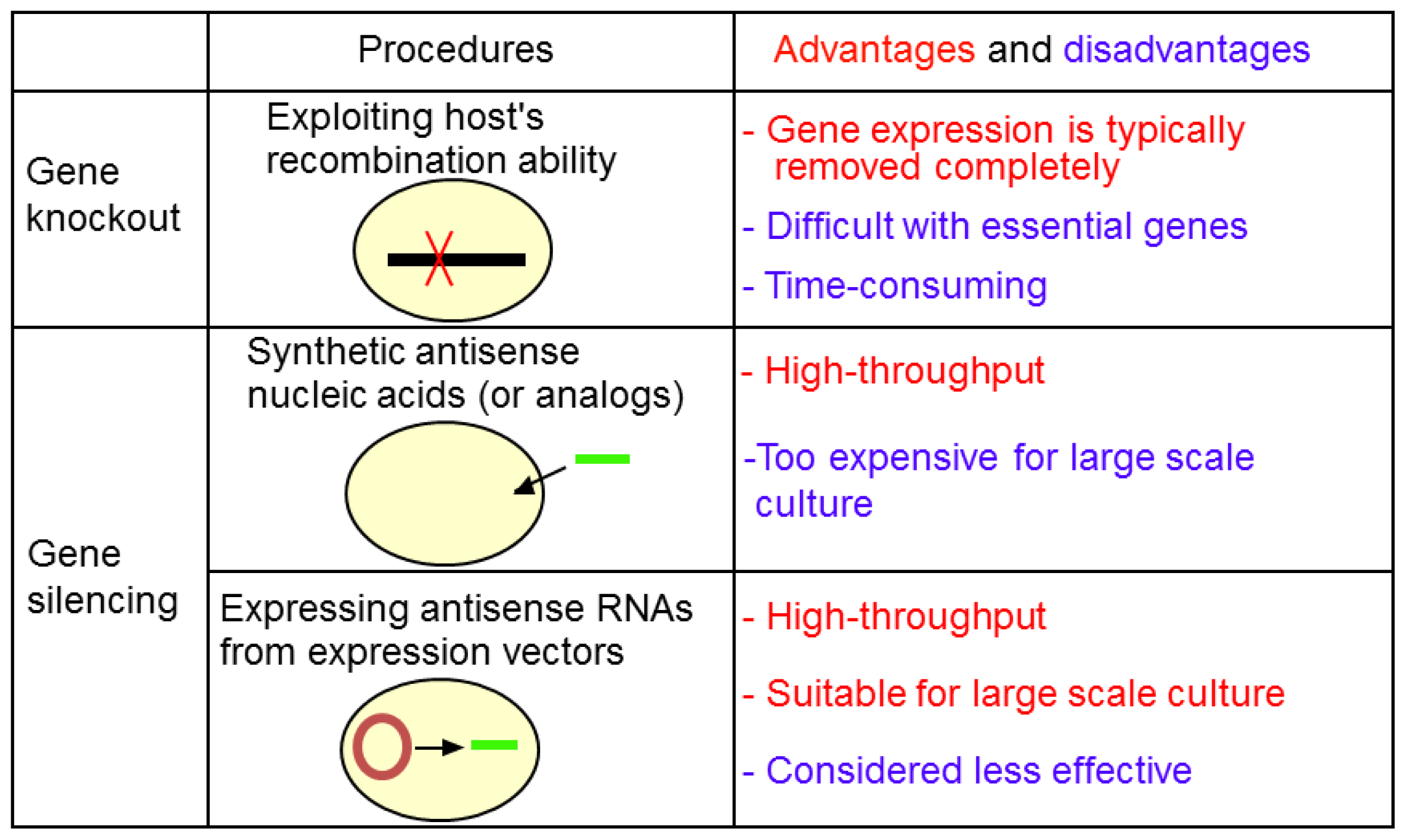
2.1. Gene Knockout
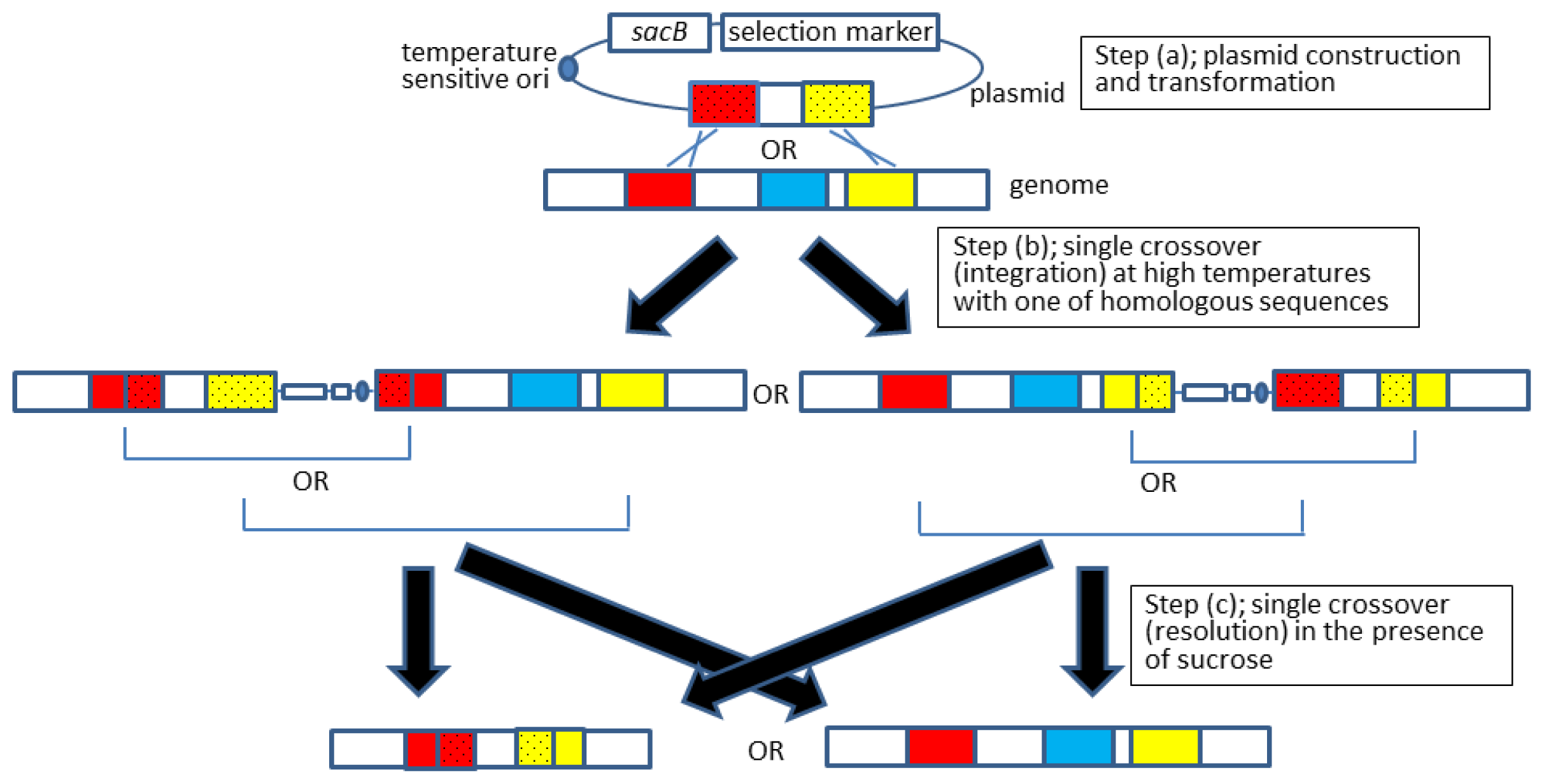
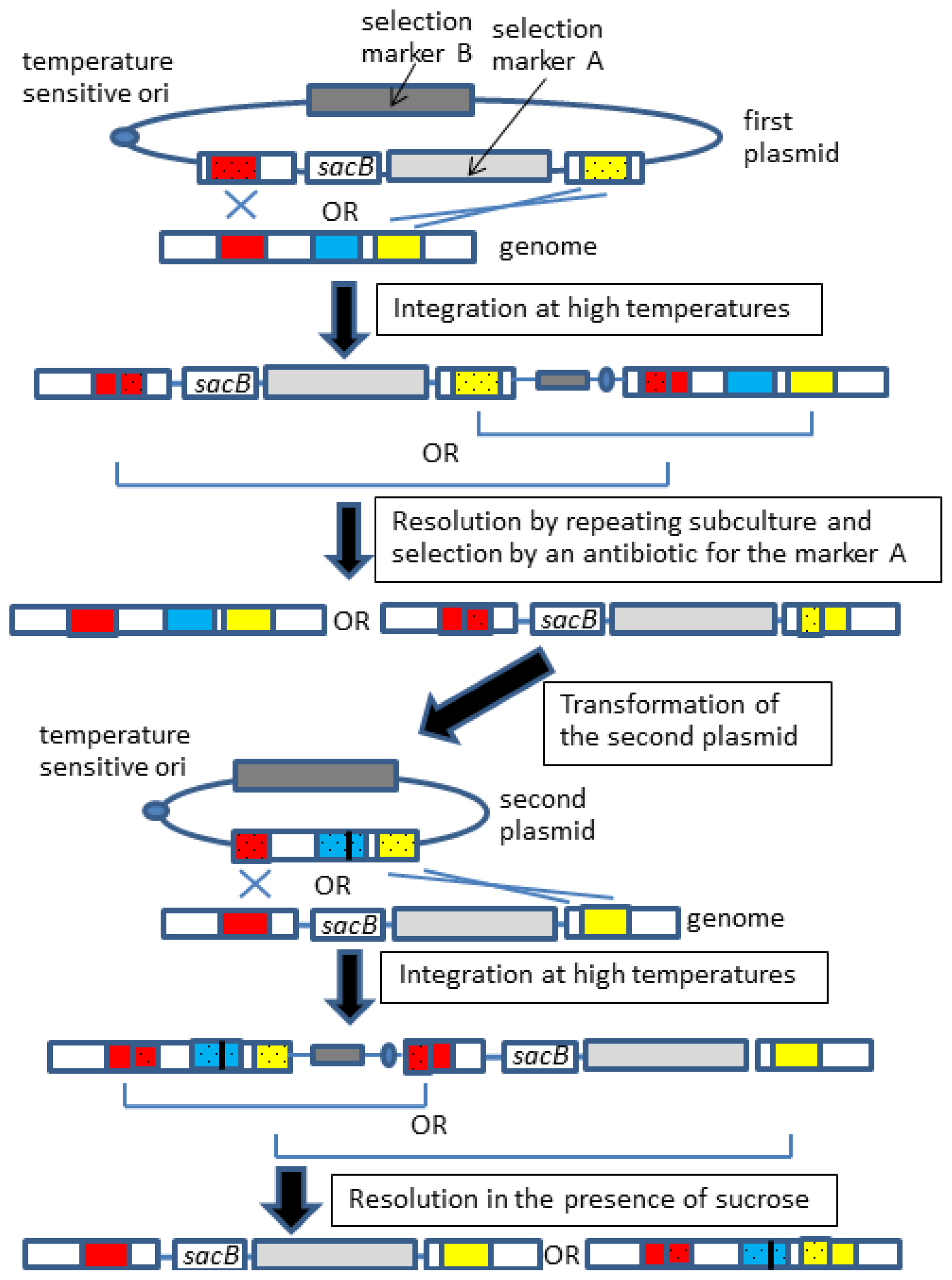
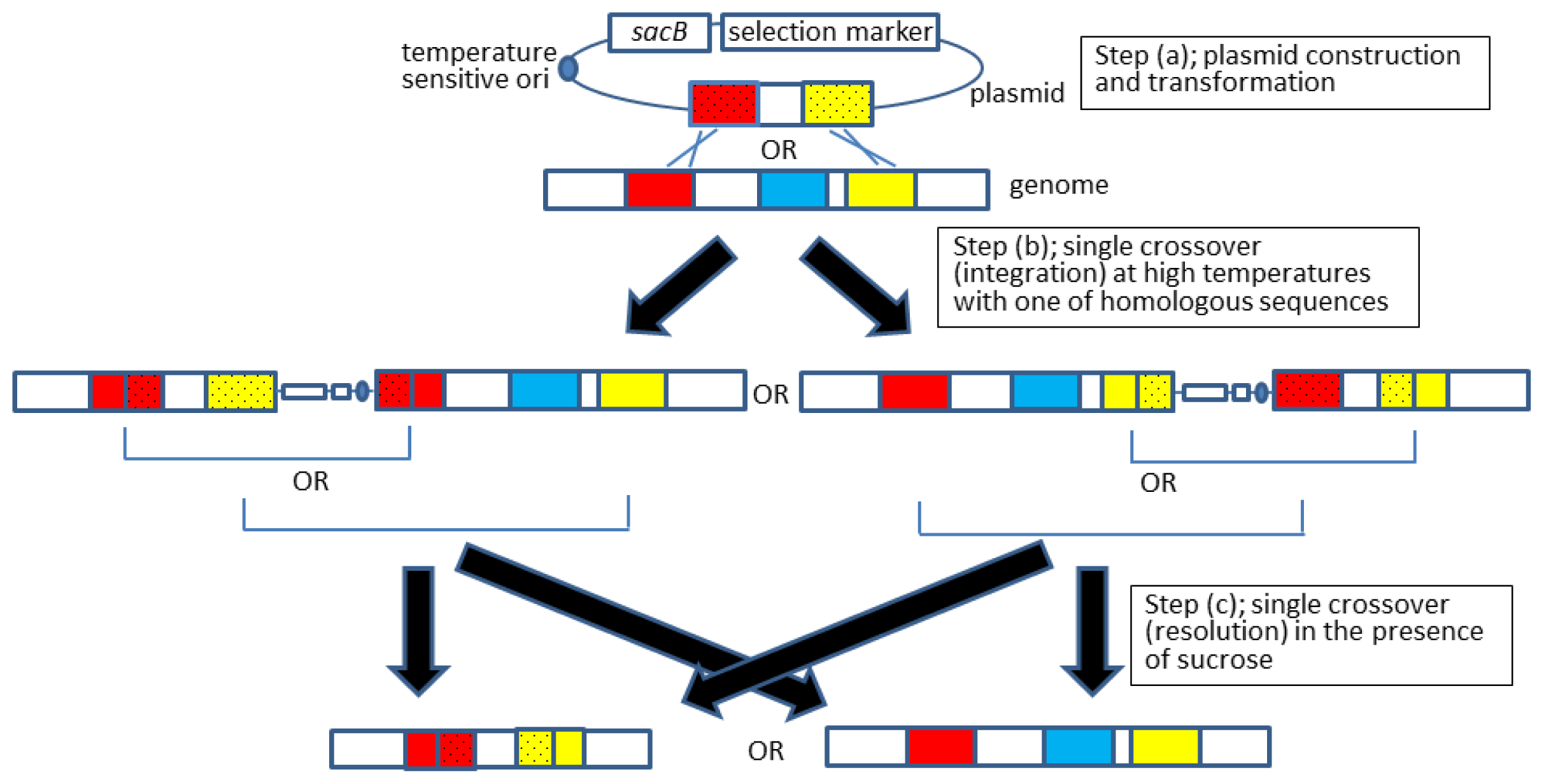
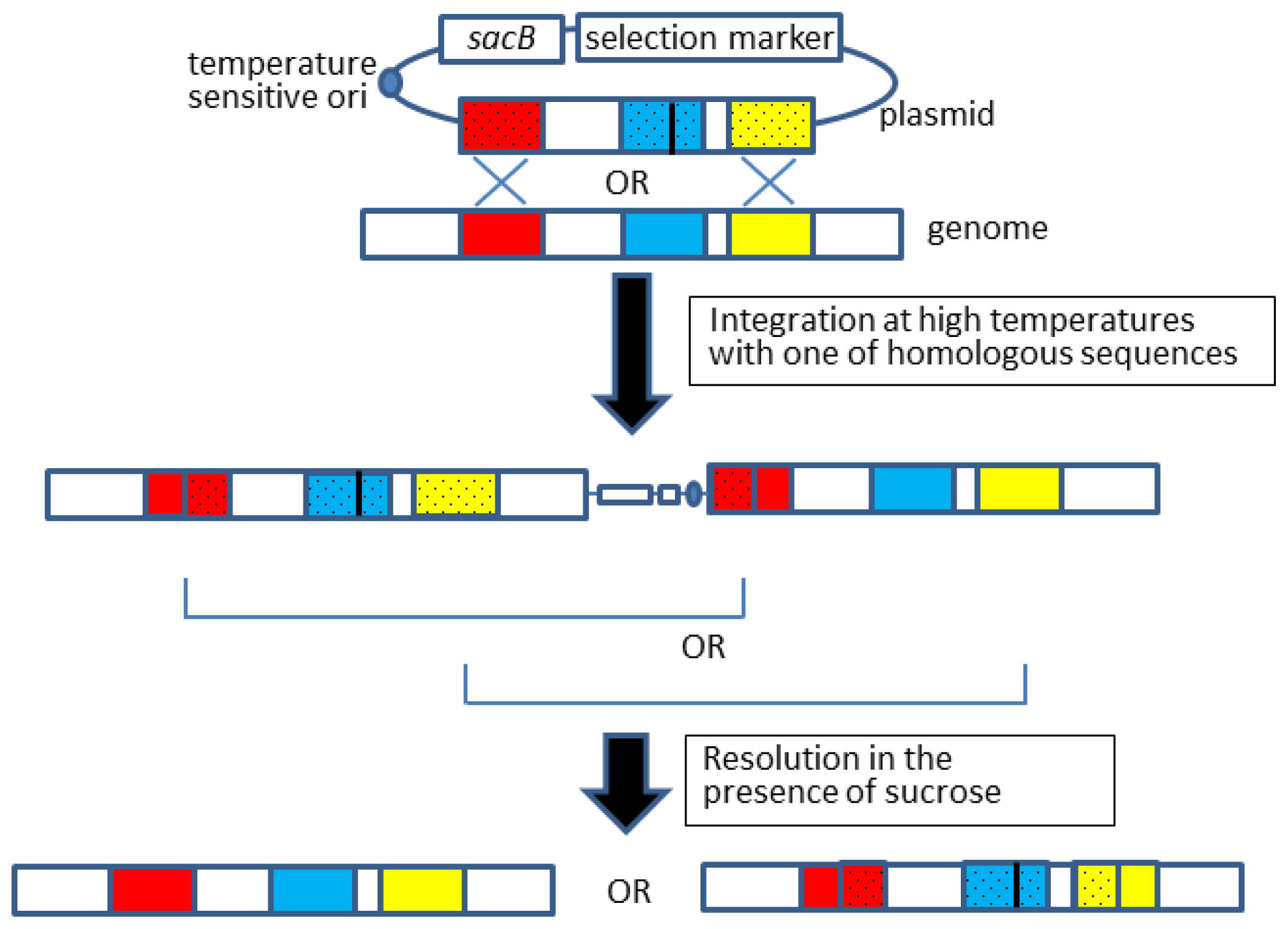
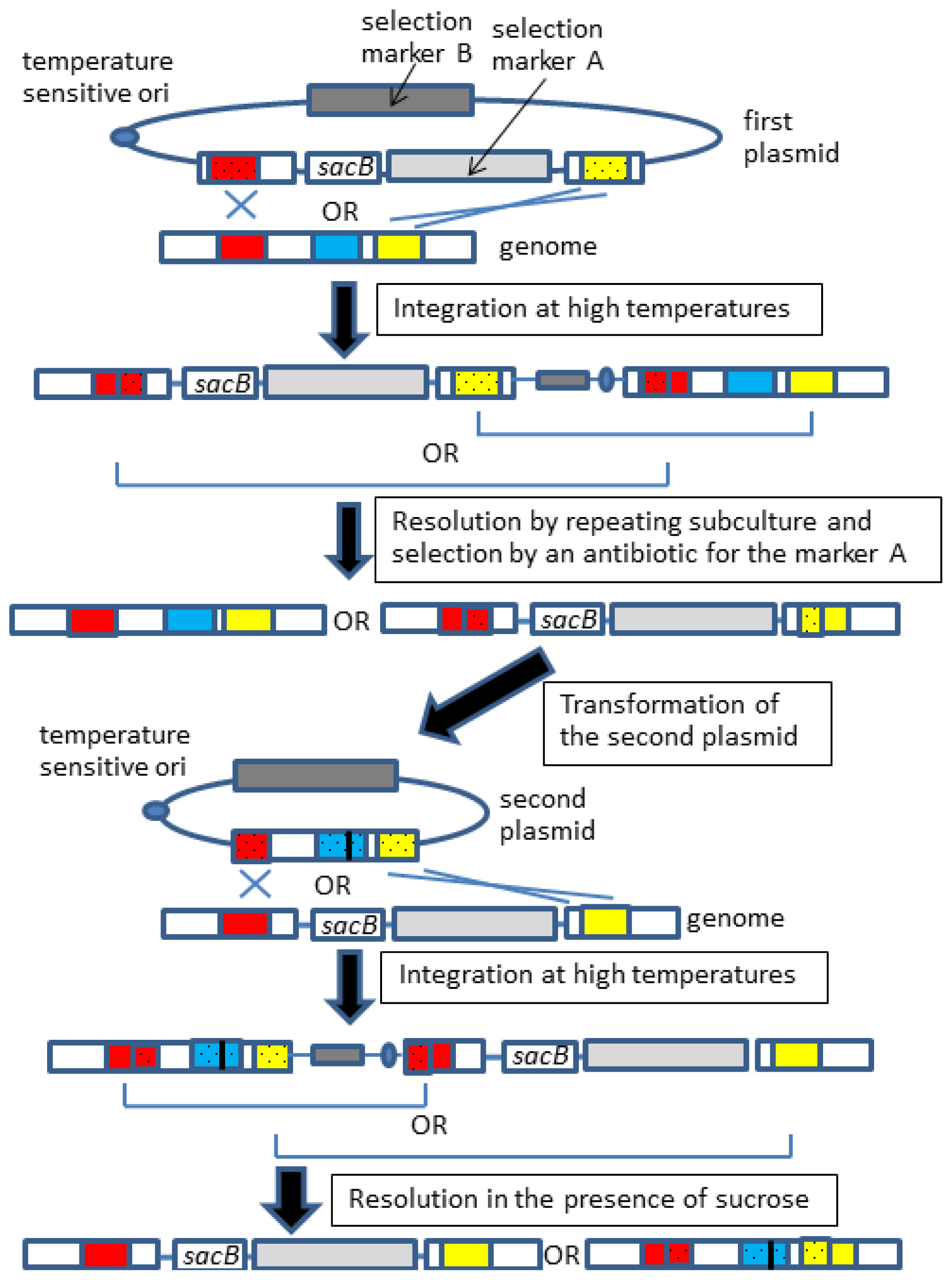
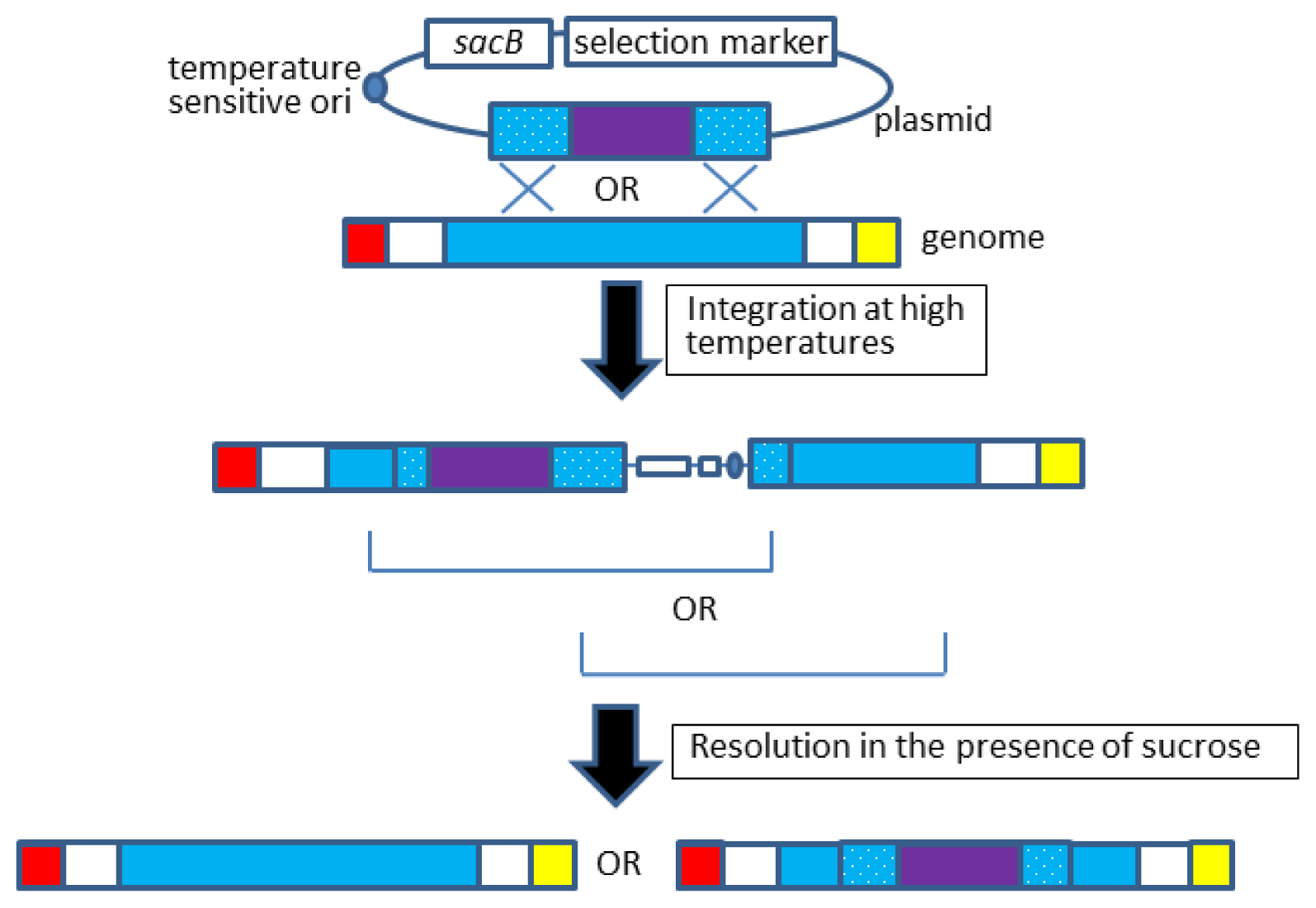
2.2. Allelic Exchange
2.3. Gene Knock-in
3. New Technologies for Genome Editing
3.1. Gene Knockout with Mobile Group II Introns
3.2. RNA Guided-, Artificial Endonuclease Mediated-, and Peptide Nucleic Acid Stimulated-Recombination
3.3. Possibility of the Brand-New Methods for the Future
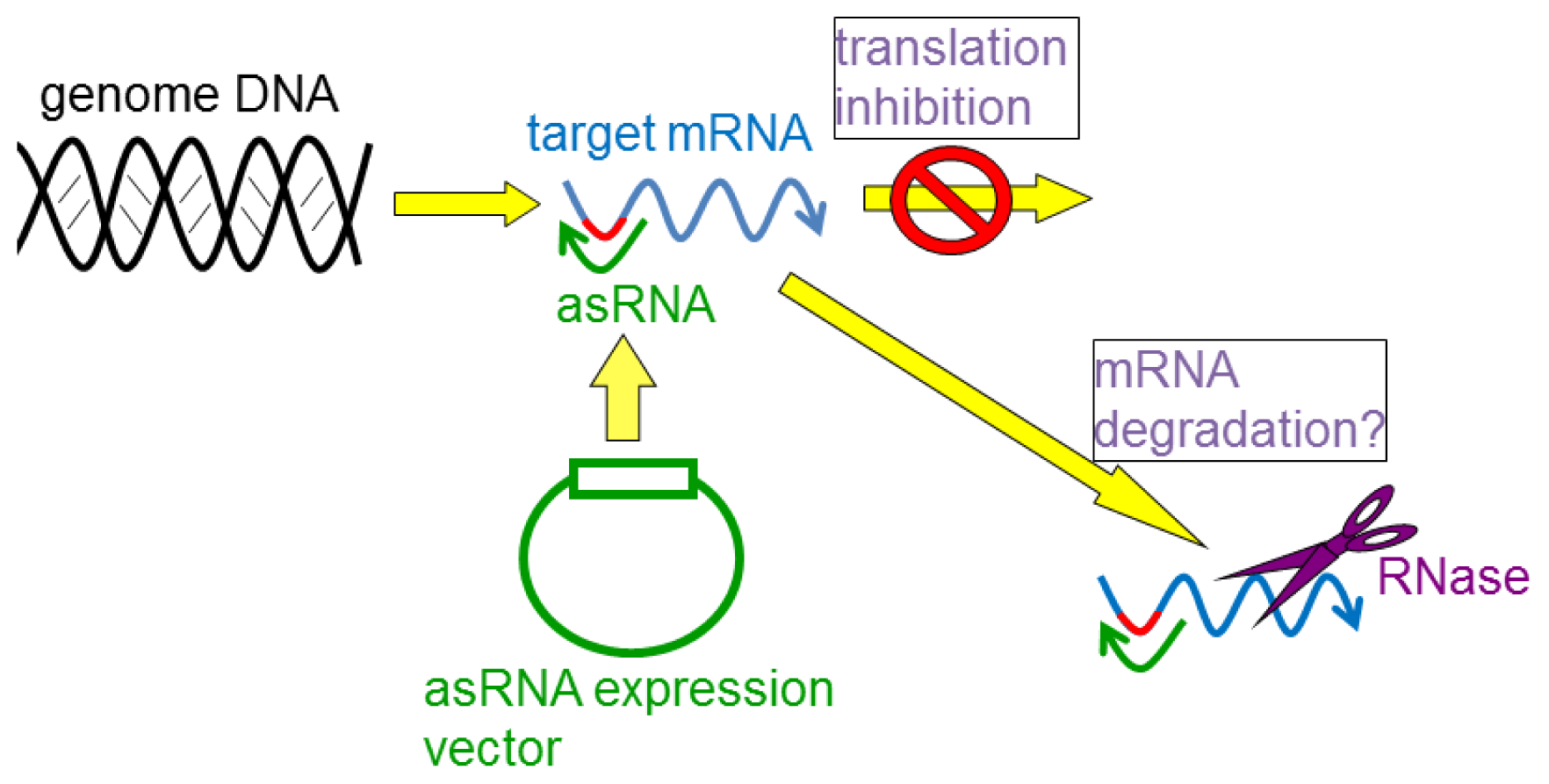
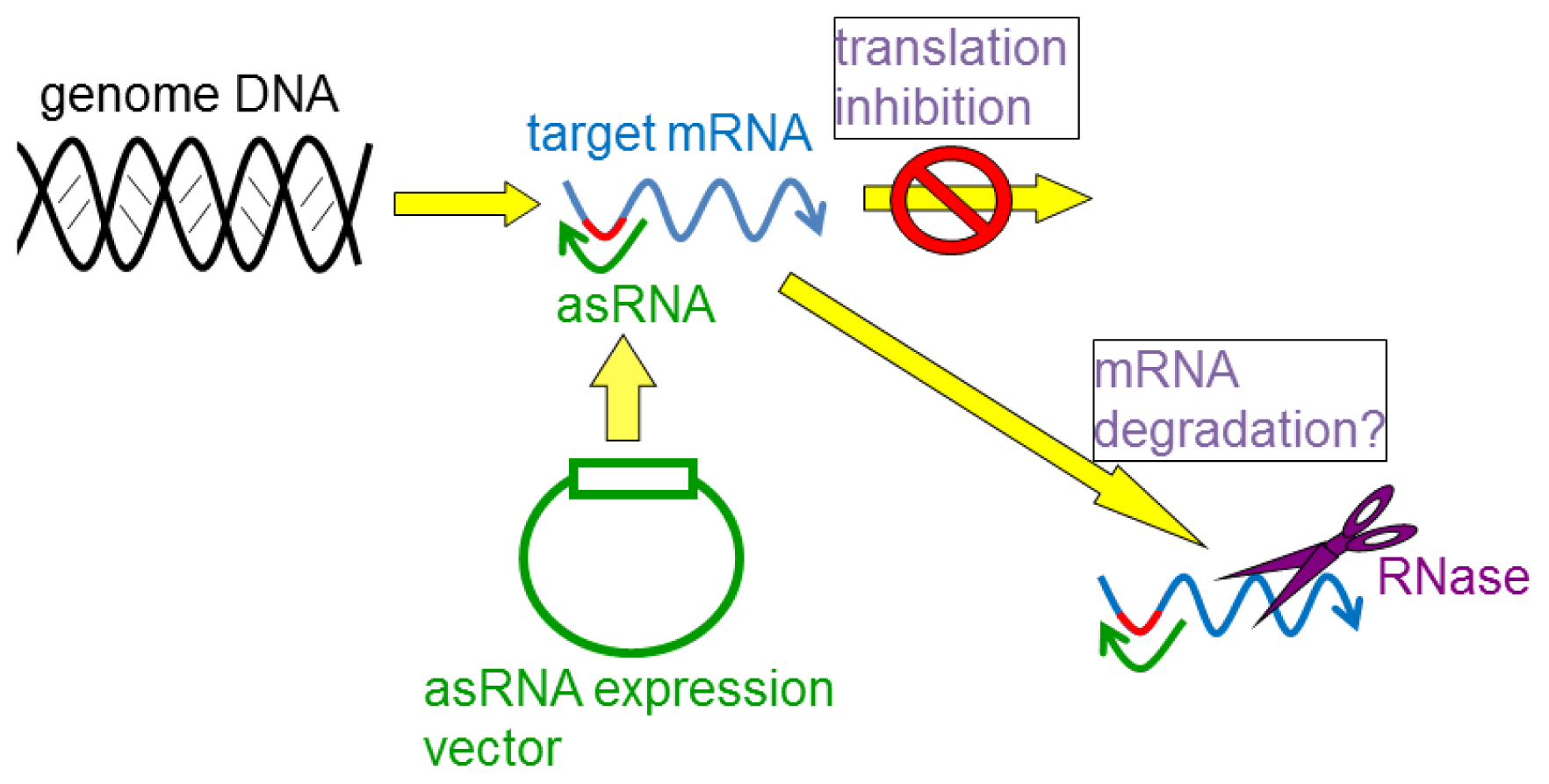
4. Gene Silencing Using asRNAs
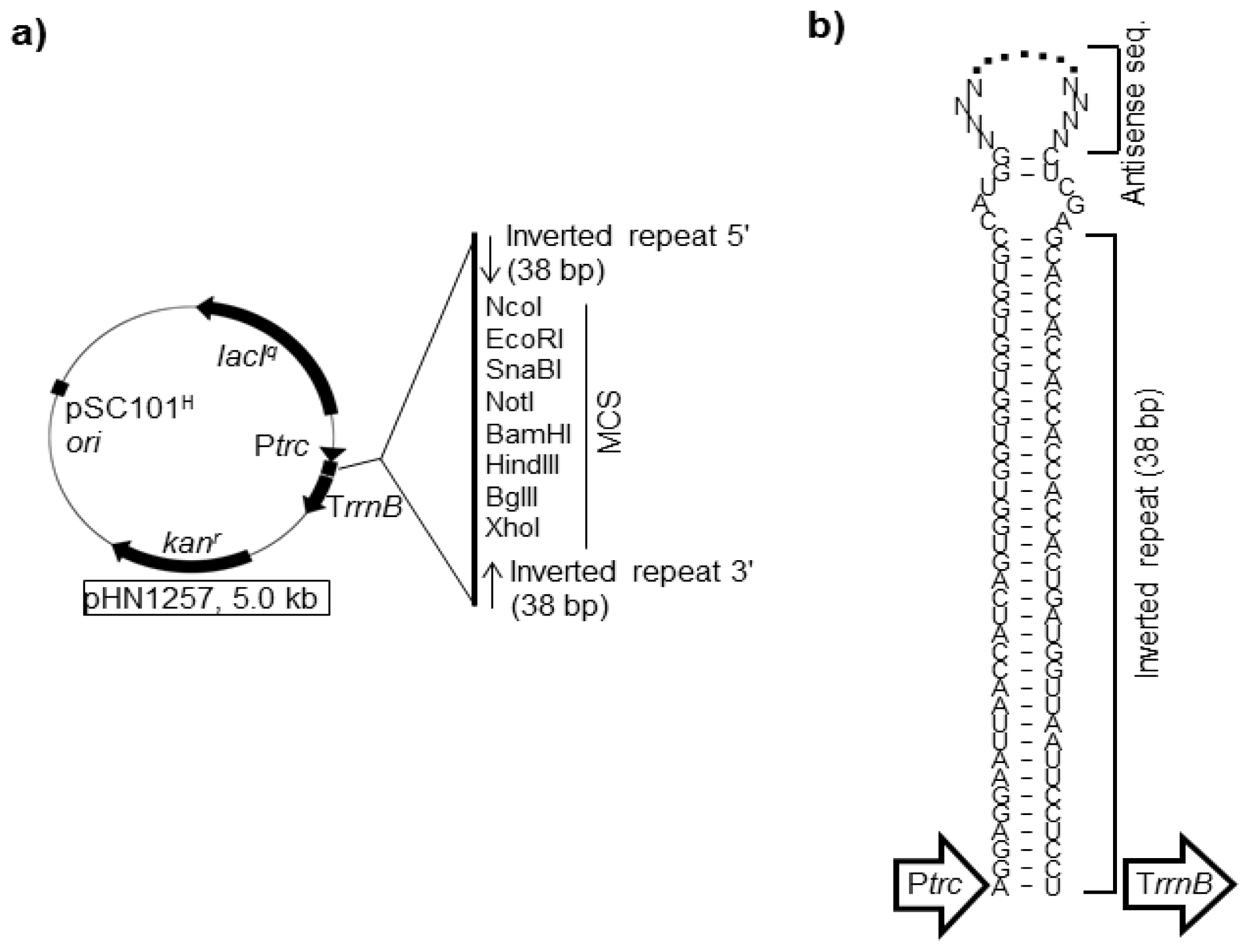
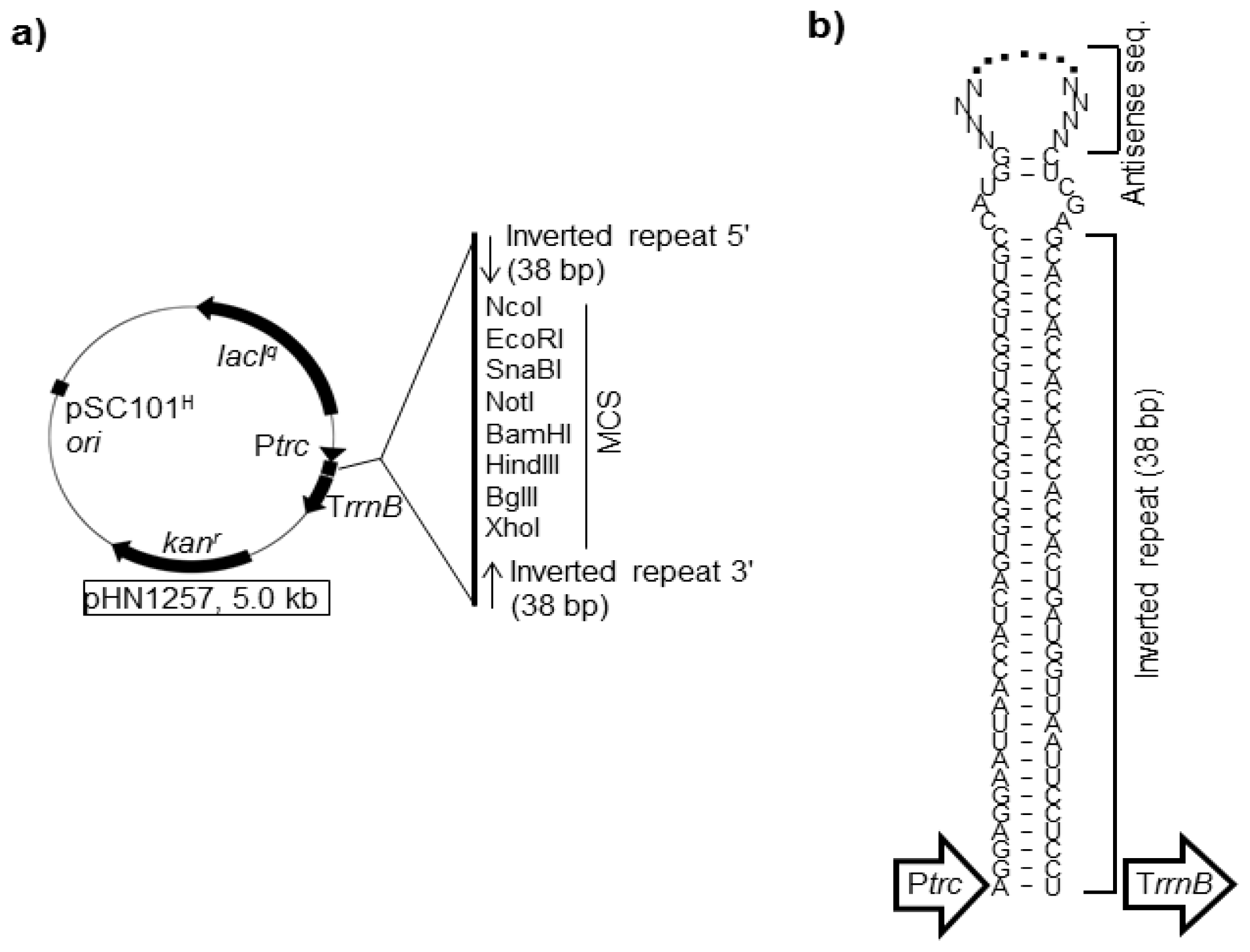
4.1. asRNAs Expressed from Expression Vectors
4.2. Antisnese Oligonucleotides Synthesized in Vitro
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Curtis, T.P.; Sloan, W.T.; Scannell, J.W. Estimating prokaryotic diversity and its limits. Proc. Natl. Acad. Sci. USA 2002, 99, 10494–10499. [Google Scholar]
- Ward, B.B. How many species of prokaryotes are there? Proc. Natl. Acad. Sci. USA 2002, 99, 10234–10236. [Google Scholar]
- Hardy, S.; Legagneux, V.; Audic, Y.; Paillard, L. Reverse genetics in eukaryotes. Biol. Cell 2010, 102, 561–580. [Google Scholar] [Green Version]
- Collins, J.J.; Endy, D.; Hutchison, C.A.; Roberts, R.J. Editorial-synthetic biology. Nucleic Acids Res 2010, 38, 2513. [Google Scholar]
- Reyrat, J.M.; Pelicic, V.; Gicquel, B.; Rappuoli, R. Counterselectable markers: Untapped tools for bacterial genetics and pathogenesis. Infect. Immun 1998, 66, 4011–4017. [Google Scholar]
- Hamilton, C.M.; Aldea, M.; Washburn, B.K.; Babitzke, P.; Kushner, S.R. New method for generating deletions and gene replacements in Escherichia coli. J. Bacteriol 1989, 171, 4617–4622. [Google Scholar]
- Emmerson, J.R.; Gally, D.L.; Roe, A.J. Generation of gene deletions and gene replacements in Escherichia coli O157: H7 using a temperature sensitive allelic exchange system. Biol. Proced. Online 2006, 8, 153–162. [Google Scholar]
- Blomfield, I.C.; Vaughn, V.; Rest, R.F.; Eisenstein, B.I. Allelic exchange in Escherichia coli using the Bacillus subtilis sacB gene and a temperature-sensitive pSC101 replicon. Mol. Microbiol 1991, 5, 1447–1457. [Google Scholar]
- Link, A.J.; Phillips, D.; Church, G.M. Methods for generating precise deletions and insertions in the genome of wild-type Escherichia coli: Application to open reading frame characterization. J. Bacteriol 1997, 179, 6228–6237. [Google Scholar]
- Wu, S.S.; Kaiser, D. Markerless deletions of pil genes in Myxococcus xanthus generated by counterselection with the Bacillus subtilis sacB gene. J. Bacteriol 1996, 178, 5817–5821. [Google Scholar]
- Okibe, N.; Suzuki, N.; Inui, M.; Yukawa, H. Efficient markerless gene replacement in Corynebacterium glutamicum using a new temperature-sensitive plasmid. J. Microbiol. Methods 2011, 85, 155–163. [Google Scholar]
- Van der Geize, R.; Hessels, G.I.; van Gerwen, R.; Vrijbloed, J.W.; van der Meijden, P.; Dijkhuizen, L. Targeted disruption of the kstD gene encoding a 3-ketosteroid Δ1-dehydrogenase isoenzyme of Rhodococcus erythropolis strain SQ1. Appl. Environ. Microbiol 2000, 66, 2029–2036. [Google Scholar]
- Graf, N.; Altenbuchner, J. Development of a method for markerless gene deletion in Pseudomonas putida. Appl. Environ. Microbiol 2011, 77, 5549–5552. [Google Scholar]
- Datsenko, K.A.; Wanner, B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar]
