Design and Synthesis of Chiral Zn2+ Complexes Mimicking Natural Aldolases for Catalytic C–C Bond Forming Reactions in Aqueous Solution


Abstract
:
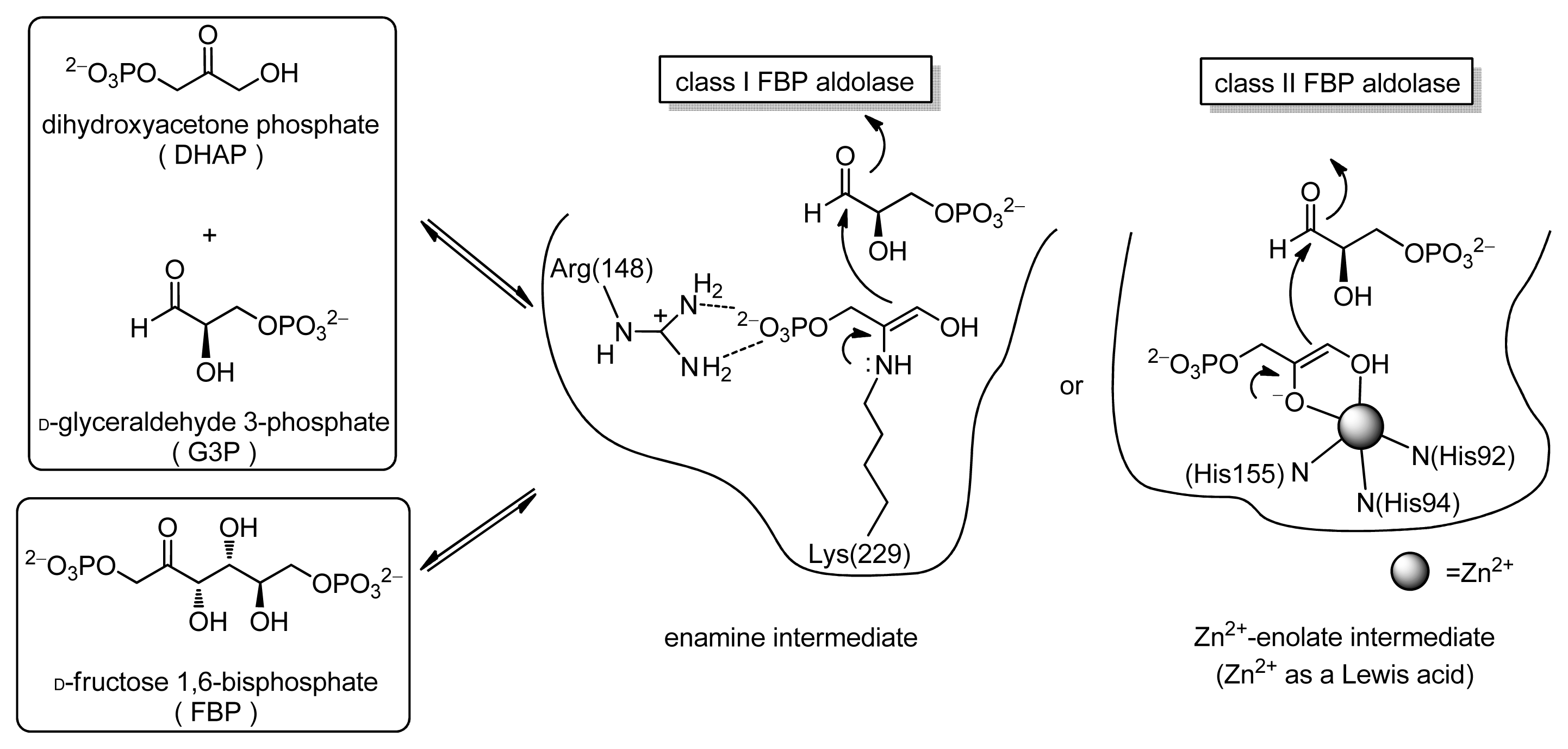
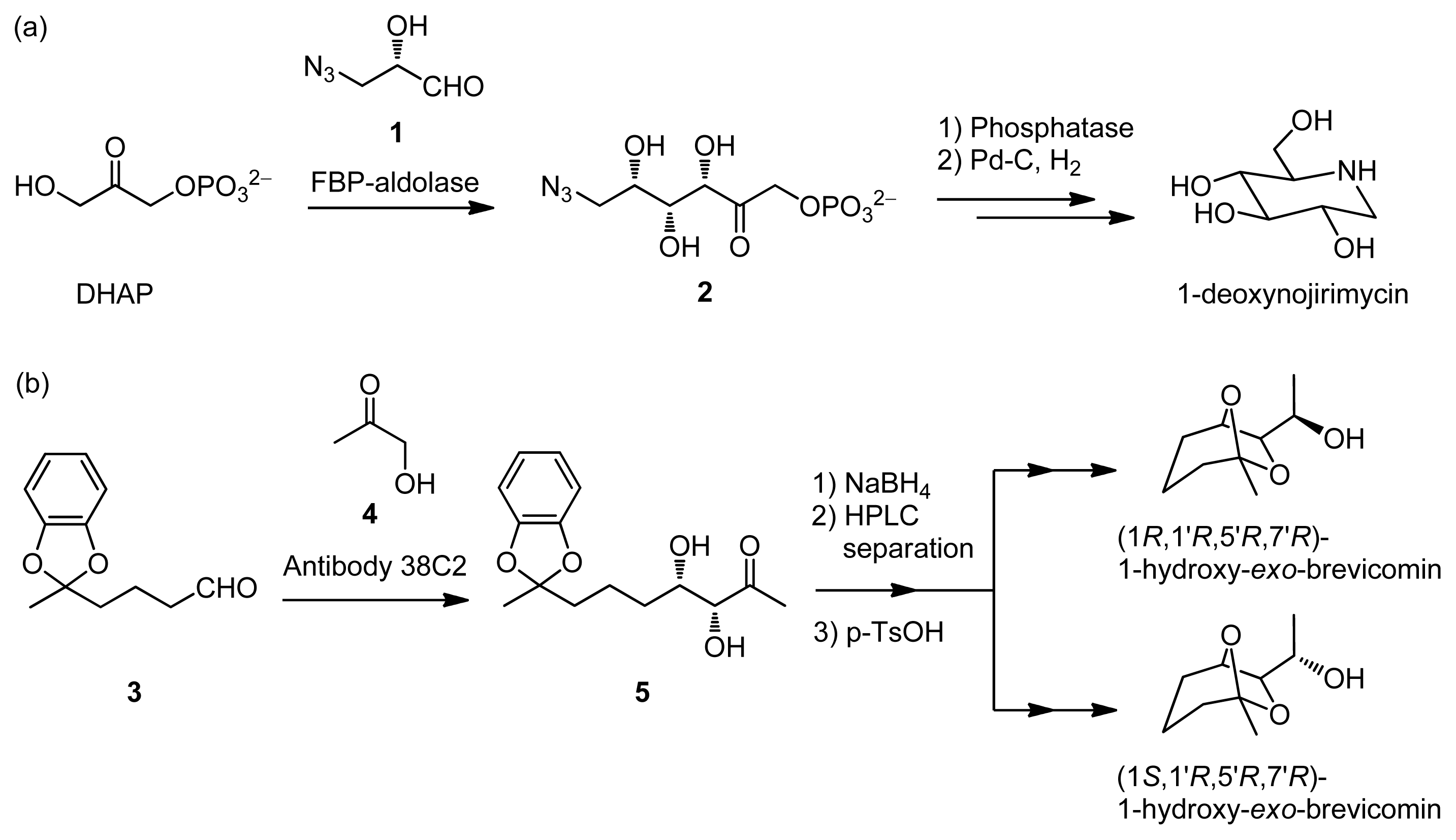
1. Introduction
2. Results and Discussion
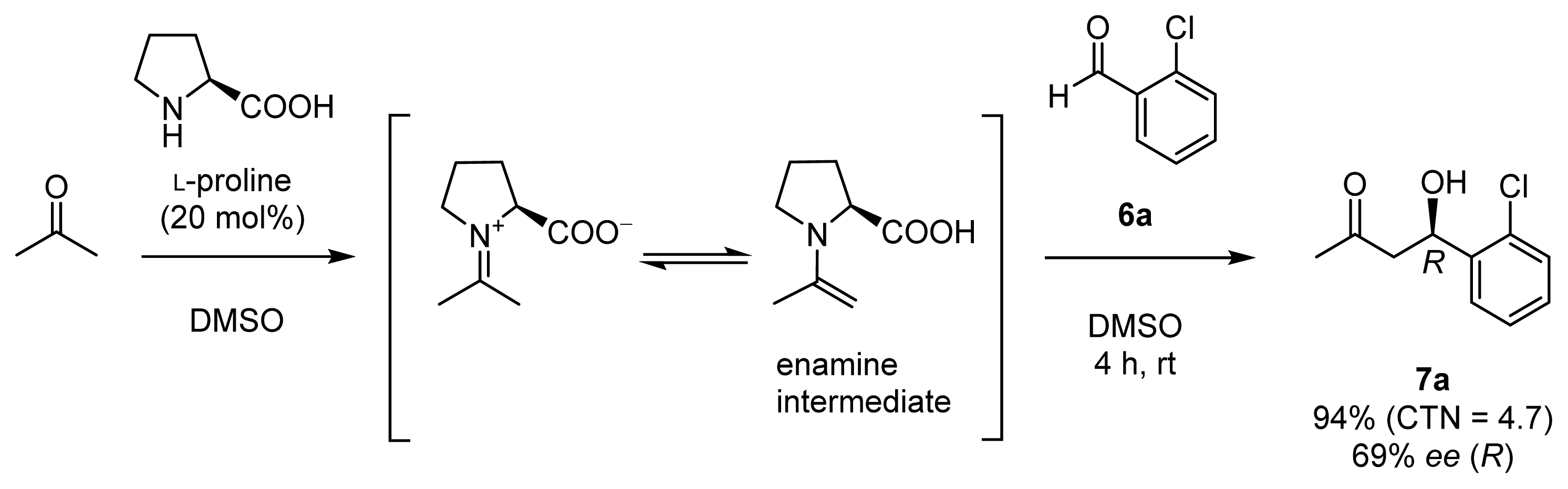
2.1. Chiral Catalysts that Are Dually Functionalized with Amino Acid and Zn2+ Complex Components for Direct Enantioselective Aldol Reactions Inspired by Natural Aldolases
2.1.1. Design and Synthesis of Chiral Zn2+ Complexes
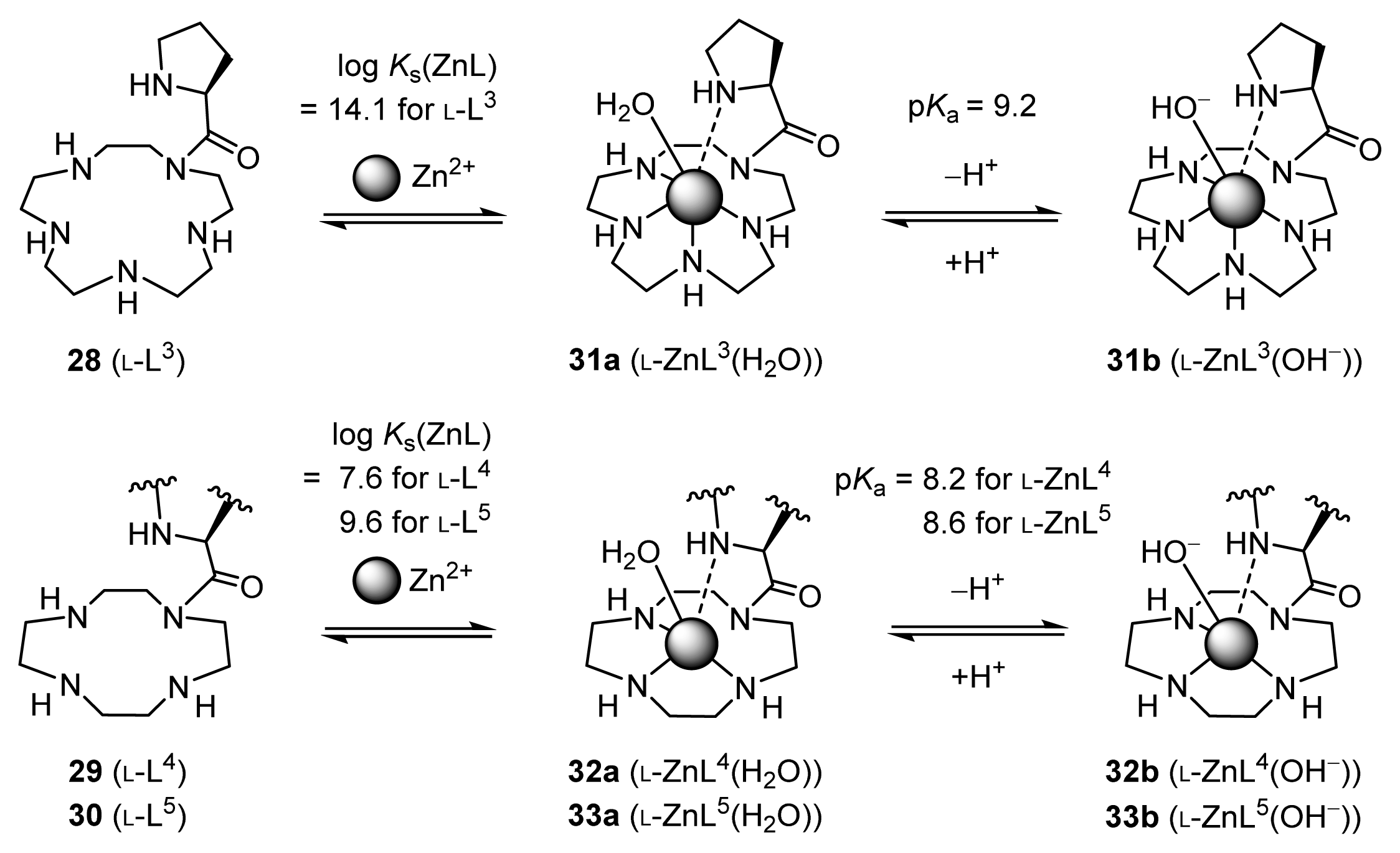
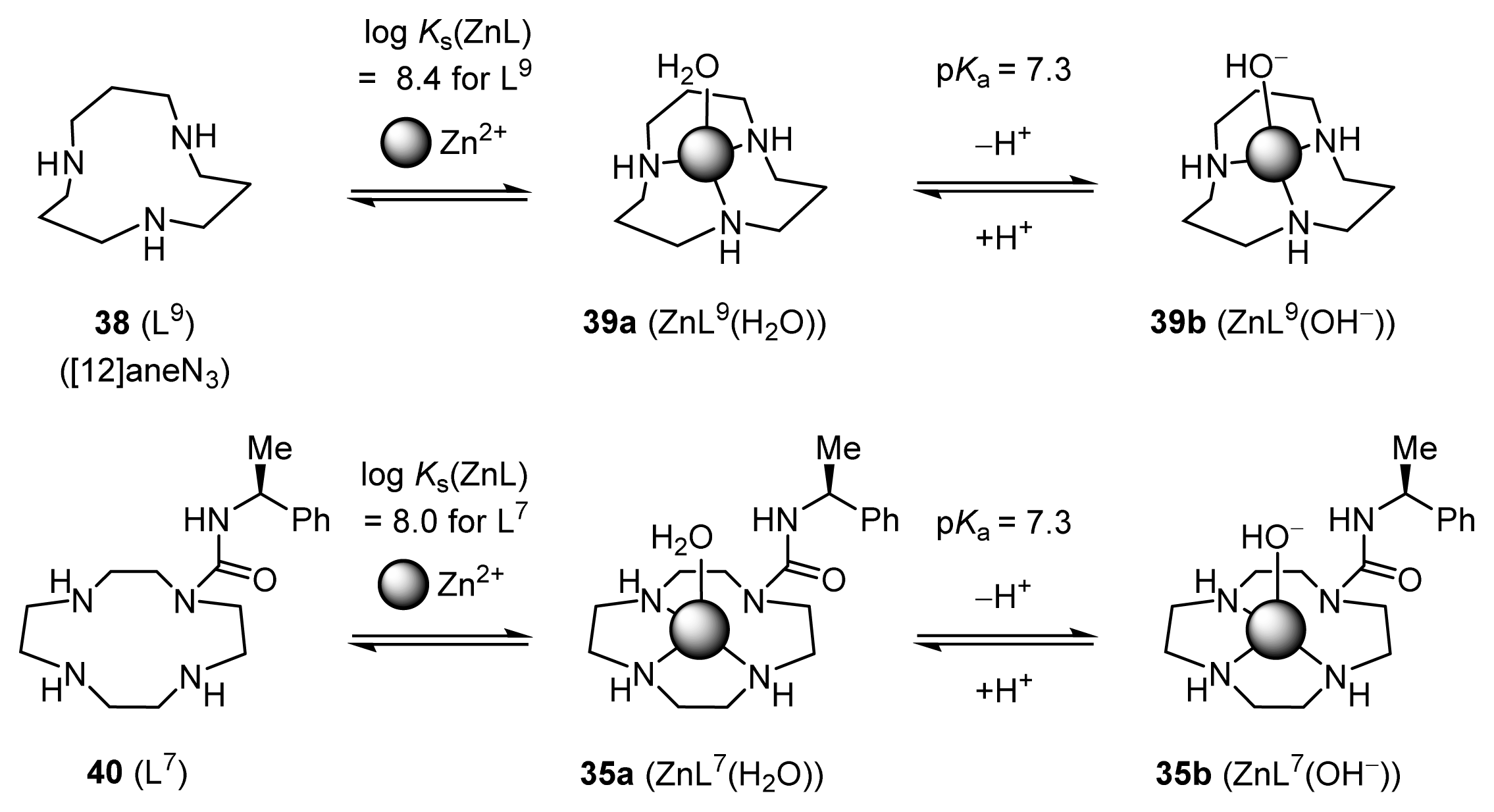
2.1.2. Complexation Properties of Chiral Zn2+ Complexes
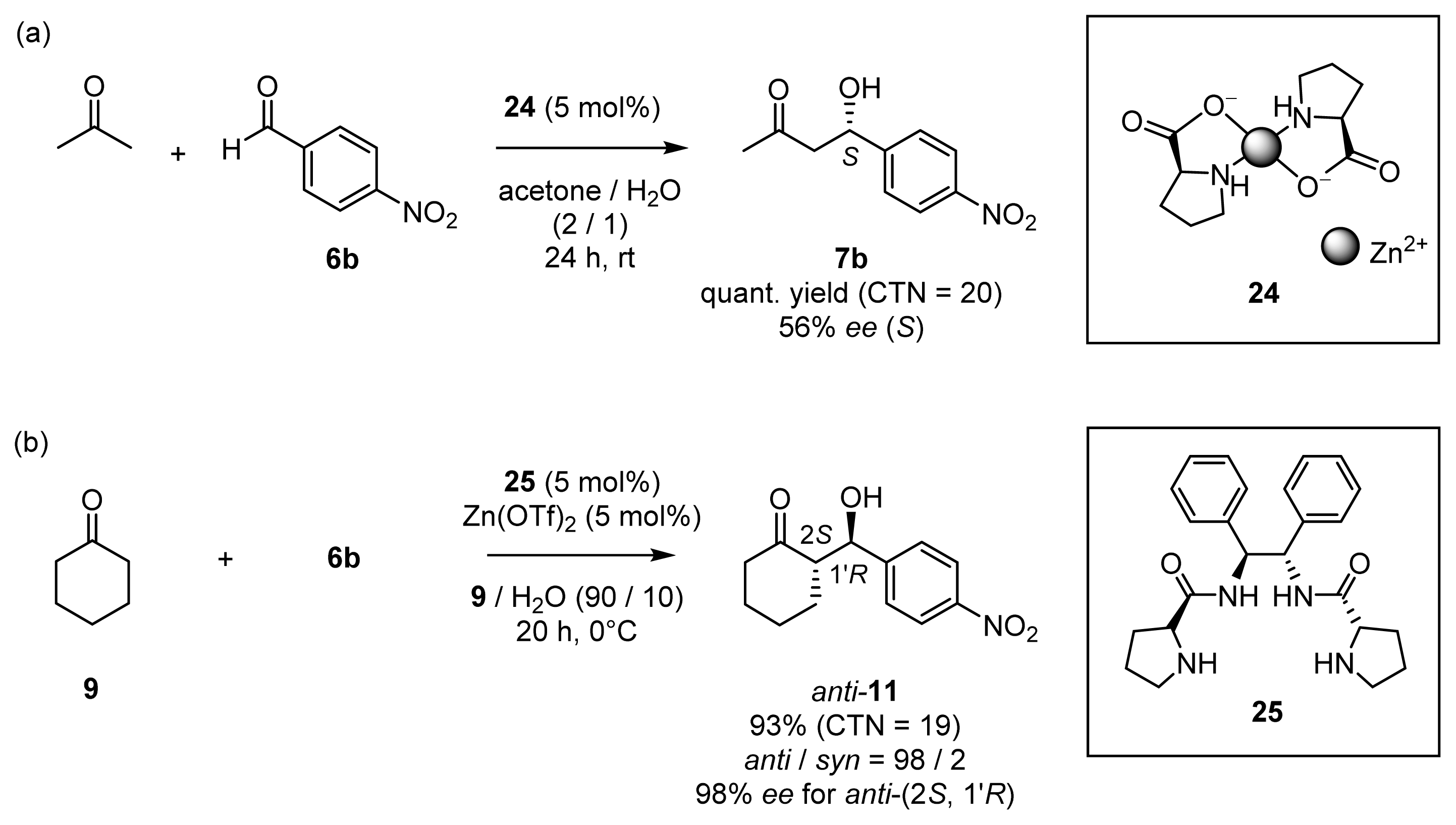
2.1.3. Enantioselective Aldol Reactions in Aqueous Media Catalyzed by Chiral Zn2+ Complexes
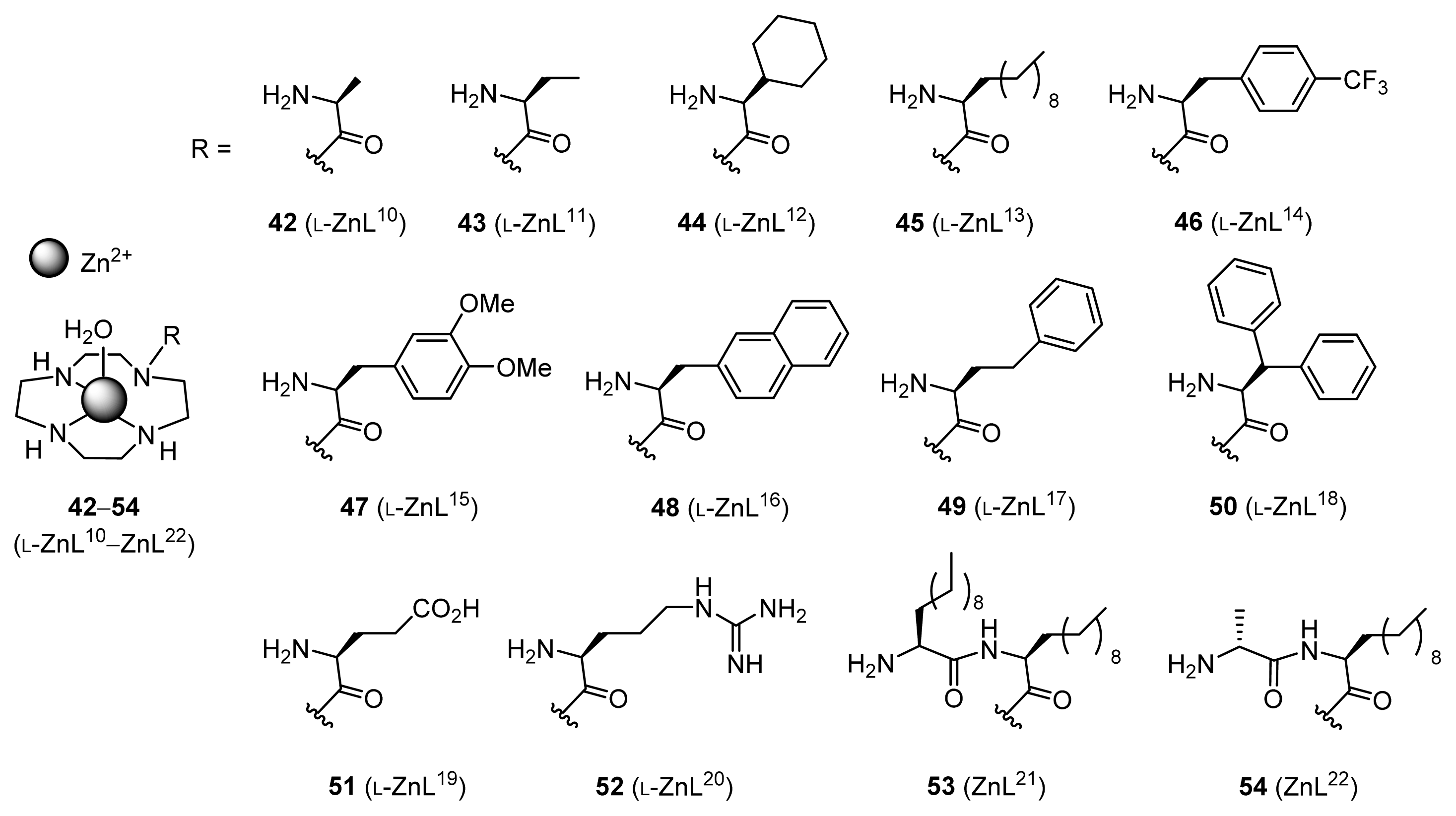
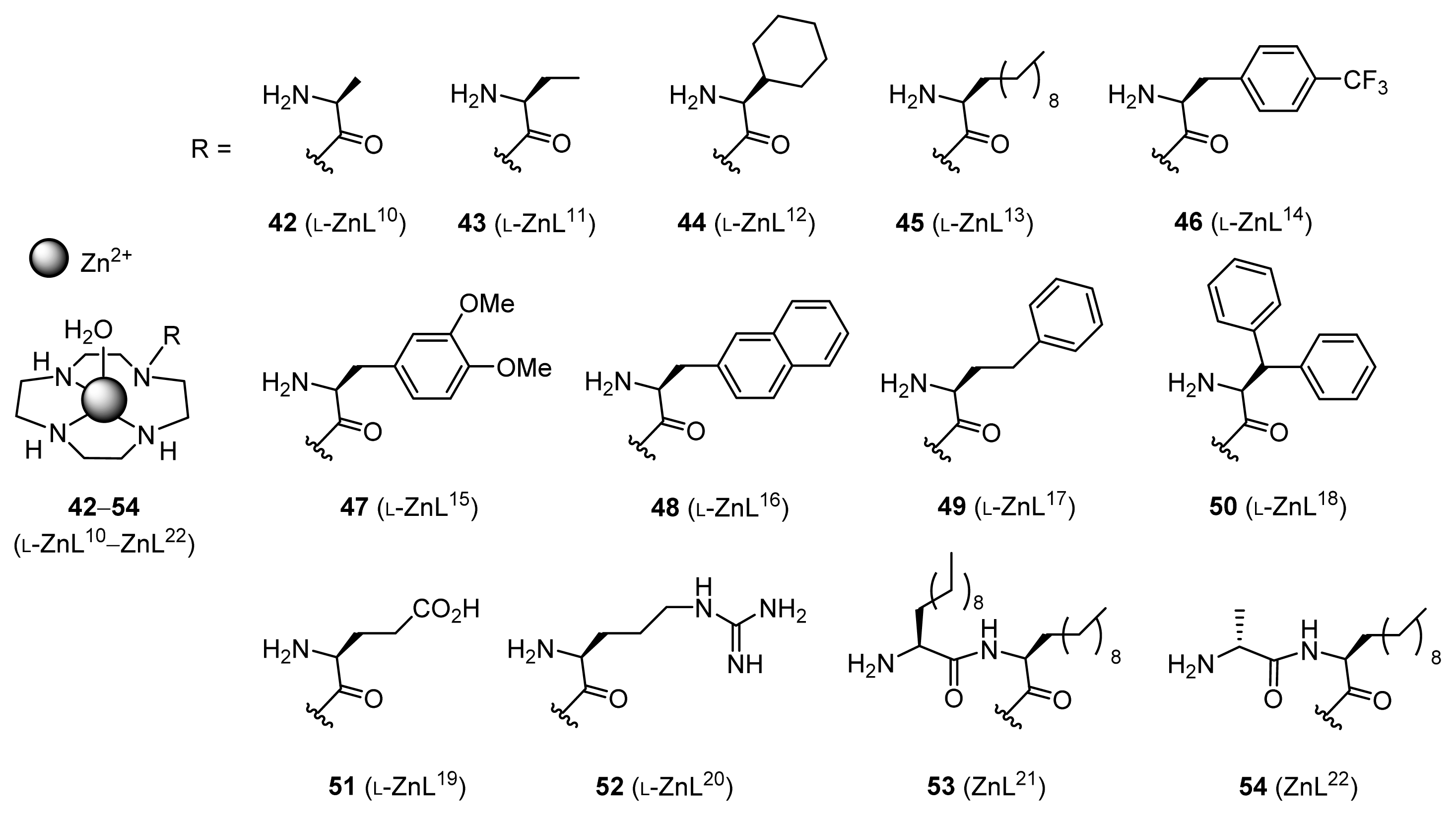
2.1.4. Fine Tuning of Structure of Chiral Zn2+ Complexes
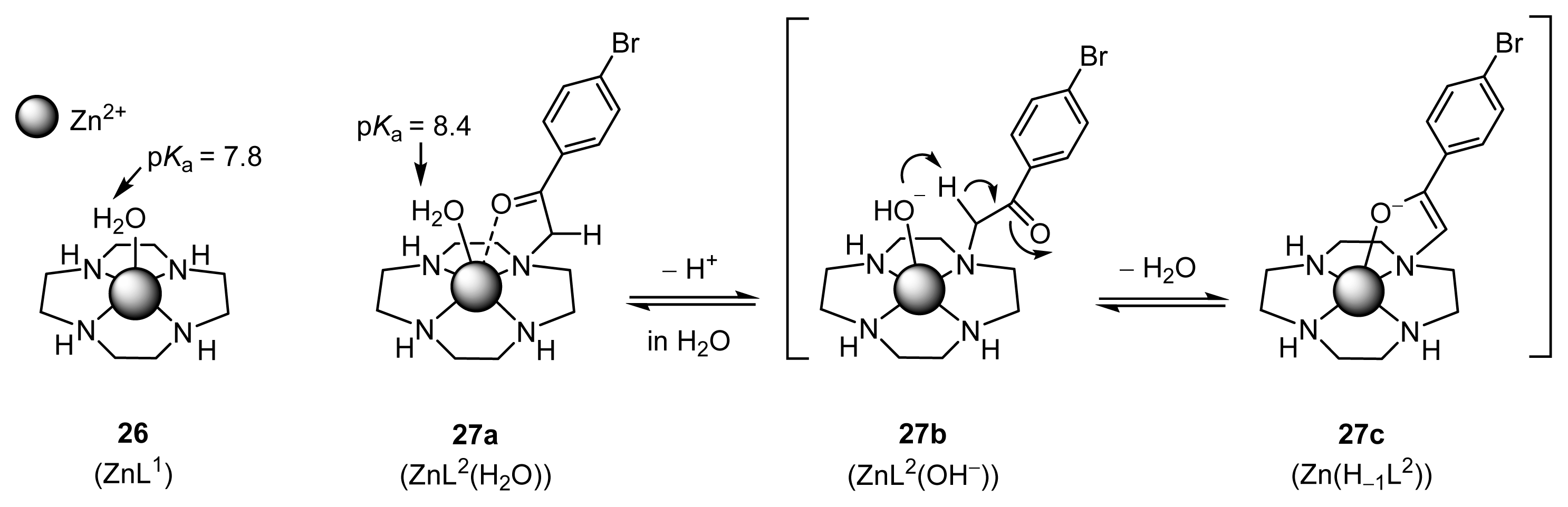
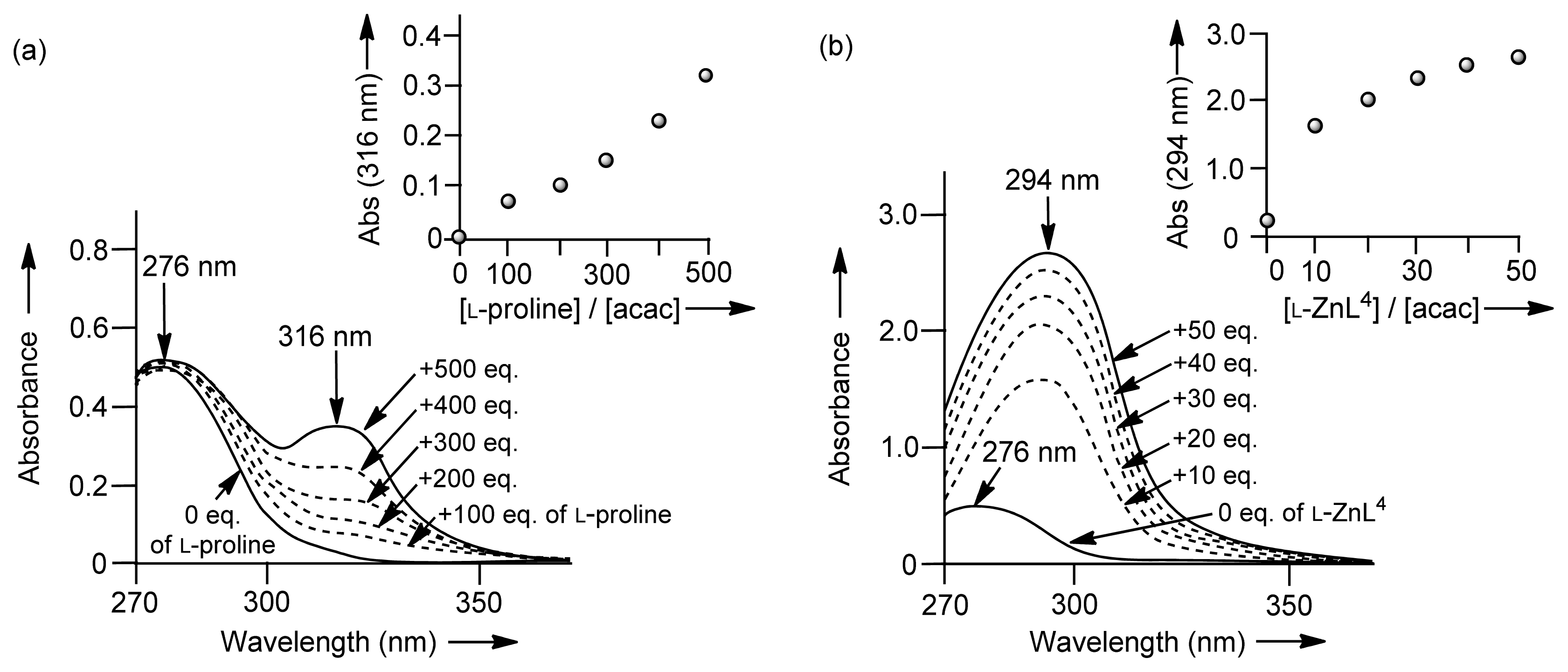
2.1.5. UV Titrations of Acetylacetone (Acac) with l-Proline, l-Valine, Zn(OTf)2, and Zn2+ Complexes (32 (l-ZnL4), 33 (l-ZnL5), 35 (ZnL7), and 26 (ZnL1)) in a Mechanistic Study
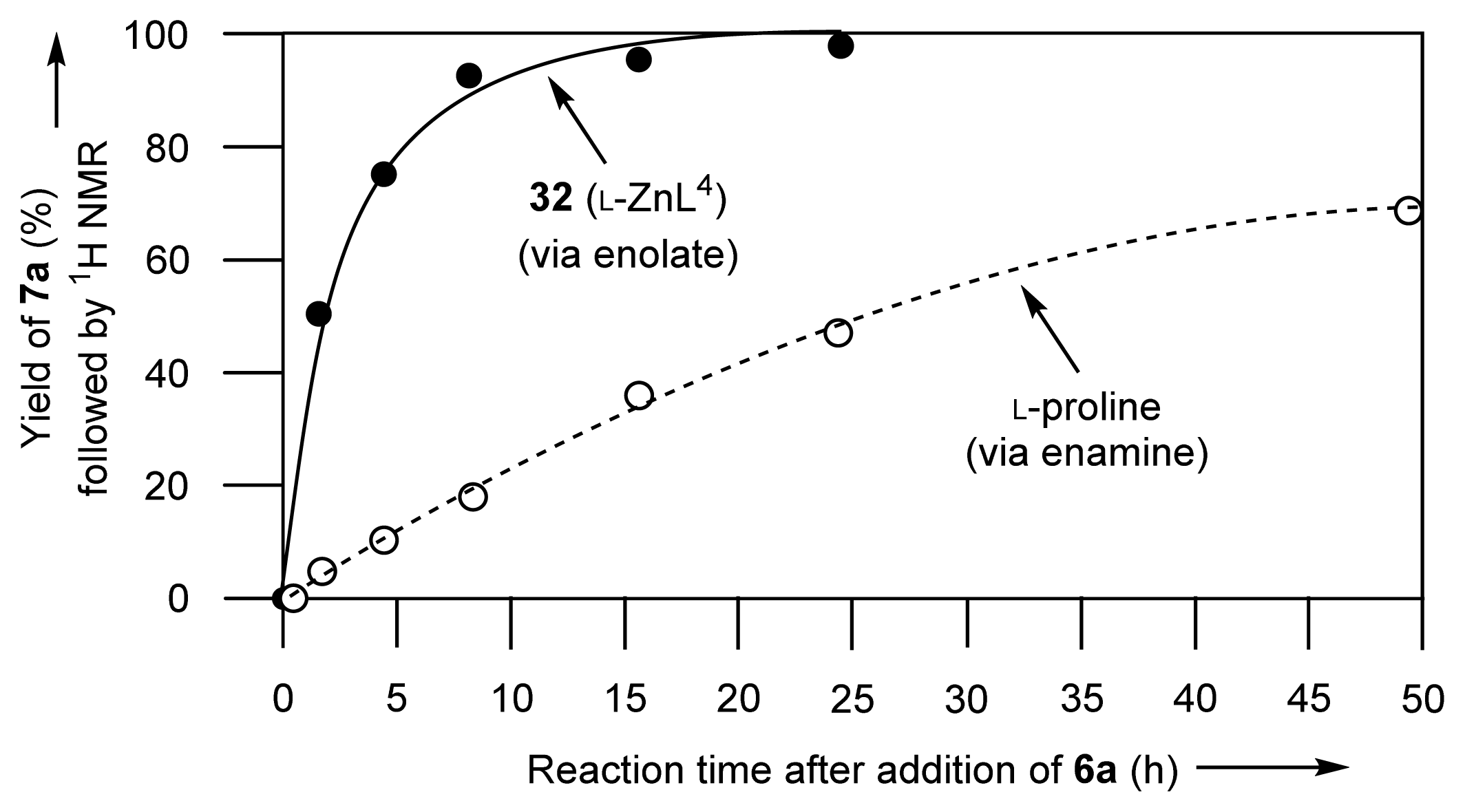
2.1.6. Stopped-Flow Experiments to Determine the Rates of ZnL-(acac)− Complexation
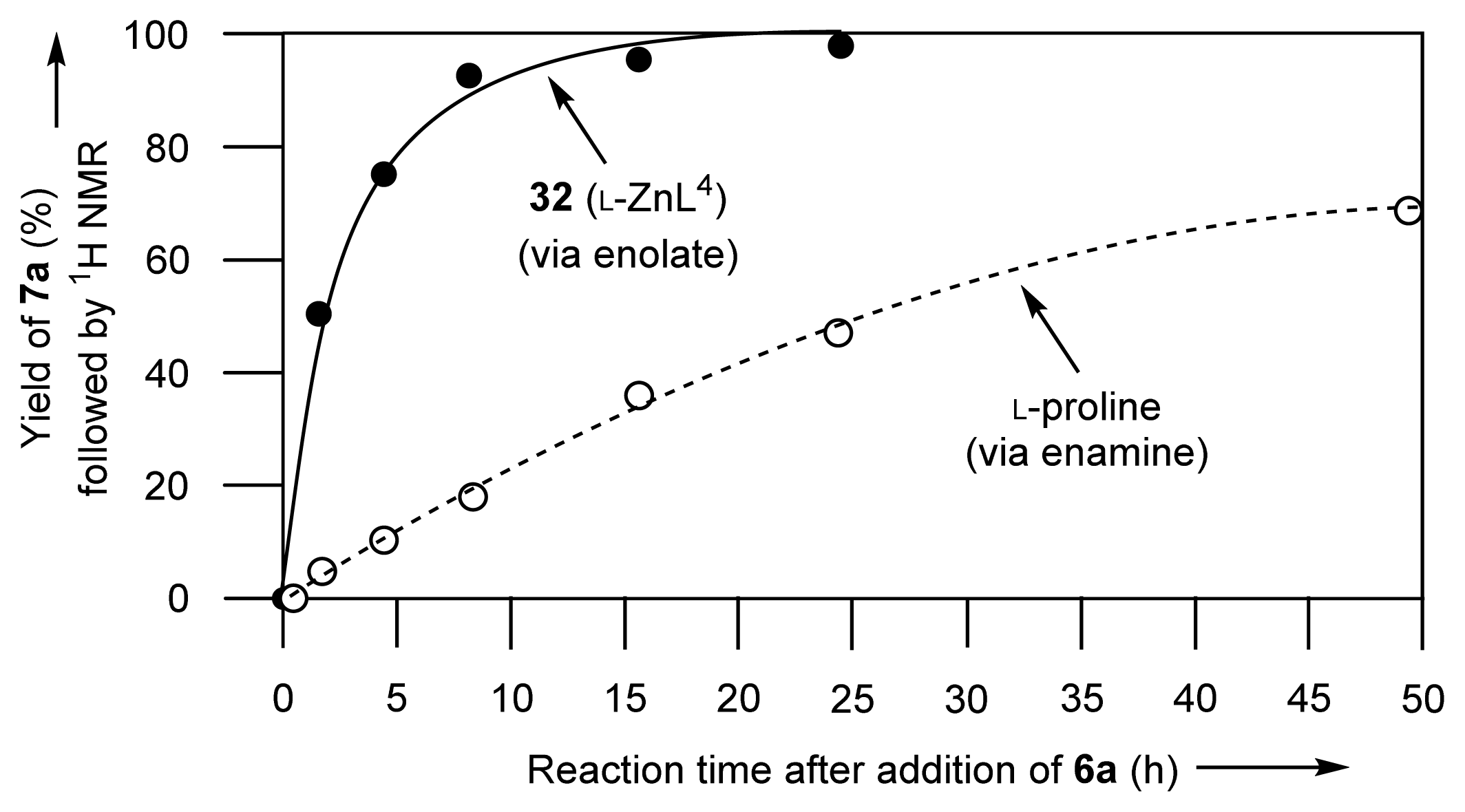
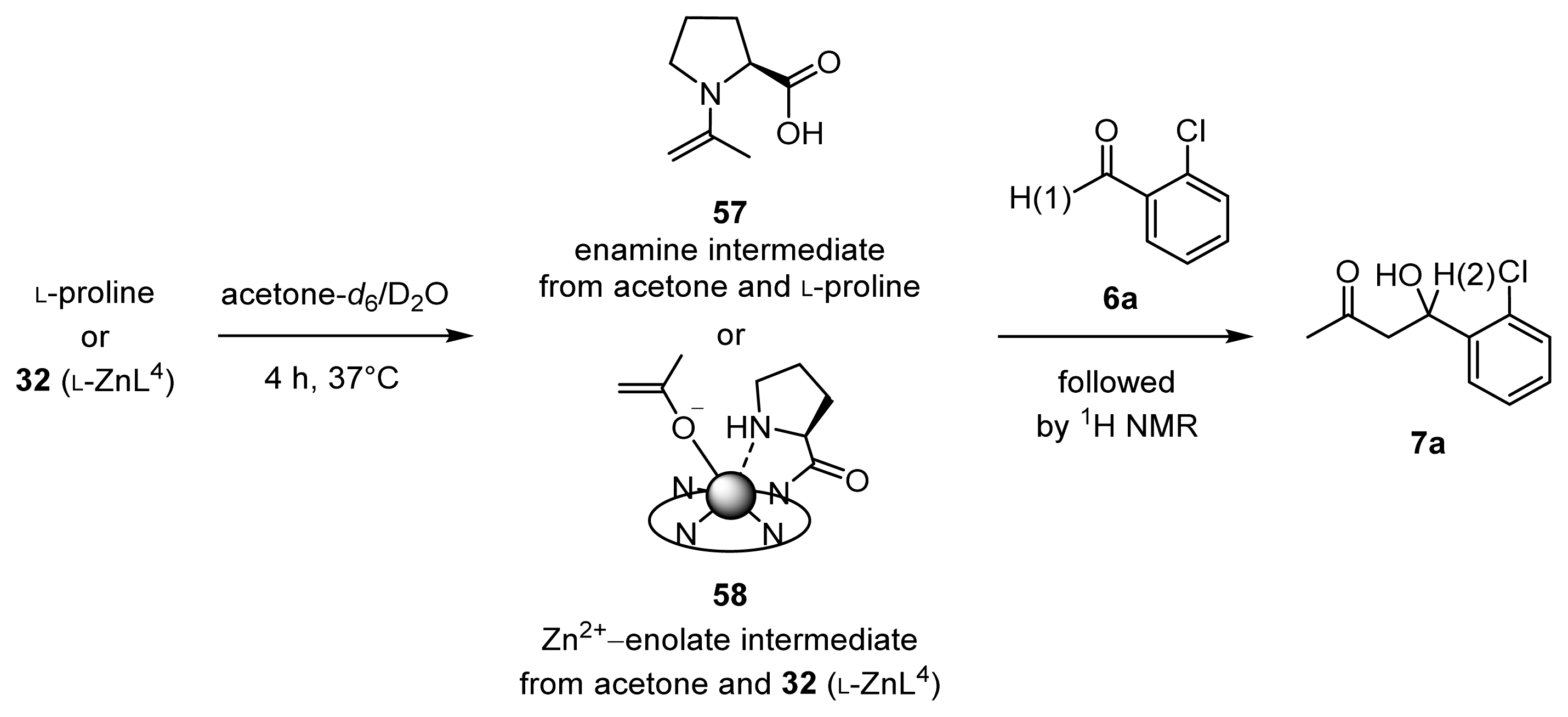
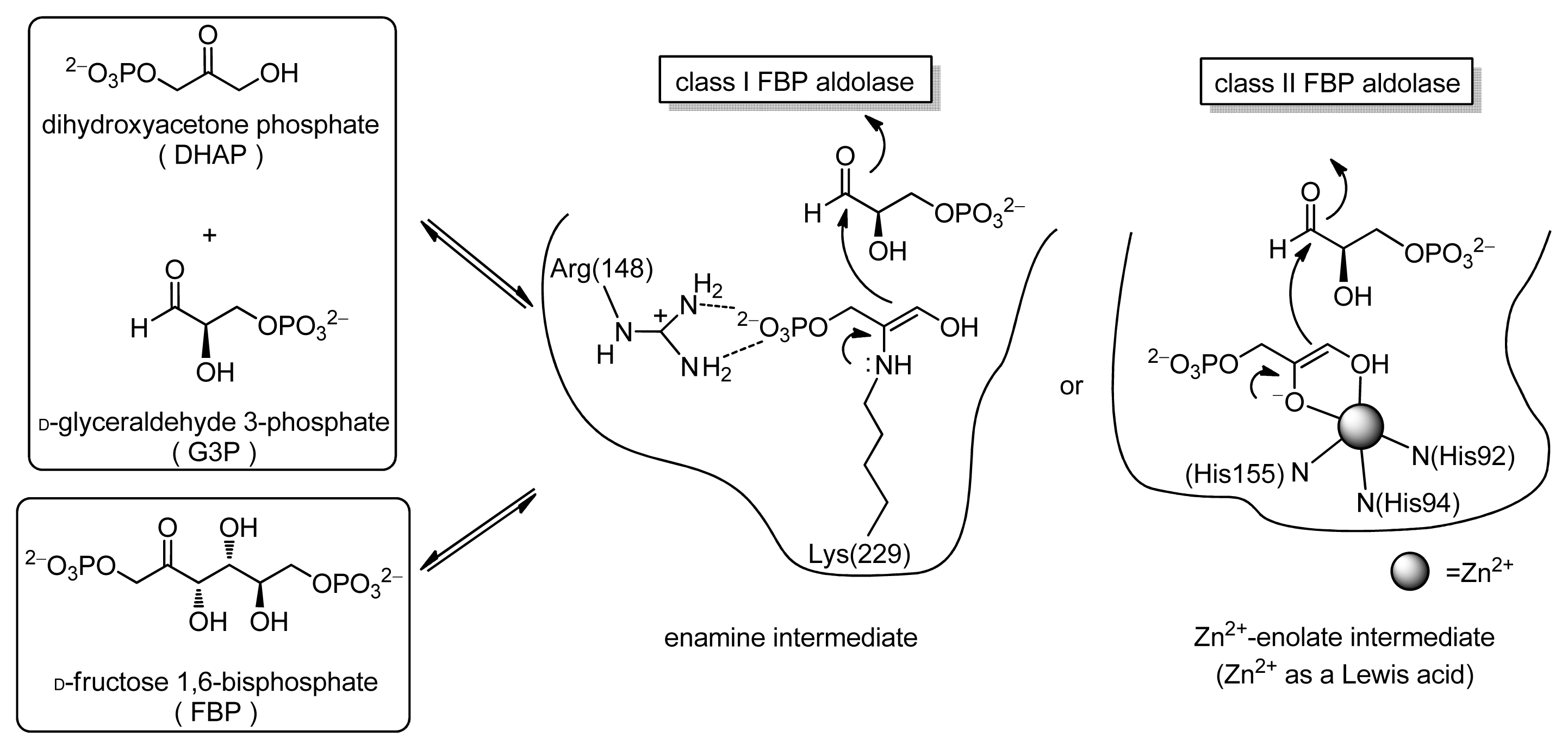
2.1.7. Comparison between Reactivity of Enamine and Zn2+-Enolate Intermediates
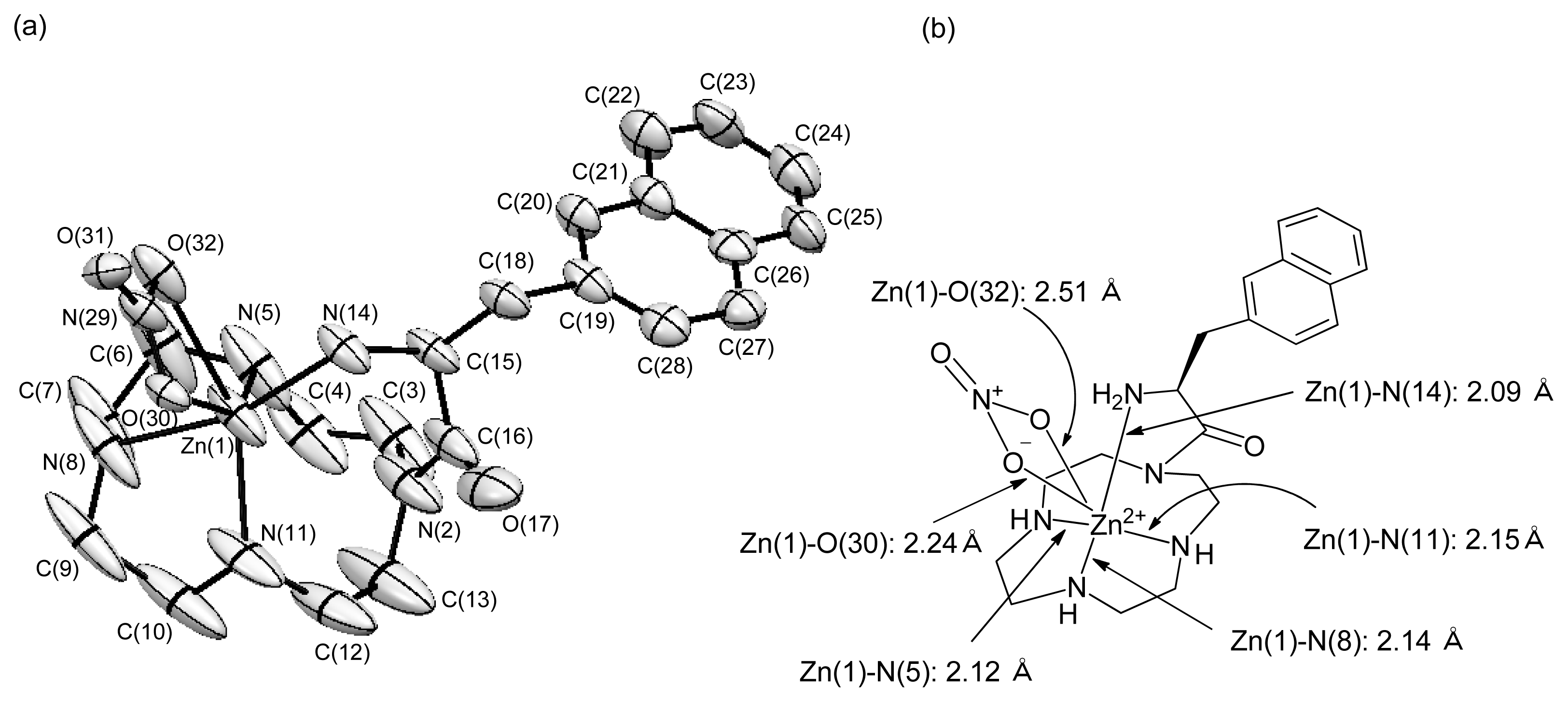
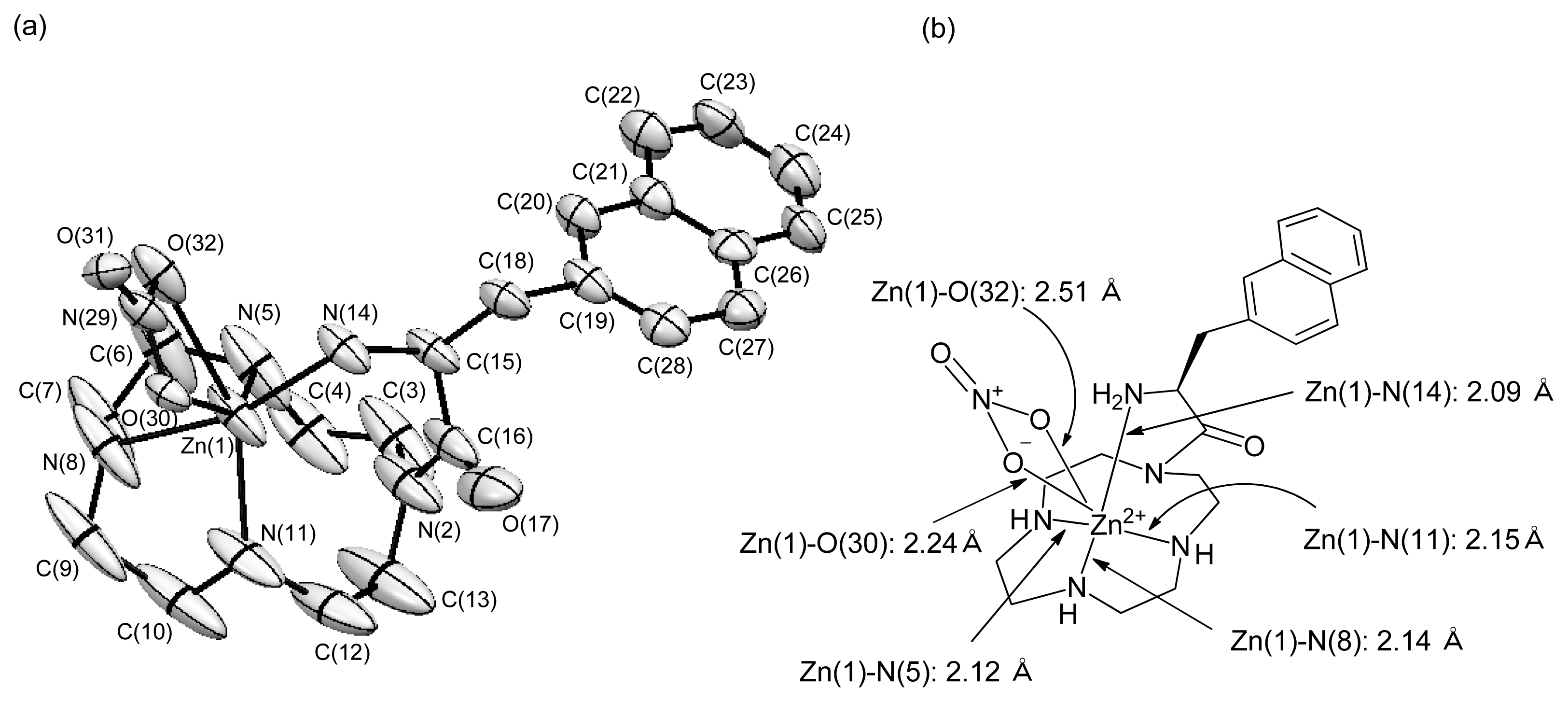
2.1.8. X-ray Crystal Structure of 48 (l-ZnL16)
2.1.9. Proposed Mechanism for the Aldol Reaction of Acetone Catalyzed by Chiral Zn2+ Complexes
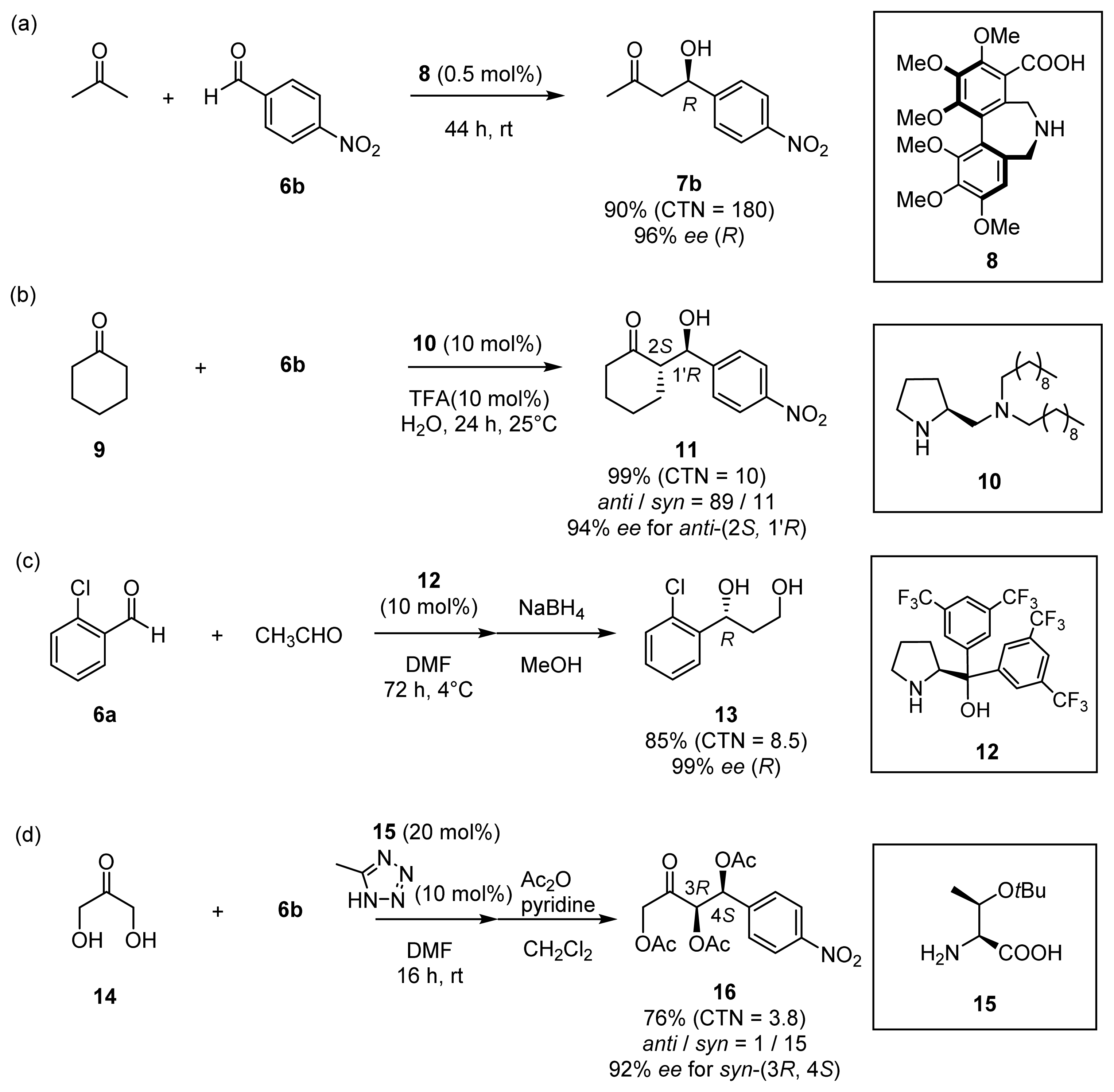
2.2. Asymmetric Aldol Reactions of Hydroxylated Acetones and Cyclic Ketones
2.2.1. Asymmetric Aldol Reactions of Hydroxylated Acetones (Hydroxyacetone 4 and Dihydroxyacetone 14)
2.2.2. Asymmetric Aldol Reactions of Cyclic Ketones (Cyclohexanone 9 and Cyclopentanone 68)
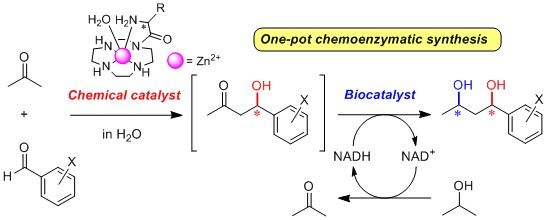
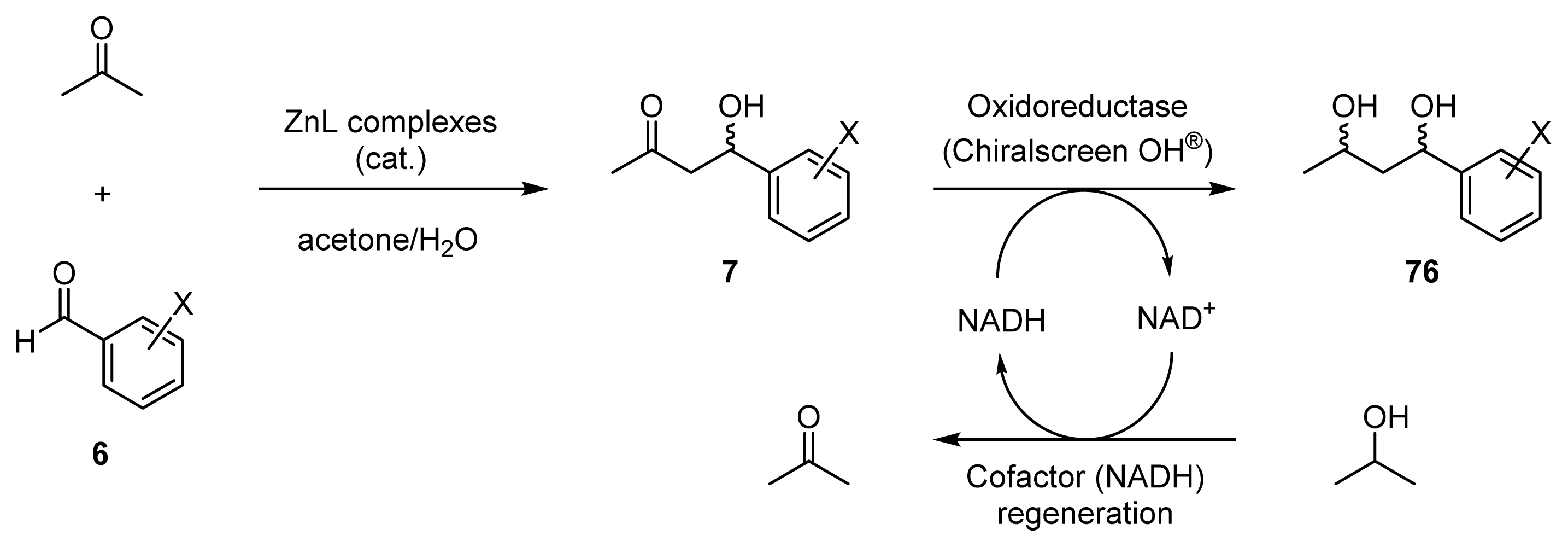
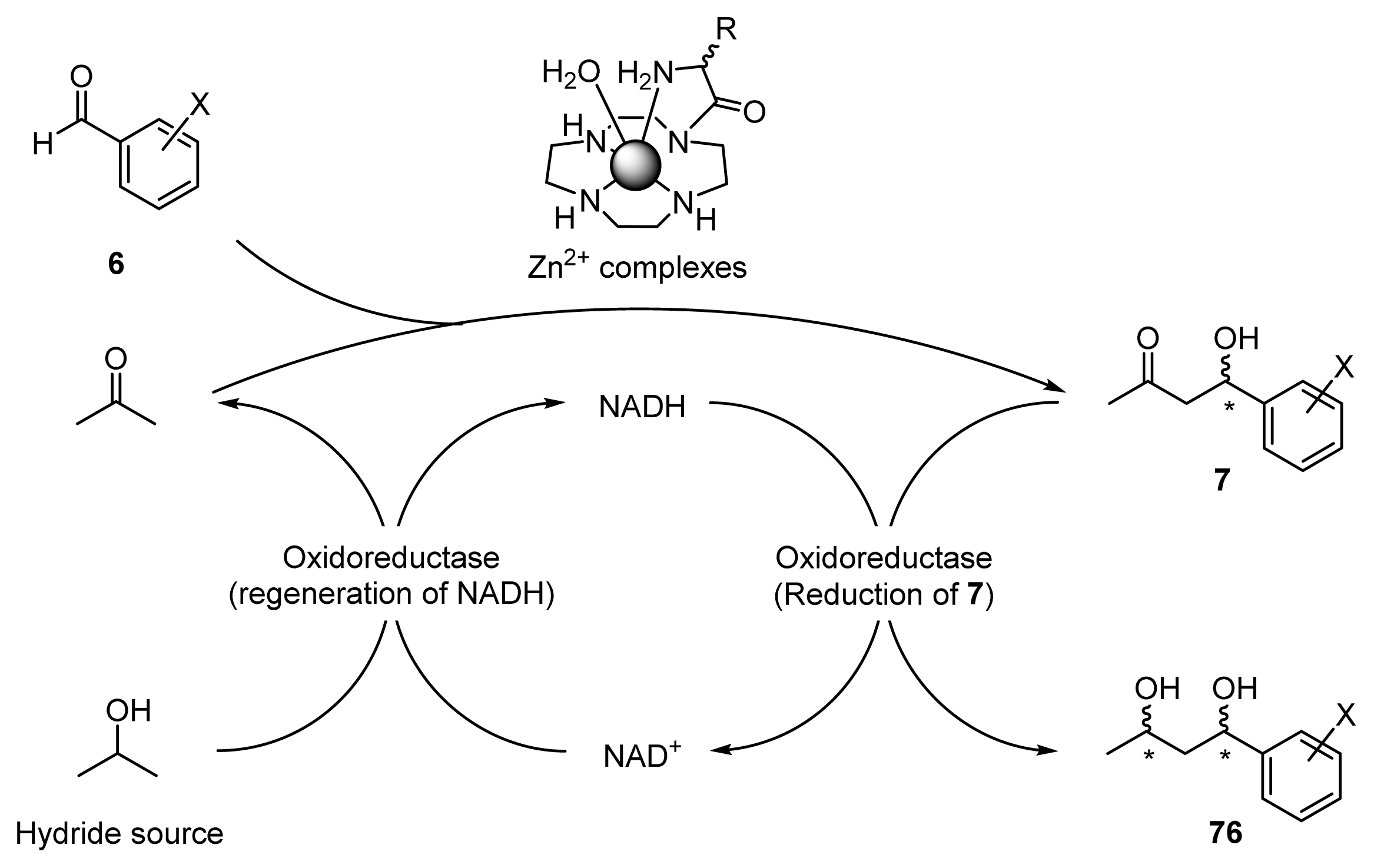
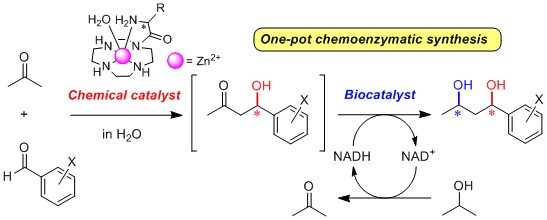
2.3. One-Pot Chemoenzymatic Synthesis of Chiral 1,3-Diols Using an Enantioselective Aldol Reaction and Enzymatic Reduction
2.3.1. Introduction for One-Pot Chemoenzymatic Synthesis
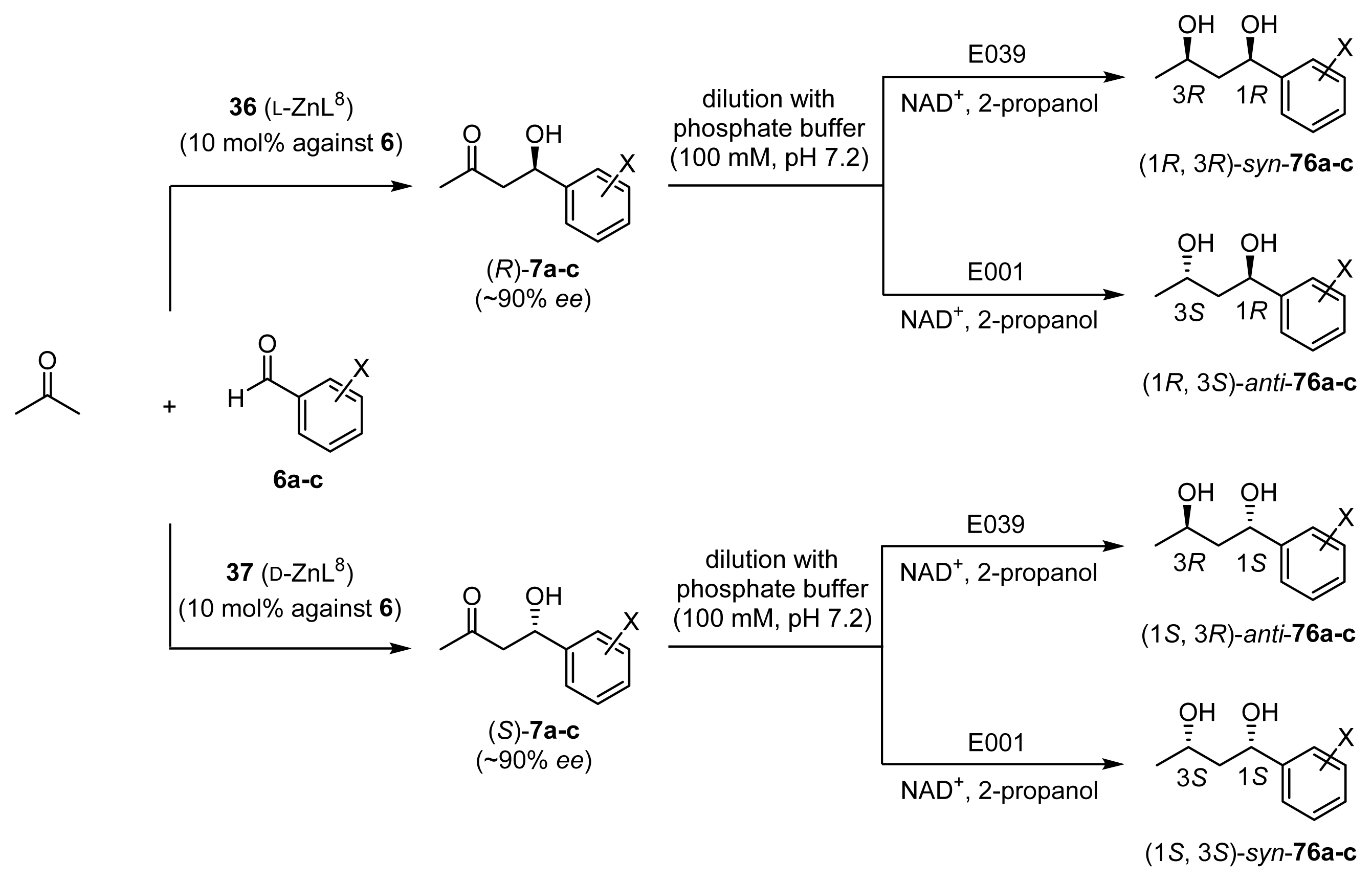
2.3.2. Enantioselective Reductions of β-Hydroxyketones 7a–c Using Oxidoreductase
2.3.3. One-Pot Chemoenzymatic Synthesis of Optically Active 1,3-Diols 76a–c from Acetone and Benzaldehydes 6a–c
3. Conclusions
Acknowledgments
Conflicts of Interest
References