- Solano, C.; Garcia, B.; Valle, J.; Berasain, C.; Ghigo, J.M.; Gamazo, C.; Lasa, I. Genetic analysis of Salmonella enteritidis biofilm formation: Critical role of cellulose. Mol. Microbiol 2002, 43, 793–808. [Google Scholar]
- Van Kessel, J.C.; Hatfull, G.F. Recombineering in Mycobacterium tuberculosis. Nat. Methods 2007, 4, 147–152. [Google Scholar]
- Gust, B.; Chandra, G.; Jakimowicz, D.; Tian, Y.Q.; Bruton, C.J.; Chater, K.F. λ red-mediated genetic manipulation of antibiotic-producing Streptomyces. Adv. Appl. Microbiol 2004, 54, 107–128. [Google Scholar]
- Wang, Y.; Weng, J.; Waseem, R.; Yin, X.H.; Zhang, R.F.; Shen, Q.R. Bacillus subtilis genome editing using ssDNA with short homology regions. Nucleic Acids Res 2012, 40, e91. [Google Scholar]
- Blank, K.; Hensel, M.; Gerlach, R.G. Rapid and highly efficient method for scarless mutagenesis within the Salmonella enterica chromosome. PLoS One 2011, 6, e15763. [Google Scholar]
- Geng, S.Z.; Jiao, X.A.; Pan, Z.M.; Chen, X.J.; Zhang, X.M.; Chen, X. An improved method to knock out the asd gene of Salmonella enterica serovar Pullorum. J. Biomed. Biotechnol 2009, 2009, 646380. [Google Scholar]
- Murphy, K.C.; Campellone, K.G.; Poteete, A.R. PCR-mediated gene replacement in Escherichia coli. Gene 2000, 246, 321–330. [Google Scholar]
- Sun, W.; Wang, S.; Curtiss, R., III. Highly efficient method for introducing successive multiple scarless gene deletions and markerless gene insertions into the Yersinia pestis chromosome. Appl. Environ. Microbiol 2008, 74, 4241–4245. [Google Scholar]
- Ried, J.L.; Collmer, A. An nptI-sacB-sacR cartridge for constructing directed, unmarked mutations in gram-negative bacteria by marker exchange-eviction mutagenesis. Gene 1987, 57, 239–246. [Google Scholar]
- Nakashima, N.; Tamura, T. A new carbon catabolite repression mutation of Escherichia coli, mlc*, and its use for producing isobutanol. J. Biosci. Bioeng 2012, 114, 38–44. [Google Scholar]
- Costantino, N.; Court, D.L. Enhanced levels of λ Red mediated recombinants in mismatch repair mutants. Proc. Natl. Acad. Sci. USA 2003, 100, 15748–15753. [Google Scholar]
- Wang, H.H.; Xu, G.; Vonner, A.J.; Church, G. Modified bases enable high-efficiency oligonucleotide-mediated allelic replacement via mismatch repair evasion. Nucleic Acids Res 2011, 39, 7336–7347. [Google Scholar]
- Wang, H.H.; Isaacs, F.J.; Carr, P.A.; Sun, Z.Z.; Xu, G.; Forest, C.R.; Church, G.M. Programming cells by multiplex genome engineering and accelerated evolution. Nature 2009, 460, 894–898. [Google Scholar]
- Isaacs, F.J.; Carr, P.A.; Wang, H.H.; Lajoie, M.J.; Sterling, B.; Kraal, L.; Tolonen, A.C.; Gianoulis, T.A.; Goodman, D.B.; Reppas, N.B.; et al. Precise manipulation of chromosomes in vivo enables genome-wide codon replacement. Science 2011, 333, 348–353. [Google Scholar]
- Van Kessel, J.C.; Hatfull, G.F. Efficient point mutagenesis in mycobacteria using single-stranded DNA recombineering: Characterization of antimycobacterial drug targets. Mol. Microbiol 2008, 67, 1094–1107. [Google Scholar]
- Swingle, B.; Bao, Z.; Markel, E.; Chambers, A.; Cartinhour, S. Recombineering using RecTE from Pseudomonas syringae. Appl. Environ. Microbiol 2010, 76, 4960–4968. [Google Scholar]
- Van Pijkeren, J.P.; Britton, R.A. High efficiency recombineering in lactic acid bacteria. Nucleic Acids Res 2012, 40, e76. [Google Scholar]
- Nakashima, N.; Tamura, T. Gene silencing in Escherichia coli using antisense RNAs expressed from doxycycline-inducible vectors. Lett. Appl. Microbiol 2013, 56, 436–442. [Google Scholar]