- Kleemann, A.; Engels, J.; Kutscher, B.; Reichert, D. Pharmaceutical Substances: Syntheses, Patents, Applications, 4th ed.; Thieme: Stuttgart, Baden-Württemberg, Germany, 2001. [Google Scholar]
- Denard, C.A.; Hartwig, J.F.; Zhao, H. Multistep one-pot reactions combining biocatalysts and chemical catalysts for asymmetric synthesis. ACS Catal 2013, 3, 2856–2864. [Google Scholar]
- McMurry, J. Organic Chemistry, A Biological Approach; Thomson Brooks/Cole: Belmont, Boston, MA, USA, 2007. [Google Scholar]
- Dean, S.M.; Greenberg, W.A.; Wong, C.-H. Recent advances in aldolase-catalyzed asymmetric synthesis. Adv. Synth. Catal 2007, 349, 1308–1320. [Google Scholar]
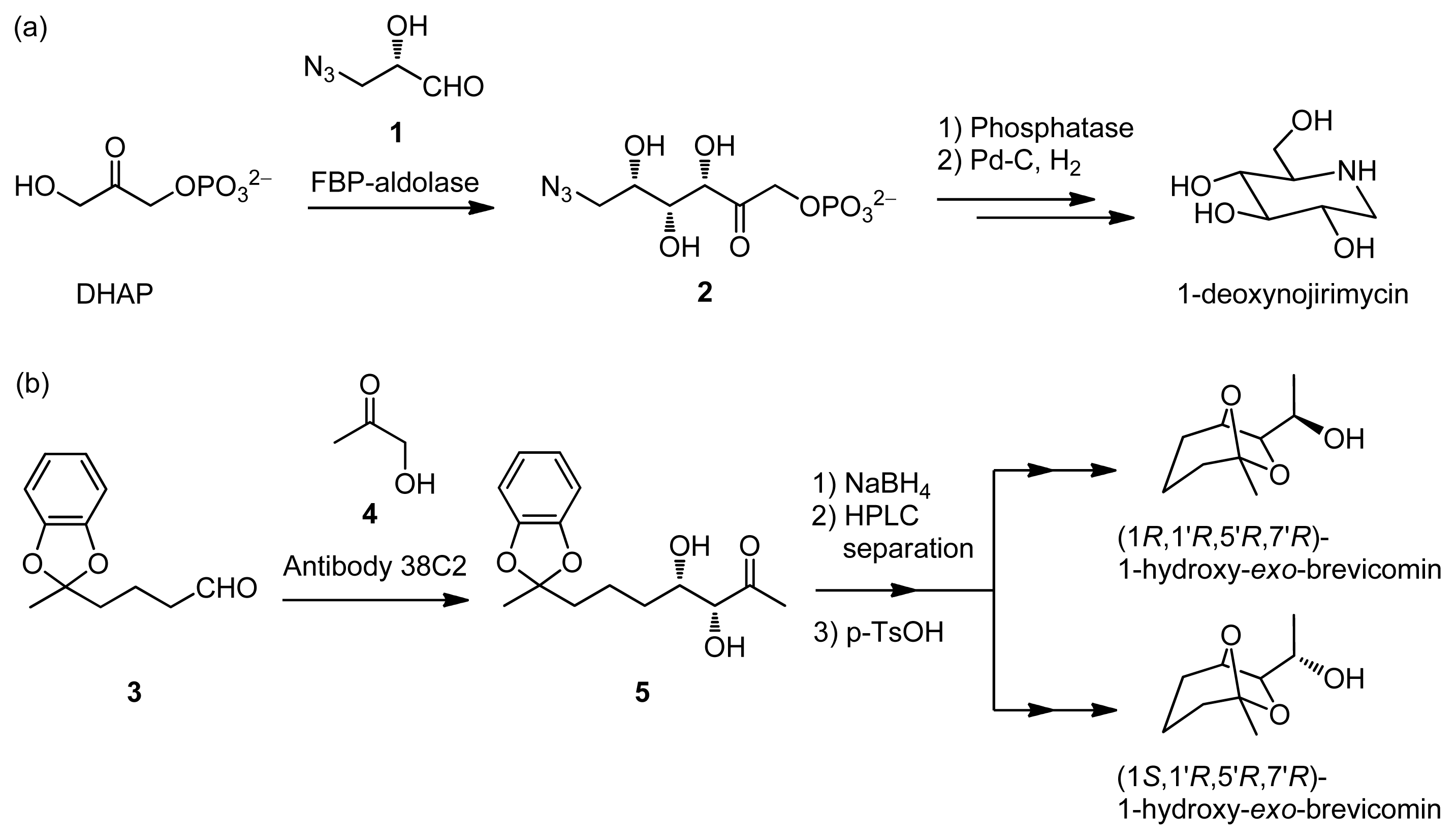
- Osten, C.-H.; Sinskey, A.J.; Barbas, C.F., III; Pederson, R.L.; Wang, Y.-F.; Wong, C.-H. Use of a recombinant bacterial fructose-1,6-dephosphate aldolase in aldol reactions: Proparative syntheses of 1-deoxynojirimycin, 1-deoxymannojirimycin, 1,4-dideoxy-1,4-imino-d-arabinitol, and fagomine. J. Am. Chem. Soc 1989, 111, 3924–3927. [Google Scholar]
- Wagner, J.; Lerner, R.A.; Barbas, C.F., III. Efficient aldolase catalytic antibodies that use the enamine mechanism of natural enzymes. Science 1995, 270, 1797–1800. [Google Scholar]
- Barbas, C.F., III; Heine, A.; Zhong, G.; Hoffmann, T.; Gramatikova, S.; Björnestedt, R.; List, B.; Anderson, J.; Stura, E.A.; Wilson, I.A.; et al. Immune versus natural selection: Antibody aldolases with enzymic rates but broader scope. Science 1997, 278, 2085–2092. [Google Scholar]