- Stolworthy, T.S.; Krabbenhoft, E.; Black, M.E. A novel Escherichia coli strain allows functional analysis of guanylate kinase drug resistance and sensitivity. Anal. Biochem 2003, 322, 40–47. [Google Scholar]
- Yamada, J.; Yamasaki, S.; Hirakawa, H.; Hayashi-Nishino, M.; Yamaguchi, A.; Nishino, K. Impact of the RNA chaperone Hfq on multidrug resistance in Escherichia coli. J. Antimicrob. Chemother 2010, 65, 853–858. [Google Scholar]
- Tyo, K.E.; Ajikumar, P.K.; Stephanopoulos, G. Stabilized gene duplication enables long-term selection-free heterologous pathway expression. Nat. Biotechnol 2009, 27, 760–765. [Google Scholar]
- Karberg, M.; Guo, H.T.; Zhong, J.; Coon, R.; Perutka, J.; Lambowitz, A.M. Group II introns as controllable gene targeting vectors for genetic manipulation of bacteria. Nat. Biotechnol 2001, 19, 1162–1167. [Google Scholar]
- Simon, D.M.; Clarke, N.A.C.; McNeil, B.A.; Johnson, I.; Pantuso, D.; Dai, L.X.; Chai, D.G.; Zimmerly, S. Group II introns in eubacteria and archaea: ORF-less introns and new varieties. RNA 2008, 14, 1704–1713. [Google Scholar]
- Martínez-Abarca, F.; Toro, N. Group II introns in the bacterial world. Mol. Microbiol 2000, 38, 917–926. [Google Scholar]
- Michel, F.; Ferat, J.L. Structure and activities of group-II introns. Annu. Rev. Biochem 1995, 64, 435–461. [Google Scholar]
- Quiroga, C.; Kronstad, L.; Ritlop, C.; Filion, A.; Cousineau, B. Contribution of base-pairing interactions between group II intron fragments during trans-splicing in vivo. RNA 2011, 17, 2212–2221. [Google Scholar]
- Plante, I.; Cousineau, B. Restriction for gene insertion within the Lactococcus lactis Ll.LtrB group II intron. RNA 2006, 12, 1980–1992. [Google Scholar]
- Guo, H.T.; Zimmerly, S.; Perlman, P.S.; Lambowitz, A.M. Group II intron endonucleases use both RNA and protein subunits for recognition of specific sequences in double-stranded DNA. EMBO J 1997, 16, 6835–6848. [Google Scholar]
- Perutka, J.; Wang, W.J.; Goerlitz, D.; Lambowitz, A.M. Use of computer-designed group II introns to disrupt Escherichia coli DExH/D-box protein and DNA helicase genes. J. Mol. Biol 2004, 336, 421–439. [Google Scholar]
- Belhocine, K.; Mak, A.B.; Cousineau, B. Trans-splicing of the Ll.LtrB group II intron in Lactococcus lactis. Nucleic Acids Res 2007, 35, 2257–2268. [Google Scholar]
- Jones, J.P., III; Kierlin, M.N.; Coon, R.G.; Perutka, J.; Lambowitz, A.M.; Sullenger, B.A. Retargeting mobile group II introns to repair mutant genes. Mol. Ther 2005, 11, 687–694. [Google Scholar]
- Heap, J.T.; Kuehne, S.A.; Ehsaan, M.; Cartman, S.T.; Cooksley, C.M.; Scott, J.C.; Minton, N.P. The ClosTron: Mutagenesis in Clostridium refined and streamlined. J. Microbiol. Methods 2010, 80, 49–55. [Google Scholar]
- Yao, J.; Lambowitz, A.A. Gene targeting in gram-negative bacteria by use of a mobile group II intron (“targetron”) expressed from a broad-host-range vector. Appl. Environ. Microbiol 2007, 73, 2735–2743. [Google Scholar]
- Frazier, C.L.; San Filippo, J.; Lambowitz, A.M.; Mills, D.A. Genetic manipulation of Lactococcus lactis by using targeted group II introns: Generation of stable insertions without selection. Appl. Environ. Microbiol 2003, 69, 1121–1128. [Google Scholar]
- Mohr, G.; Hong, W.; Zhang, J.; Cui, G.Z.; Yang, Y.; Cui, Q.; Liu, Y.J.; Lambowitz, A.M. A targetron system for gene targeting in thermophiles and its application in Clostridium thermocellum. PLoS One 2013, 8, e69032. [Google Scholar]
- Chen, Y.; McClane, B.A.; Fisher, D.J.; Rood, J.I.; Gupta, P. Construction of an alpha toxin gene knockout mutant of Clostridium perfringens type A by use of a mobile group II intron. Appl. Environ. Microbiol 2005, 71, 7542–7547. [Google Scholar]
- Yao, J.; Zhong, J.; Fang, Y.; Geisinger, E.; Novick, R.P.; Lambowitz, A.M. Use of targetrons to disrupt essential and nonessential genes in Staphylococcus aureus reveals temperature sensitivity of Ll.LtrB group II intron splicing. RNA 2006, 12, 1271–1281. [Google Scholar]
- Corvaglia, A.R.; François, P.; Hernandez, D.; Perron, K.; Linder, P.; Schrenzel, J. A type III-like restriction endonuclease functions as a major barrier to horizontal gene transfer in clinical Staphylococcus aureus strains. Proc. Natl. Acad. Sci. USA 2010, 107, 11954–11958. [Google Scholar]
- King, N.P.; Beatson, S.A.; Totsika, M.; Ulett, G.C.; Alm, R.A.; Manning, P.A.; Schembri, M.A. UafB is a serine-rich repeat adhesin of Staphylococcus saprophyticus that mediates binding to fibronectin, fibrinogen and human uroepithelial cells. Microbiology 2011, 157, 1161–1175. [Google Scholar]
- Upadhyay, A.; Srivastava, S. Phenazine-1-carboxylic acid is a more important contributor to biocontrol Fusarium oxysporum than pyrrolnitrin in Pseudomonas fluorescens strain Psd. Microbiol. Res 2011, 166, 323–335. [Google Scholar]
- Malhotra, M.; Srivastava, S. An ipdC gene knock-out of Azospirillum brasilense strain SM and its implications on indole-3-acetic acid biosynthesis and plant growth promotion. Antonie Van Leeuwen 2008, 93, 425–433. [Google Scholar]
- Rodriguez, S.A.; Yu, J.J.; Davis, G.; Arulanandam, B.P.; Klose, K.E. Targeted inactivation of Francisella tularensis genes by group II introns. Appl. Environ. Microbiol 2008, 74, 2619–2626. [Google Scholar]
- Alonzo, F.; Port, G.C.; Cao, M.; Freitag, N.E. The posttranslocation chaperone PrsA2 contributes to multiple facets of Listeria monocytogenes pathogenesis. Infect. Immun 2009, 77, 2612–2623. [Google Scholar]
- Zarschler, K.; Janesch, B.; Zayni, S.; Schäffer, C.; Messner, P. Construction of a gene knockout system for application in Paenibacillus alvei CCM 2051T, exemplified by the S-layer glycan biosynthesis initiation enzyme WsfP. Appl. Environ. Microbiol 2009, 75, 3077–3085. [Google Scholar]
- Steen, J.A.; Steen, J.A.; Harrison, P.; Seemann, T.; Wilkie, I.; Harper, M.; Adler, B.; Boyce, J.D. Fis is essential for capsule production in Pasteurella multocida and regulates expression of other important virulence factors. PLoS Pathog 2010, 6, e1000750. [Google Scholar]
- Park, J.M.; Jang, Y.S.; Kim, T.Y.; Lee, S.Y. Development of a gene knockout system for Ralstonia eutropha H16 based on the broad-host-range vector expressing a mobile group II intron. FEMS Microbiol. Rev 2010, 309, 193–200. [Google Scholar]
- Palonen, E.; Lindström, M.; Karttunen, R.; Somervuo, P.; Korkeala, H. Expression of signal transduction system encoding genes of Yersinia pseudotuberculosis IP32953 at 28 and 3 °C. PLoS One 2011, 6, e25063. [Google Scholar]
- Palonen, E.; Lindström, M.; Somervuo, P.; Johansson, P.; Björkroth, J.; Korkeala, H. Requirement for RNA helicase CsdA for growth of Yersinia pseudotuberculosis IP32953 at low temperatures. Appl. Environ. Microbiol 2012, 78, 1298–1301. [Google Scholar]
- Maltz, M.A.; Weiss, B.L.; O’Neill, M.; Wu, Y.; Aksoy, S. OmpA-mediated biofilm formation is essential for the commensal bacterium Sodalis glossinidius to colonize the tsetse fly gut. Appl. Environ. Microbiol 2012, 78, 7760–7768. [Google Scholar]
- Akhtar, P.; Khan, S.A. Two independent replicons can support replication of the anthrax toxin-encoding plasmid pXO1 of Bacillus anthracis. Plasmid 2012, 67, 111–117. [Google Scholar]
- Zhong, J.; Karberg, M.; Lambowitz, A.M. Targeted and random bacterial gene disruption using a group II intron (targetron) vector containing a retrotransposition-activated selectable marker. Nucleic Acids Res 2003, 31, 1656–1664. [Google Scholar]