- List, B.; Shabat, D.; Barbas, C.F., III; Lerner, R.A. Enantioselective total synthesis of some brevicomins using aldolase antibody 38C2. Chem. Eur. J 1998, 4, 881–885. [Google Scholar]
- Mukherjee, S.; Yang, J.W.; Hoffmann, S.; List, B. Asymmetric enamine catalysis. Chem. Rev 2007, 107, 5471–5569. [Google Scholar]
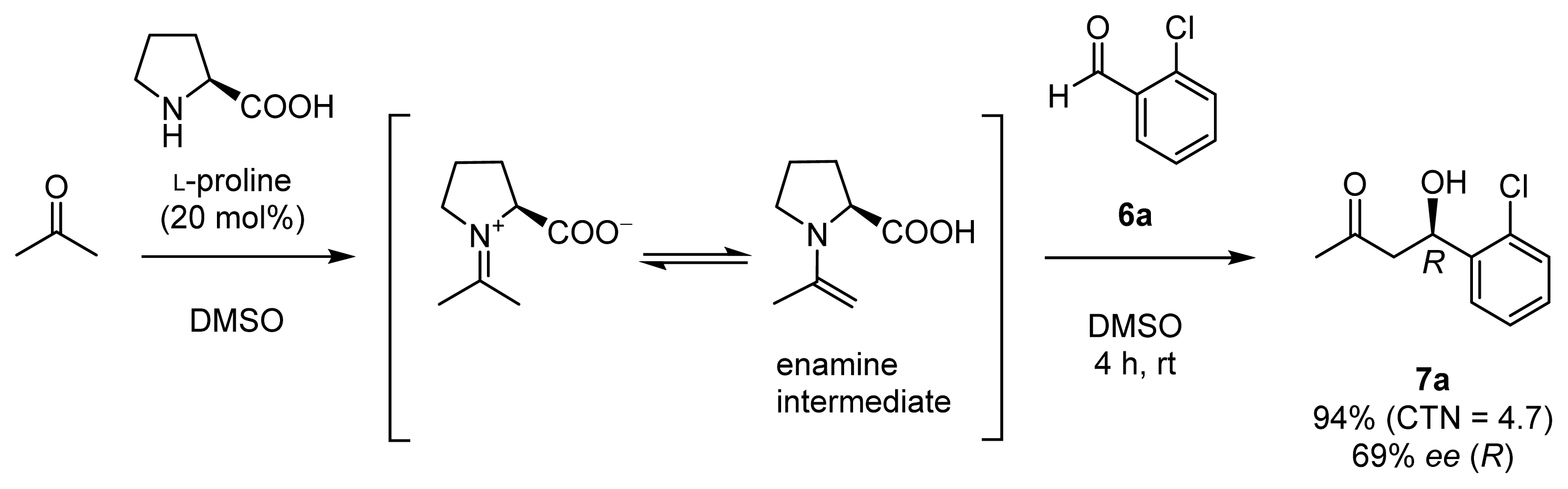
- List, B.; Lerner, R.A.; Barbas, C.F., III. Proline-catalyzed direct asymmetric aldol reactions. J. Am. Chem. Soc 2000, 122, 2395–2396. [Google Scholar]
- Allemann, C.; Gordillo, R.; Clemente, F.R.; Cheong, P.H.-Y.; Houk, K.N. Theory of asymmetric organocatalysis of aldol and related reactions: Rationalizations and predictions. Acc. Chem. Res 2004, 37, 558–569. [Google Scholar]
- Kotsuki, H.; Ikishima, H.; Okuyama, A. Organocatalytic asymmetric synthesis using proline and related molecules. Part 1. Heterocycles 2008, 75, 493–529. [Google Scholar]
- Kano, T.; Tokuda, O.; Maruoka, K. Synthesis of a biphenyl-based axially chiral amino acid as a highly efficient catalyst for the direct asymmetric aldol reaction. Tetrahedron Lett 2006, 47, 7423–7426. [Google Scholar]
- Mase, N.; Nakai, Y.; Ohara, N.; Yoda, H.; Takabe, K.; Tanaka, F.; Barbas, C.F., III. Organocatalytic direct asymmetric aldol reactions in water. J. Am. Chem. Soc 2006, 128, 734–735. [Google Scholar]