- Enyeart, P.J.; Chirieleison, S.M.; Dao, M.N.; Perutka, J.; Quandt, E.M.; Yao, J.; Whitt, J.T.; Keatinge-Clay, A.T.; Lambowitz, A.M.; Ellington, A.D. Generalized bacterial genome editing using mobile group II introns and Cre-lox. Mol. Syst. Biol. 2013, 9, 685. [Google Scholar]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar]
- Hruscha, A.; Krawitz, P.; Rechenberg, A.; Heinrich, V.; Hecht, J.; Haass, C.; Schmid, B. Efficient CRISPR/Cas9 genome editing with low off-target effects in zebrafish. Development 2013, 140, 4982–4987. [Google Scholar]
- Chen, K.; Gao, C. Targeted genome modification technologies and their applications in crop improvements. Plant Cell Rep 2013. [Google Scholar] [CrossRef]
- Malina, A.; Mills, J.R.; Cencic, R.; Yan, Y.; Fraser, J.; Schippers, L.M.; Paquet, M.; Dostie, J.; Pelletier, J. Repurposing CRISPR/Cas9 for in situ functional assays. Genes Dev 2013, 27, 2602–2614. [Google Scholar]
- Beumer, K.J.; Carroll, D. Targeted genome engineering techniques in Drosophila. Methods 2014. [Google Scholar] [CrossRef]
- Wang, T.; Wei, J.J.; Sabatini, D.M.; Lander, E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014, 343, 80–84. [Google Scholar]
- Jiang, W.; Bikard, D.; Cox, D.; Zhang, F.; Marraffini, L.A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol 2013, 31, 233–239. [Google Scholar]
- Wood, A.J.; Lo, T.W.; Zeitler, B.; Pickle, C.S.; Ralston, E.J.; Lee, A.H.; Amora, R.; Miller, J.C.; Leung, E.; Meng, X.D.; et al. Targeted genome editing across species using ZFNs and TALENs. Science 2011, 333, 307–307. [Google Scholar]
- Urnov, F.D.; Rebar, E.J.; Holmes, M.C.; Zhang, H.S.; Gregory, P.D. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet 2010, 11, 636–646. [Google Scholar]
- Gupta, A.; Christensen, R.G.; Rayla, A.L.; Lakshmanan, A.; Stormo, G.D.; Wolfe, S.A. An optimized two-finger archive for ZFN-mediated gene targeting. Nat. Methods 2012, 9, 588–590. [Google Scholar]
- Cermak, T.; Doyle, E.L.; Christian, M.; Wang, L.; Zhang, Y.; Schmidt, C.; Baller, J.A.; Somia, N.V.; Bogdanove, A.J.; Voytas, D.F. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res 2011, 39, e82. [Google Scholar]
- Curtin, S.J.; Voytas, D.F.; Stupar, R.M. Genome engineering of crops with designer nucleases. Plant Genome 2012, 5, 42–50. [Google Scholar]
- Mussolino, C.; Cathomen, T. TALE nucleases: Tailored genome engineering made easy. Curr. Opin. Biotechnol 2012, 23, 644–650. [Google Scholar]
- Christian, M.L.; Demorest, Z.L.; Starker, C.G.; Osborn, M.J.; Nyquist, M.D.; Zhang, Y.; Carlson, D.F.; Bradley, P.; Bogdanove, A.J.; Voytas, D.F. Targeting G with TAL effectors: A comparison of activities of TALENs constructed with NN and NK repeat variable di-Residues. PLoS One 2012, 7, e45383. [Google Scholar]
- Li, T.; Huang, S.; Zhao, X.F.; Wright, D.A.; Carpenter, S.; Spalding, M.H.; Weeks, D.P.; Yang, B. Modularly assembled designer TAL effector nucleases for targeted gene knockout and gene replacement in eukaryotes. Nucleic Acids Res 2011, 39, 6315–6325. [Google Scholar]
- Good, L.; Nielsen, P.E. Antisense inhibition of gene expression in bacteria by PNA targeted to mRNA. Nat. Biotechnol 1998, 16, 355–358. [Google Scholar]
- Good, L.; Awasthi, S.K.; Dryselius, R.; Larsson, O.; Nielsen, P.E. Bactericidal antisense effects of peptide-PNA conjugates. Nat. Biotechnol 2001, 19, 360–364. [Google Scholar]
- Lundin, K.E.; Good, L.; Strömberg, R.; Gräslund, A.; Smith, C.I.E. Biological activity and biotechnological aspects of peptide nucleic acid. Adv. Genet 2006, 56, 1–51. [Google Scholar]
- Hansen, M.E.; Bentin, T.; Nielsen, P.E. High-affinity triplex targeting of double stranded DNA using chemically modified peptide nucleic acid oligomers. Nucleic Acids Res 2009, 37, 4498–4507. [Google Scholar]
- McNeer, N.A.; Chin, J.Y.; Schleifman, E.B.; Fields, R.J.; Glazer, P.M.; Saltzman, W.M. Nanoparticles deliver triplex-forming PNAs for site-specific genomic recombination in CD34+ human hematopoietic progenitors. Mol. Ther 2011, 19, 172–180. [Google Scholar]
- Schleifman, E.B.; Bindra, R.; Leif, J.; del Campo, J.; Rogers, F.A.; Uchil, P.; Kutsch, O.; Shultz, L.D.; Kumar, P.; Greiner, D.L.; et al. Targeted disruption of the CCR5 gene inhuman hematopoietic stem cells stimulated by peptide nucleic acids. Chem. Biol 2011, 18, 1189–1198. [Google Scholar]
- Osiak, A.; Radecke, F.; Guhl, E.; Radecke, S.; Dannemann, N.; Lütge, F.; Glage, S.; Rudolph, C.; Cantz, T.; Schwarz, K.; et al. Selection-independent generation of gene knockout mouse embryonic stem cells using zinc-finger nucleases. PLoS One 2011, 6, e28911. [Google Scholar]
- Vos, M.; Didelot, X. A comparison of homologous recombination rates in bacteria and archaea. ISME J 2009, 3, 199–208. [Google Scholar]
- Cradick, T.J.; Ambrosini, G.; Iseli, C.; Bucher, P.; McCaffrey, A.P. ZFN-site searches genomes for zinc finger nuclease target sites and off-target sites. BMC Bioinf 2011, 12, 152. [Google Scholar]
- Sawitzke, J.A.; Thomason, L.C.; Costantino, N.; Bubunenko, M.; Datta, S.; Court, D.L. Recombineering: In vivo genetic engineering in E. coli, S. enterica, and beyond. Methods Enzymol 2007, 421, 171–199. [Google Scholar]
- Nakashima, N.; Goh, S.; Good, L.; Tamura, T. Multiple-gene silencing using antisense RNAs in Escherichia coli. Methods Mol. Biol 2012, 815, 307–319. [Google Scholar]
- Nakashima, N.; Tamura, T. Conditional gene silencing of multiple genes with antisense RNAs and generation of a mutator strain of Escherichia coli. Nucleic Acids Res 2009, 37, e103. [Google Scholar]
- Nakashima, N.; Tamura, T.; Good, L. Paired termini stabilize antisense RNAs and enhance conditional gene silencing in Escherichia coli. Nucleic Acids Res 2006, 34, e138. [Google Scholar]
- Brantl, S. Antisense-RNA regulation and RNA interference. Biochim. Biophys. Acta 2002, 1575, 15–25. [Google Scholar]
- Sharma, V.; Yamamura, A.; Yokobayashi, Y. Engineering artificial small RNAs for conditional gene silencing in Escherichia coli. ACS Synth. Biol 2012, 1, 6–13. [Google Scholar]
- Stefan, A.; Schwarz, F.; Bressanin, D.; Hochkoeppler, A. Shine-Dalgarno sequence enhances the efficiency of lacZ repression by artificial anti-lac antisense RNAs in Escherichia coli. J. Biosci. Bioeng 2010, 110, 523–528. [Google Scholar]
- Goh, S.; Boberek, J.M.; Nakashima, N.; Stach, J.; Good, L. Concurrent growth rate and transcript analyses reveal essential gene stringency in Escherichia coli. PLoS One 2009, 4, e6061. [Google Scholar]
- Pestka, S.; Daugherty, B.L.; Jung, V.; Hotta, K.; Pestka, R.K. Anti-mRNA; specific inhibition of translation of single mRNA molecules. Proc. Natl. Acad. Sci. USA 1984, 81, 7525–7528. [Google Scholar]
- Srivastava, R.; Cha, H.J.; Peterson, M.S.; Bentley, W.E. Antisense downregulation of σ32 as a transient metabolic controller in Escherichia coli: Effects on yield of active organophosphorus hydrolase. Appl. Environ. Microbiol 2000, 66, 4366–4371. [Google Scholar]
- Krylov, A.A.; Airich, L.G.; Kiseleva, E.M.; Minaeva, N.I.; Biryukova, I.V.; Mashko, S.V. Conditional silencing of the Escherichia coli pykF gene results from artificial convergent transcription protected from rho-dependent termination. J. Mol. Microbiol. Biotechnol 2010, 18, 1–13. [Google Scholar]