- Hayashi, Y.; Itoh, T.; Aratake, S.; Ishikawa, H. A diarylprolinol in an asymmetric catalytic, and direct crossed-aldol reaction of acetaldehyde. Angew. Chem. Int. Ed 2008, 47, 2082–2084. [Google Scholar]
- Ramasastry, S.S.V.; Albertshofer, K.; Utsumi, N.; Tanaka, F.; Barbas, C.F., III. Mimicking fructose and rhamnulose aldolases: Organocatalytic syn-aldol reactions with unprotected dihydroxyacetone. Angew. Chem. Int. Ed 2007, 46, 5572–5575. [Google Scholar]
- Modern Aldol Reactions; Mahrwald, R. (Ed.) Wiley-VCH: Weinheim, Baden-Württemberg, Germany, 2004.
- Kumagai, N.; Matsunaga, S.; Kinoshita, T.; Harada, S.; Okada, S.; Sakamoto, S.; Yamaguchi, K.; Shibasaki, M. Direct catalytic asymmetric aldol reaction of hydroxyketones: Asymmetric Zn catalysis with a Et2Zn/linked-BINOL complex. J. Am. Chem. Soc 2003, 125, 2169–2178. [Google Scholar]
- Trost, B.M.; Ito, H.; Silcoff, E.R. Asymmetric Aldol reaction via a dinuclear zinc catalyst: α-hydroxyketones as donors. J. Am. Chem. Soc 2001, 123, 3367–3368. [Google Scholar]
- Darbre, T.; Machuqueiro, M. Zn-proline catalyzed direct aldol reaction in aqueous media. Chem. Commun 2003, 1090–1091. [Google Scholar]
- Kofoed, J.; Darbre, T.; Reymond, J.-L. Dual mechanism of zinc-proline catalyzed aldol reactions in water. Chem. Commun 2006, 1482–1484. [Google Scholar]