- Chen, H.; Ferbeyre, G.; Cedergren, R. Efficient hammerhead ribozyme and antisense RNA targeting in a slow ribosome Escherichia coli mutant. Nat. Biotechnol 1997, 15, 432–435. [Google Scholar]
- Kim, J.Y.H.; Cha, H.J. Down-regulation of acetate pathway through antisense strategy in Escherichia coli: Improved foreign protein production. Biotechnol. Bioeng 2003, 83, 841–853. [Google Scholar]
- Boberek, J.M.; Stach, J.; Good, L. Genetic evidence for inhibition of bacterial division protein FtsZ by berberine. PLoS One 2010, 5, e13745. [Google Scholar]
- Meng, J.; Kanzaki, G.; Meas, D.; Lam, C.K.; Crummer, H.; Tain, J.; Xu, H.H. A genome-wide inducible phenotypic screen identifies antisense RNA constructs silencing Escherichia coli essential genes. FEMS Microbiol. Lett 2012, 329, 45–53. [Google Scholar]
- Setoyama, D.; Ito, R.; Takagi, Y.; Sekiguchi, M. Molecular actions of Escherichia coli MutT for control of spontaneous mutagenesis. Mutat. Res 2011, 707, 9–14. [Google Scholar]
- Ji, Y.D.; Zhang, B.; van Horn, S.F.; Warren, P.; Woodnutt, G.; Burnham, M.K.R.; Rosenberg, M. Identification of critical staphylococcal genes using conditional phenotypes generated by antisense RNA. Science 2001, 293, 2266–2269. [Google Scholar]
- Desai, R.P.; Papoutsakis, E.T. Antisense RNA strategies for metabolic engineering of Clostridium acetobutylicum. Appl. Environ. Microbiol 1999, 65, 936–945. [Google Scholar]
- Biedendieck, R.; Malten, M.; Barg, H.; Bunk, B.; Martens, J.H.; Deery, E.; Leech, H.; Warren, M.J.; Jahn, D. Metabolic engineering of cobalamin (vitamin B12) production in Bacillus megaterium. Microb. Biotechnol 2010, 3, 24–37. [Google Scholar]
- Uguru, G.C.; Mondhe, M.; Goh, S.; Hesketh, A.; Bibb, M.J. Synthetic RNA silencing of actinorhodin biosynthesis in Streptomyces coelicolor A3(2). PLoS One 2013, 8, e67509. [Google Scholar]
- Bouazzaoui, K.; LaPointe, G. Use of antisense RNA to modulate glycosyltransferase gene expression and exopolysaccharide molecular mass in Lactobacillus rhamnosus. J. Microbiol. Methods 2006, 65, 216–225. [Google Scholar]
- Kaur, P.; Agarwal, S.; Datta, S. Delineating bacteriostatic and bactericidal targets in Mycobacteria using IPTG inducible antisense expression. PLoS One 2009, 4, e5923. [Google Scholar]
- Parish, T.; Stoker, N.G. Development and use of a conditional antisense mutagenesis system in Mycobacteria. FEMS Microbiol. Lett 1997, 154, 151–157. [Google Scholar]
- Thomason, M.K.; Storz, G. Bacterial antisense RNAs: How many are there, and what are they doing? Annu. Rev. Genet 2010, 44, 167–188. [Google Scholar]
- Nakashima, N.; Ohno, S.; Yoshikawa, K.; Shimizu, H.; Tamura, T. A vector library for silencing central carbon metabolism genes with antisense RNAs in Escherichia coli. Appl. Environ. Microbiol 2014, 80, 564–573. [Google Scholar]
- Na, D.; Yoo, S.M.; Chung, H.; Park, H.; Park, J.H.; Lee, S.Y. Metabolic engineering of Escherichia coli using synthetic small regulatory RNAs. Nat. Biotechnol 2013, 31, 170–174. [Google Scholar]
- Stach, J.E.M.; Good, L. Synthetic RNA silencing in bacteria-antimicrobial discovery and resistance breaking. Front. Microbiol 2011, 2, 185. [Google Scholar]
- Bistué, A.; Martín, F.A.; Vozza, N.; Ha, H.; Joaquín, J.C.; Zorreguieta, A.; Tolmasky, M.E. Inhibition of aac(6′)-Ib-mediated amikacin resistance by nuclease-resistant external guide sequences in bacteria. Proc. Natl. Acad. Sci. USA 2009, 106, 13230–13235. [Google Scholar]