- Pradowska, J.; Pasternak, M.; Gut, B.; Gryzlo, B.; Mlynarski, J. Direct asymmetric aldol reactions inspired by two types of natural aldolases: Water-compatible organocatalysts and ZnII complexes. J. Org. Chem 2012, 77, 173–187. [Google Scholar]
- Aoki, S.; Kimura, E. Comprehensive Coordination Chemistry II; Que, L., Jr., Tolman, W.B., Eds.; Elsevier: Amsterdam, The Netherlands, 2004; Volume 8, pp. 601–640. [Google Scholar]
- Kimura, E. Evolution of macrocyclic polyamines from molecular science to supramolecular science. Bull. Jpn. Soc. Coord. Chem 2012, 59, 26–47. [Google Scholar]
- Kimura, E.; Gotoh, T.; Koike, T.; Shiro, M. Dynamic enolate recognition in aqueous solution by zinc(II) in a phenacyl-pendant cyclen complex: Implications for the role of zinc(II) in class II aldolases. J. Am. Chem. Soc 1999, 121, 1267–1274. [Google Scholar]
- Kan, S.B.J.; Ng, K.K.-H.; Paterson, I. The impact of the mukaiyama aldol reaction in total synthesis. Angew. Chem. Int. Ed 2013, 52, 9097–9108. [Google Scholar]
- Arya, P.; Qin, H. Advances in asymmetric enolate methodology. Tetrahedron 2000, 56, 917–947. [Google Scholar]
- Li, C.-J.; Chan, T.-H. Organic Reactions in Aqueous Media; Wiley: New York, NY, USA, 1997. [Google Scholar]
- Organic Synthesis in Water; Grieco, P.A. (Ed.) Blacky Academic and Professional: London, UK, 1998.
- Mlynarski, J.; Baś, S. Catalytic asymmetric aldol reactions in aqueous media—A 5 year update. Chem. Soc. Rev 2014, 43, 577–587. [Google Scholar]
- Itoh, S.; Kitamura, M.; Yamada, Y.; Aoki, S. Chiral catalysts dually functionalized with amino acids and zinc(II) complexes for enantioselective direct aldol reactions inspired by natural aldolases: Design, synthesis, complexation properties, catalytic activity, and mechanistic study. Chem. Eur. J 2009, 15, 10570–10584. [Google Scholar]
- Kimura, E.; Shiota, T.; Koike, T.; Shiro, M.; Kodama, M. A zinc(II) complex of 1,5,9-triazacyclododecane ([12]aneN3) as a model for carbonic anhydrase. J. Am. Chem. Soc 1990, 112, 5805–5811. [Google Scholar]
- Aoki, S.; Sakurama, K.; Matsuo, N.; Yamada, Y.; Takasawa, R.; Tanuma, S.; Shiro, M.; Takeda, K.; Kimura, E. A New Fluorescent Probe for zinc(II), a 8-Hydroxy-5-N,N-dimethylaminosulfonylquinoline-pendant 1,4,7,10-Tetraazacyclododecane. Chem. Eur. J 2006, 12, 9066–9080. [Google Scholar]
- Sonoike, S.; Itakura, T.; Kitamura, M.; Aoki, S. One-pot chemoenzymatic synthesis of chiral 1,3-diols using an enantioselective aldol reaction with chiral Zn2+ complex catalysts and enzymatic reduction using oxidoreductases with cofactor regeneration. Chem. Asian J 2012, 7, 64–74. [Google Scholar]
- Itoh, S.; Tokunaga, T.; Sonike, S.; Kitamura, M.; Yamano, A.; Aoki, S. Asymmetric aldol reactions between acetone and benzaldehydes catalyzed by chiral Zn2+ complexes of aminoacyl 1,4,7,10-tetraazacyclododecane: Fine-tuning of the amino-acid side chains and revised reaction mechanism. Chem. Asian J 2013, 8, 2125–2135. [Google Scholar]
- Zimmerman, H.E.; Traxler, M.D. The stereochemisty of the ivanov and reformatsky reactions. I. J. Am. Chem. Soc 1957, 79, 1920–1923. [Google Scholar]
- Gao, J.; Bai, S.; Gao, Q.; Liu, Y.; Yang, Q. Acid controlled diastereoselectivity in asymmetric aldol reaction of cycloketones with aldehydes using enamine-based organocatalysts. Chem. Commun 2011, 47, 6716–6718. [Google Scholar]
- Moteki, S.A.; Han, J.; Arimitsu, S.; Akakura, M.; Nakayama, K.; Maruoka, K. An achiral-acid-induced switch in the enantioselectivity of a chiral cis-diamine-based organocatalyst for asymmetric aldol and mannich reactions. Angew. Chem. Int. Ed. Engl 2012, 51, 1187–1190. [Google Scholar]
- Itoh, S.; Tokunaga, T.; Kurihara, M.; Aoki, S. Asymmetric aldol reactions between cyclic ketones and benzaldehydes catalyzed by chiral Zn2+ complexes of aminoacyl 1,4,7,10-tetraazacyclododecane: Effects of solvent and additives on the stereoselectivities of the aldol products. Tetrahedron Asymmetry 2013, 24, 1583–1590. [Google Scholar]
- Sletten, E.M.; Bertozzi, C.R. Bioorthogonal chemistry: Fishing for selectivity in a sea of functionality. Angew. Chem. Int. Ed 2009, 48, 6974–6998. [Google Scholar]
- Tietze, L.F.; Brasche, G.; Gericke, K. Domino Reactions in Organic Synthesis; Wiley: Weinheim, Baden-Württemberg, Germany, 2006. [Google Scholar]
- Bruggink, A.; Schoevaart, R.; Kieboom, T. Concepts of nature in organic synthesis: Cascade catalysis and multistep conversions in concert. Org. Process Res. Dev 2003, 7, 622–640. [Google Scholar]
- Wong, C.-H.; Whitesides, G.M. Enzymes in Synthetic Organic Chemistry; Pergamon: Oxford, UK, 1994. [Google Scholar]
- Weiß, M.; Brinkmann, T.; Gröger, H. Towards a greener synthesis of (S)-3-aminobutanoic acid: Process development and environmental assessment. Green Chem 2010, 12, 1580–1588. [Google Scholar]
- Baer, K.; Kraußer, M.; Burda, E.; Hummel, W.; Berkessel, A.; Gröger, H. Sequential and modular synthesis of chiral 1,3-diols with two stereogenic centers: Access to all four stereoisomers by combination of organo- and biocatalysis. Angew. Chem. Int. Ed 2009, 48, 9355–9358. [Google Scholar]
- Burda, E.; Hummel, W.; Gröger, H. Modular chemoenzymatic one-pot syntheses in aqueous media: Combination of a palladium-catalyzed cross-coupling with an asymmetric biotransformation. Angew. Chem. Int. Ed 2008, 47, 9551–9554. [Google Scholar]
- Bode, S.E.; Wolberg, M.; Müller, M. Stereoselective synthesis of 1,3-diols. Synthesis 2006, 557–588. [Google Scholar]
- Hayashi, Y.; Aratake, S.; Okano, T.; Takahashi, J.; Sumiya, T.; Shoji, M. Combined proline-surfactant organocatalyst for the highly diastereo- and enantioselective aqueous direct cross-aldol reaction of aldehydes. Angew. Chem. Int. Ed 2006, 45, 5527–5529. [Google Scholar]
- Sinz, C.J.; Rychnovsky, S.D. 4-acetoxy- and 4-cyano-1,3-dioxanes in synthesis. Top. Curr. Chem 2001, 216, 51–92. [Google Scholar]
- Acetti, D.; Brenna, E.; Fuganti, C.; Gatti, F.G.; Serra, S. Baker’s yeast reduction of β-hydroxy ketones. Eur. J. Org. Chem 2010, 1, 142–151. [Google Scholar]
- Chen, K.; Richter, J.M.; Baran, P.S. 1,3-diol synthesis via controlled, radical-mediated C–H functionalization. J. Am. Chem. Soc 2008, 130, 7247–7249. [Google Scholar]
- Guo, Z.W.; Wu, S.H.; Chen, C.S.; Girdaukas, G.; Sih, C.J. Sequential biocatalytic kinetic resolutions. J. Am. Chem. Soc 1990, 112, 4942–4945. [Google Scholar]
- Mlynarski, J. Direct asymmetric aldol-tishchenko reaction. Eur. J. Org. Chem 2006, 4779–4786. [Google Scholar]
- Wong, C.-H.; Whitesides, G.M. Enzyme-catalyzed organic synthesis: NAD(P)H cofactor regeneration by using glucose-6-phosphate and the glucose-5-phosphate dehydrogenase from Leuconostoc mesenteroides. J. Am. Chem. Soc 1981, 103, 4890–4899. [Google Scholar]
- Brenna, E.; Fuganti, C.; Gatti, F.G.; Serra, S. Biocatalytic methods for the synthesis of enantioenriched odor active compounds. Chem. Rev 2011, 111, 4036–4072. [Google Scholar]
- Hayashi, M. Biocatalyst for asymmetric synthesis –Chiralscreen–. Synth. Org. Chem. Jpn 2011, 69, 517–525. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
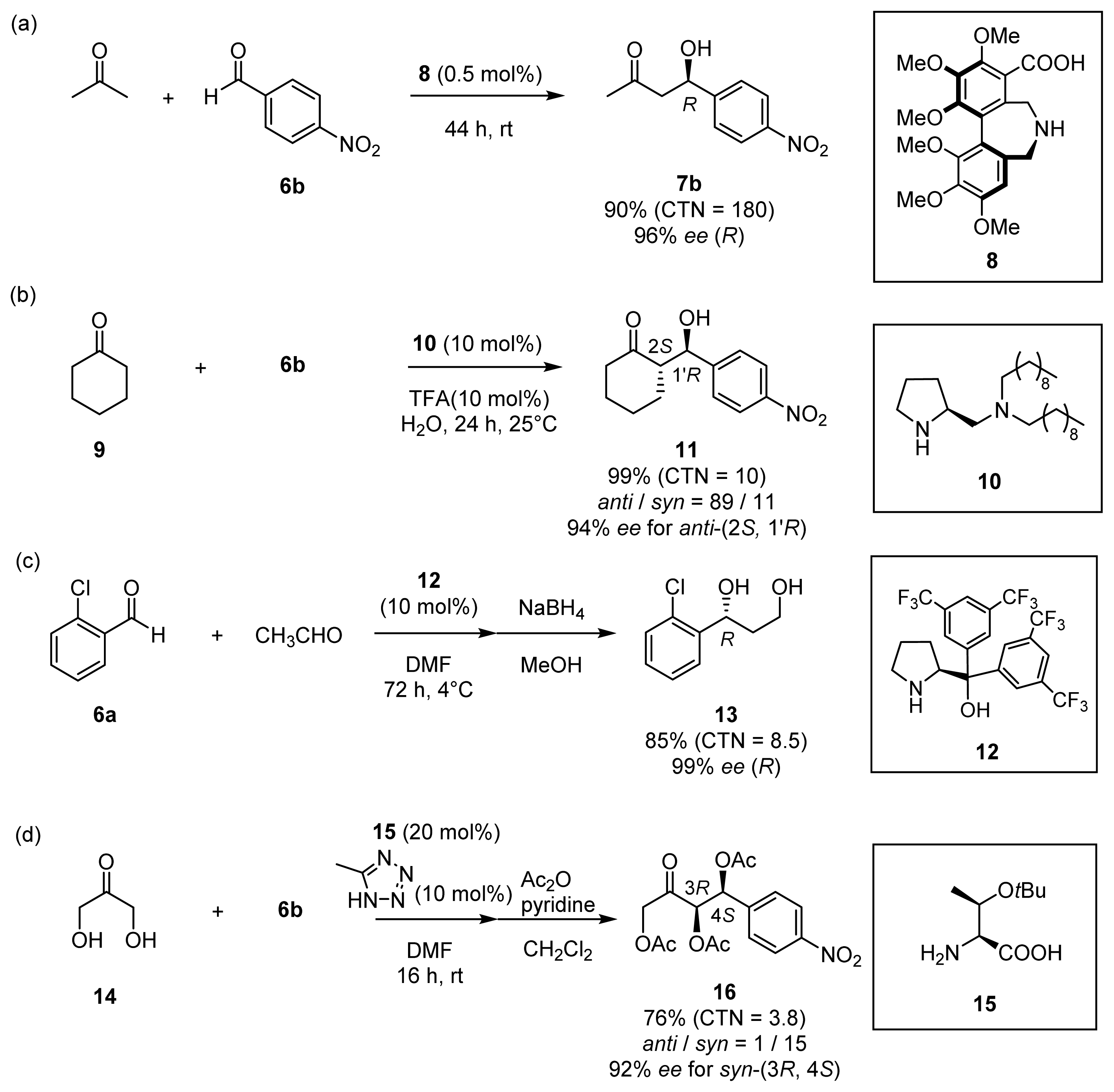{kind=link}
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst [a] (mM) | Mol % | Solvent [b] | Conditions | Yield [c] (%) | CTN [d] | ee (%) [e] |
| 1 | l-proline (20) | 20 | DMSO/acetone | 4 h, 25 °C | 85 | 4 | 67 (R) |
| 2 | 31 (l-ZnL3) [f] (10) | 10 | DMSO/acetone | 26 h, 25–50 °C | 3 | – | 1 (R) |
| 3 | 32 (l-ZnL4) [f] (10) | 10 | DMSO/acetone | 4 h, 25 °C | 12 | 1 | 34 (R) |
| 4 | l-proline (50) | 5 | acetone/H2O | 20 h, 37 °C | 22 | 4 | 48 (R) |
| 5 | 31 (l-ZnL3) [f] (50) | 5 | acetone/H2O | 20 h, 37 °C | 43 | 9 | 1 (R) |
| 6 | 32 (l-ZnL4) [g] (50) | 5 | acetone/H2O | 20 h, 37 °C | 73 | 15 | 80 (R) |
| 7 | 29 (l-L4) [h] (50) | 5 | acetone/H2O | 20 h, 37 °C | 72 | 14 | racemic |
| 8 | 41a (l-CdL4) [f] (50) | 5 | acetone/H2O | 20 h, 37 °C | 5 | 1 | 50 (R) |
| 9 | 41b (l-CuL4) [f] (50) | 5 | acetone/H2O | 20 h, 37 °C | trace | – | – |
| 10 | 33 (l-ZnL5) [f] (50) | 5 | acetone/H2O | 20 h, 37 °C | 87 | 17 | 80 (R) |
| 11 | 34 (l-ZnL6) [f] (50) | 5 | acetone/H2O | 20 h, 37 °C | 54 | 11 | 9 (R) |
| 12 | 35 (ZnL7) [g] (50) | 5 | acetone/H2O | 20 h, 37 °C | 5 | 1 | 2 (S) |
| 13 | 36 (l-ZnL8) [f] (50) | 5 | acetone/H2O | 24 h, 25 °C | 85 | 17 | 91 (R) |
| 14 | 37 (d-ZnL8) [f] (50) | 5 | acetone/H2O | 24 h, 25 °C | 85 | 17 | 91 (S) |