- Rasmussen, L.C.V.; Sperling-Petersen, H.U.; Mortensen, K.K. Hitting bacteria at the heart of the central dogma: Sequence-specific inhibition. Microb. Cell Fact 2007, 6, 24. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compared points | pSC101ts-sacB method by Blomfield et al. [8] | λ-Red recombinase method by Datsenko et al. [14] |
|---|---|---|
| Recombination via | Two times of single crossover | One time of double crossover and FLP (flippase)–FRT recombination |
| Enzymes for recombination | endogenous enzymes | λ Gam, Bet, Exo, and flippase |
| Reliability | Low due to resolution of original gene organizations | High |
| Host requirements | Only recombination-proficient hosts | Any |
| Plasmid construction | Necessary | unnecessary |
| Transformation efficiency required | Low | High |
| Transformation procedures required | Once | Twice |
| Marker gene used for integration | Not retained | Not retained |
| Unnecessary genome arrangement | No | Yes, leaving an 81–85-bp “scar” sequence [19] |
| Bacteria proven to be applicable | E. coli [6–9], M. xanthus [10], C. glutamicum [11], Rhodococcus spp. [12], and P. putida [13] | E. coli [14], Salmonella spp. [15], M. tuberculosis [16], Streptomyces spp. [17], and B. subtilis [18] |
| Host bacteria | Method used | Efficiency | Reference |
|---|---|---|---|
| E. coli | λ-red recombinase method, double stranded DNA | 103 to 104 recombinants per 108 viable cells a | [92] |
| E. coli | λ-red recombinase method, single stranded DNA | ~107 recombinants per 108 viable cells | [92] |
| E. coli | λ-red recombinase method, single stranded DNA | 25% b | [25] |
| L. reuteri | λ-red recombinase method, single stranded DNA | 0.4%–19% b | [31] |
| E. coli | mobile group II introns | 1%–80% b | [43] |
| C. thermocellum | mobile group II introns | 67%–100% b | [49] |
| S. aureus | mobile group II introns | 37%–100% b | [51] |
| S. pneumoniae | CRISPR-Cas9 system | 100% b | [74] |
| E. coli | CRISPR-Cas9 system | 65% b | [74] |
| Gene name | Gene product | Silencing efficacy a | Observed phenotypes upon expression of HPasRNAs |
|---|---|---|---|
| lacZ | β-galactosidase | 88% [93] | – |
| ackA | Acetate kinase | 78% [94] | Reduced acetate production, no growth on minimal acetate media [95] |
| aceE | Pyruvate dehydrogenase component | Acetate auxotroph, accumulation of pyruvate [93] | |
| ftsZ | Tubulin-like protein | Severe growth (essential gene), elongated cell [105] | |
| fusA | Elongation factor G | Severe growth (essential gene), sensitization 12-fold to fusidic acid [106] | |
| (Many growth essential genes) | Construction of a shotgun genomic library expressing HPasRNAs, identification of growth essential genes [106] | ||
| mutT | Protein for maintaining DNA replication fidelity | >90% [106] | Protein level control in a stepwise fashion by changing concentration of expression inducer (IPTG) [107] |
| mutS, mutD, ndk (triple silencing) | Proteins for maintaining DNA replication fidelity | Increased mutation rate by 2000-fold over wild-type cells [94] |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Nakashima, N.; Miyazaki, K. Bacterial Cellular Engineering by Genome Editing and Gene Silencing. Int. J. Mol. Sci. 2014, 15, 2773-2793. https://doi.org/10.3390/ijms15022773
Nakashima N, Miyazaki K. Bacterial Cellular Engineering by Genome Editing and Gene Silencing. International Journal of Molecular Sciences. 2014; 15(2):2773-2793. https://doi.org/10.3390/ijms15022773
Chicago/Turabian StyleNakashima, Nobutaka, and Kentaro Miyazaki. 2014. "Bacterial Cellular Engineering by Genome Editing and Gene Silencing" International Journal of Molecular Sciences 15, no. 2: 2773-2793. https://doi.org/10.3390/ijms15022773