| 15 | 26 (ZnL1) [g] (50) | 5 | acetone/H2O | 20 h, 37 °C | trace | – | – |
| 16 | l-proline + 26 (ZnL1) [g] (50) | 5 | acetone/H2O | 20 h, 37 °C | trace | – | – |
 | ||||||||
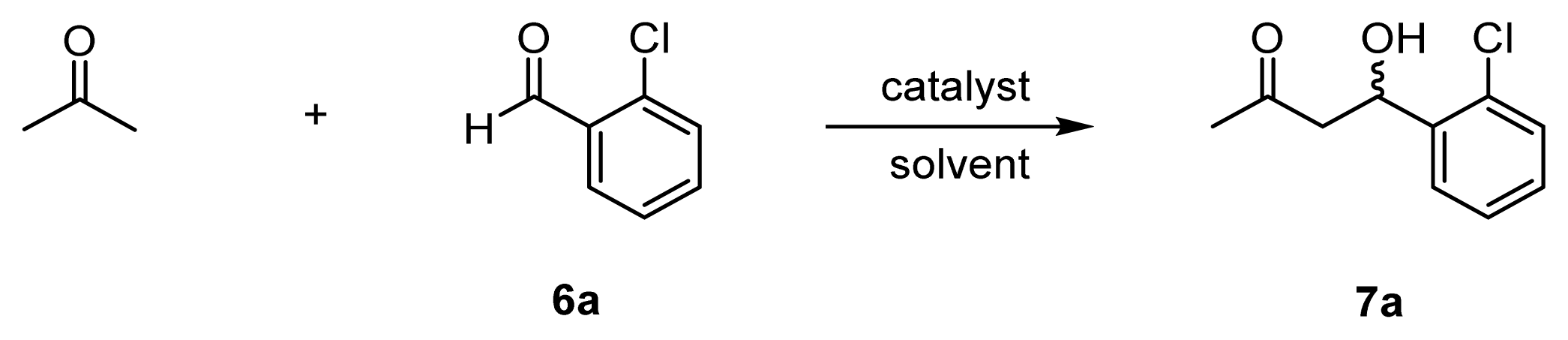
|---|---|---|---|---|---|---|---|---|
| Entry | Substrate | Catalyst [a] | mol % | Conditions | Product | Yield (%) [b] | CTN [c] | ee (%) [d] |
| 1 | 6b | 32 (l-ZnL4) | 5 | 20 h, 37 °C | 7b | 78 | 16 | 63 (R) |
| 2 | 6b | 33 (l-ZnL5) | 5 | 20 h, 37 °C | 7b | 86 | 17 | 86 (R) |
| 3 | 6b | 36 (l-ZnL8) | 10 | 24 h, 30 °C | 7b | quant | 10 | 90 (R) |
| 4 | 6b | 37 (d-ZnL8) | 10 | 24 h, 30 °C | 7b | quant | 10 | 90 (S) |
| 5 | 6c | 36 (l-ZnL8) | 10 | 72 h, 30 °C | 7c | quant | 10 | 90 (R) |
| 6 | 6c | 37 (d-ZnL8) | 10 | 72 h, 30 °C | 7c | quant | 10 | 90 (S) |
| 7 | 6d | 32 (l-ZnL4) | 10 | 20 h, 37 °C | 7d | 70 | 7 | 52 (R) |
| 8 | 6d | 33 (l-ZnL5) | 10 | 20 h, 37 °C | 7d | 49 | 5 | 75 (R) |
| 9 | 6e | 32 (l-ZnL4) | 10 | 120 h, 37 °C | 7e | 59 | 6 | 57 (R) |
| 10 | 6e | 33 (l-ZnL5) | 10 | 120 h, 37 °C | 7e | 55 | 6 | 83 (R) |
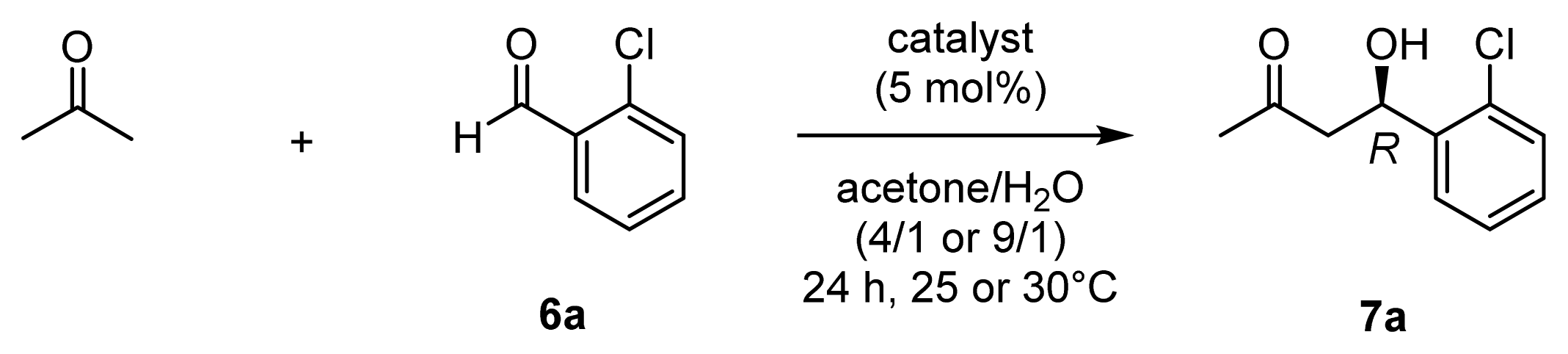 | ||||
|---|---|---|---|---|
| Entry [a] | Catalyst [b] | Yield (%) [c] | CTN [d] | ee (%) [e] |
| 1 | 42 (l-ZnL10) | 30 | 6 | 91 (R) |
| 2 | 43 (l-ZnL11) | 89 | 18 | 94 (R) |
| 3 | 44 (l-ZnL12) | 84 | 17 | 86 (R) |
| 4 | 45 (l-ZnL13) | 91 | 18 | 94 (R) |
| 5 | 46 (l-ZnL14) | 67 | 13 | 87 (R) |
| 6 | 47 (l-ZnL15) | 86 | 17 | 78 (R) |
| 7 | 48 (l-ZnL16) | 93 | 19 | 93 (R) |
| 8 | 49 (l-ZnL17) | 95 | 19 | 94 (R) |
| 9 | 50 (l-ZnL18) | 54 | 11 | 88 (R) |
| 10 | 51 (l-ZnL19) | 44 | 9 | 78 (R) |
| 11 | 52 (l-ZnL20) | 79 | 16 | 92 (R) |
| 12 | 43 (l-ZnL11) | 90 | 18 | 96 (R) |
| 13 | 45 (l-ZnL13) | 85 | 17 | 95 (R) |
| 14 | 49 (l-ZnL17) | 96 | 19 | 96 (R) |
| 15 | 53 (ZnL21) | 24 | 5 | 9 (R) |
| 16 | 54 (ZnL22) | 19 | 4 | 33 (S) |
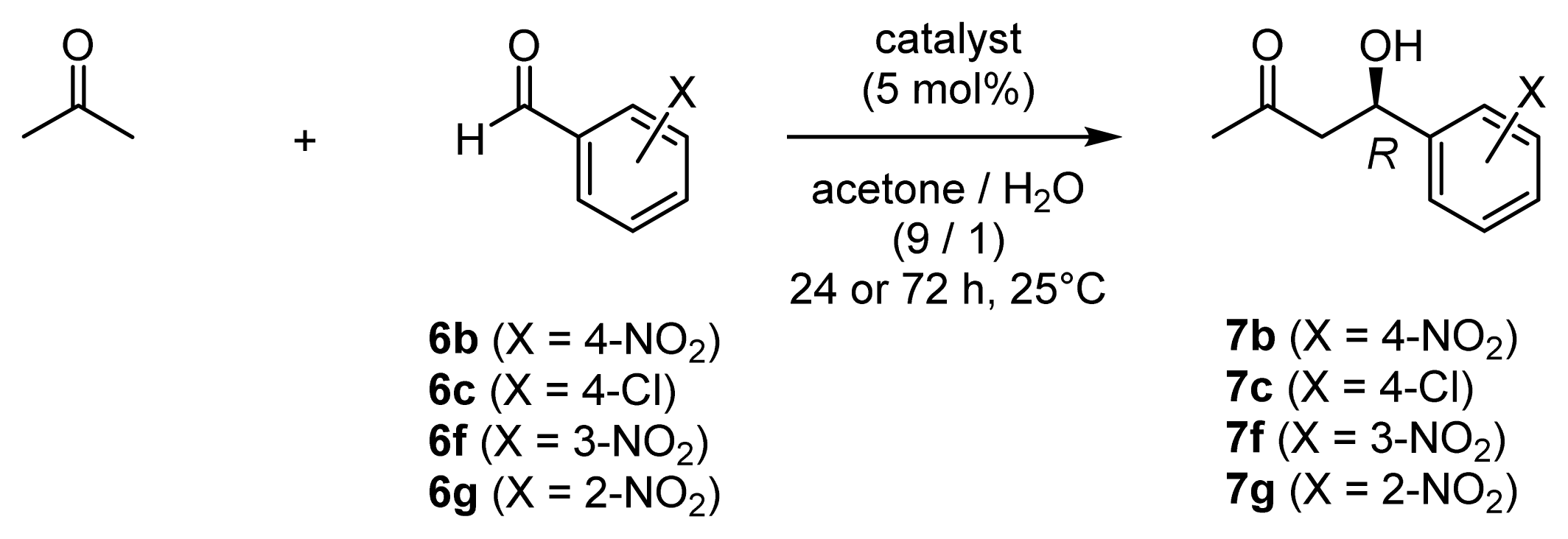 | ||||||
|---|---|---|---|---|---|---|
| Entry [a] | Substrate | Catalyst [b] | Product | Yield (%) [c] | CTN [d] | ee (%) [e] |
| 1 | 6b | 45 (l-ZnL13) | 7b | 92 | 18 | 96 (R) |
| 2 | 6b | 49 (l-ZnL17) | 7b | 92 | 18 | 95 (R) |
| 3 | 6c | 45 (l-ZnL13) | 7c | 74 | 15 | 94 (R) |
| 4 | 6c | 49 (l-ZnL17) | 7c | 83 | 17 | 95 (R) |
| 5 | 6f | 45 (l-ZnL13) | 7f | 92 | 18 | 95 (R) |
| 6 | 6f | 49 (l-ZnL17) | 7f | 97 | 19 | 95 (R) |
| 7 | 6g | 45 (l-ZnL13) | 7g | 96 | 19 | 90 (R) |
| 8 | 6g | 49 (l-ZnL17) | 7g | 89 | 18 | 92 (R) |
| l-proline | l-valine | Zn(OTf)2 | 32 (l-ZnL4) | 33 (l-ZnL5) | 35 (ZnL7) | 26 (ZnL1) | |
|---|---|---|---|---|---|---|---|
| λmax | 316 nm (enaminone) | 316 nm (enaminone) | 289 nm (Zn2+-enolate) | 294 nm (Zn2+-enolate) | 292 nm (Zn2+-enolate) | 295 nm (Zn2+-enolate) | – |
| Kapp | ~10 (62 [a]) | ~10 | 157 | 212 | 139 | 589 (49 [b]) | n. i. [c] |
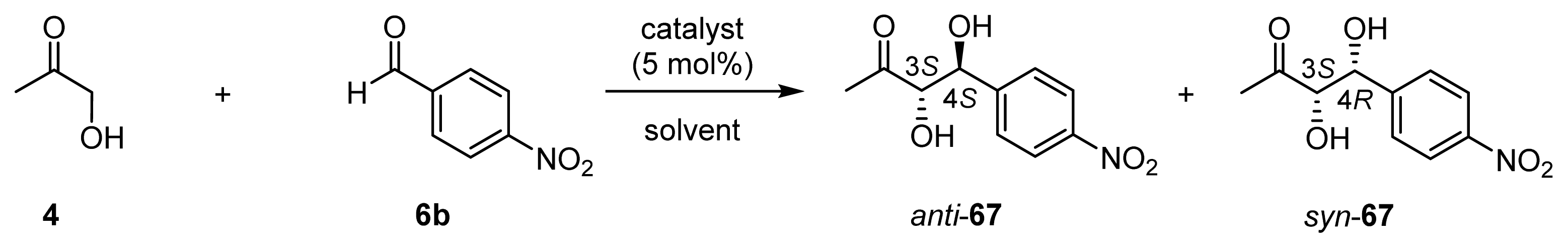 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst [a] (mM) | Solvent | Conditions | Yield [b] (%) | CTN [c] | d.r. [d] (anti/syn) | ee [%] [e] (anti/syn) |
| 1 | 32 (l-ZnL4) (25) | THF/4/H2O (5/4/1) | 24 h, 37 °C | 83 | 17 | 75/25 | racemic/racemic |
| 2 | 32 (l-ZnL4) (50) | DMSO/4/H2O (21/1.1/1) | 20 h, 37 °C | 80 | 16 | 52/48 | racemic/4 (3S, 4R) |
| 3 | 32 (l-ZnL4) (25) | 4/CH3CN (1/1) | 24 h, 25 °C | 90 | 18 | 75/25 | 1 (3R, 4R)/8 (3S, 4R) |
| 4 | 33 (l-ZnL5) (25) | THF/4/H2O (2/1/1) | 20 h, 37 °C | 89 | 18 | 64/36 | 7 (3S, 4S)/5 (3S, 4R) |
| 5 | 33 (l-ZnL5) (25) | 4/H2O (3/1) | 20 h, 37 °C | 68 | 14 | 58/42 | 9 (3S, 4S)/14 (3S, 4R) |
| 6 | 33 (l-ZnL5) (25) | 4/NMP [f] (1/1) | 24 h, 25 °C | 96 | 19 | 63/37 | 18 (3S, 4S)/2 (3R, 4S) |
| 7 | 33 (l-ZnL5) (25) | 4/CH3CN (1/1) | 24 h, 25 °C | 91 | 18 | 37/63 | 8 (3R, 4R)/45 (3S, 4R) |
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst [b] (mM) | mol % | Solvent | Conditions (for aldol) | Yield [%] [c] (for 2 steps) | d.r. [d] (anti/syn) | ee [%] [e] (anti/syn) |
| 1 | 32 (l-ZnL4) (12) | 5 | THF/H2O (1/1) | 96 h, 37 °C | Trace | – | – |
| 2 | 32 (l-ZnL4) (6) | 5 | DMSO/H2O (3/1) | 72 h, 37 °C | 22 | 37/63 | racemic/6 (3R, 4S) |
| 3 | 32 (l-ZnL4) (100) | 10 | NMP/H2O (33/1) | 24 h, 25 °C | 4 | 26/74 | 3 (3R, 4R)/2 (3S, 4R) |
| 4 | 33 (l-ZnL5) (17) | 5 | THF/H2O (3/5) | 72 h, 37 °C | 4 | 42/58 | 3 (3S, 4S)/9 (3R, 4S) |
| 5 | 33 (l-ZnL5) (6) | 5 | DMSO/H2O (2/1) | 72 h, 37 °C | 27 | 37/63 | racemic/6 (3R, 4S) |
| 6 | 33 (l-ZnL5) (100) | 10 | NMP/H2O (33/1) | 42 h, 25 °C | 26 | 34/66 | 8 (3R, 4R)/44 (3R, 4S) |
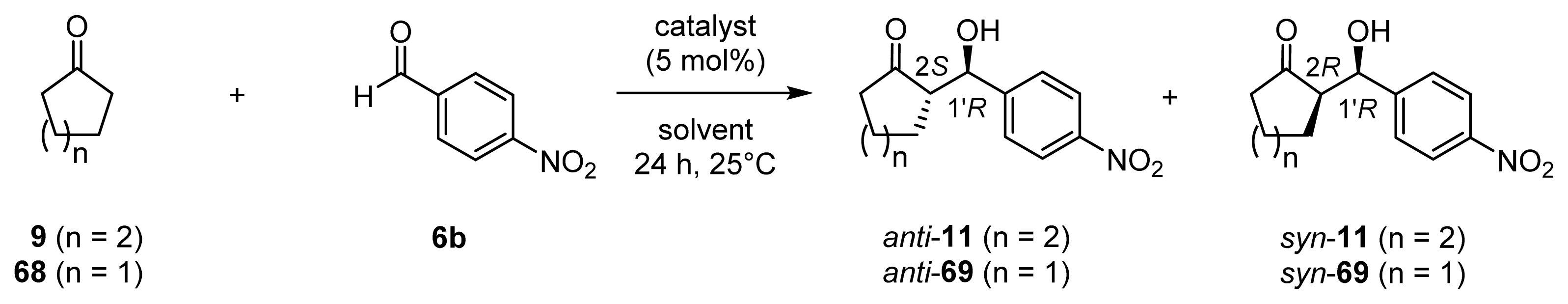 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst [a] | Solvent [b] | Product | Yield [c] (%) | CTN [d] | d.r. [e] (anti/syn) | ee [%] [f] (anti/syn) |
| 1 | 45 (l-ZnL13) | 9 | 11 | 4 | 1 | 99/1 | 28 (2S, 1′R)/19 (2R, 1′R) |
| 2 | 45 (l-ZnL13) | 9/H2O (95/5) | 11 | 79 | 16 | 49/51 | 11 (2S, 1′R)/89 (2R, 1′R) |
| 3 | 45 (l-ZnL13) [g] | 9/H2O (95/5) | 11 | 67 | 13 | 30/70 | 2 (2R, 1′S)/92 (2R, 1′R) |
| 4 | 45 (l-ZnL13) | 9/MeOH (50/50) | 11 | 81 | 16 | 82/18 | 72 (2S, 1′R)/61 (2R, 1′R) |
| 5 | 45 (l-ZnL13) | 9/EtOH (50/50) | 11 | 61 | 12 | 67/33 | 68 (2S, 1′R)/86 (2R, 1′R) |
| 6 | 45 (l-ZnL13) | 9/2-propanol (50/50) | 11 | 13 | 3 | 80/20 | 68 (2S, 1′R)/54 (2R, 1′R) |
| 7 | 45 (l-ZnL13) | 9/DMF (50/50) | 11 | 4 | 1 | 91/9 | 84 (2S, 1′R)/52 (2R, 1′R) |
| 8 | 45 (l-ZnL13) | 9/NMP (50/50) | 11 | 19 | 4 | 90/10 | 88 (2S, 1′R)/17 (2R, 1′R) |
| 9 | 45 (l-ZnL13) | 9/NMP/H2O (40/50/10) | 11 | 85 | 17 | 49/51 | 7 (2R, 1′S)/83 (2R, 1′R) |
| 10 | 45 (l-ZnL13) | 9/NMP/MeOH (40/50/10) | 11 | 96 | 19 | 88/12 | 84 (2S, 1′R)/46 (2R, 1′R) |
| 11 | 49 (l-ZnL17) | 9/H2O (95/5) | 11 | 62 | 12 | 51/49 | 6 (2S, 1′R)/90 (2R, 1′R) |
| 12 | 49 (l-ZnL17) | 9/NMP/MeOH (40/50/10) | 11 | 89 | 18 | 87/13 | 83 (2S, 1′R)/51 (2R, 1′R) |
| 13 | 49 (l-ZnL17) | 68/H2O (95/5) | 69 | 90 | 18 | 31/69 | 23 (2R, 1′S)/87 (2R, 1′R) |
| 14 | 49 (l-ZnL17) | 68/NMP/MeOH (40/50/10) | 69 | 6 | 1 | 55/45 | 37 (2R, 1′S)/7 (2R, 1′R) |
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | Oxidoreductase [a] (syn/anti) | Product [c] | Yield [%] [b] (syn/anti) | ee [%] [c] (syn/anti) | 3R/3S [c] |
| 1 | rac-7a [d] | Baker’s yeast | – | trace | – | – |
| 2 | rac-7a [d] | ADH from S. cerevisiae | – | trace | – | – |
| 3 | rac-7a [d] | ADH from L. Kefir | 76a (52/48) | quant | >99 (1R, 3R)/>99(1S, 3R) | >99/<1 |
| 4 | rac-7a [d] | E001 [e] | 76a (54/46) | quant | >99 (1S, 3S)/>99 (1R, 3S) | <1/>99 |
| 5 | rac-7a [d] | E031 [e] | 76a (47/53) | 50 | 95 (1S, 3S)/93 (1R, 3S) | 3/97 |
| 6 | rac-7a [d] | E039 [e] | 76a (48/52) | quant | >99 (1R, 3R)/>99 (1S, 3R) | >99/<1 |
| 7 | rac-7a [d] | E092 [e] | 76a (78/22) | 64 | >99 (1S, 3S)/94 (1R, 3S) | 1/99 |
| 8 | rac-7b [d] | E001 [e] | 76b (50/50) | quant | >99 (1S, 3S)/>99 (1R, 3S) | <1/>99 |
| 9 | rac-7b [d] | E039 [e] | 76b (48/52) | quant | >99 (1R, 3R)/>99 (1S, 3R) | >99/<1 |
| 10 | rac-7c [d] | E001 [e] | 76c (50/50) | quant | >99 (1S, 3S)/>99 (1R, 3S) | <1/>99 |
| 11 | rac-7c [d] | E039 [e] | 76c (49/51) | quant | >99 (1R, 3R)/>99 (1S, 3R) | >99/<1 |
| 12 | 7a (90% ee (S)) | E001 [e] | 76a (95/5) | quant | >99 (1S, 3S)/>99 (1R, 3S) | <1/>99 |
| Entry | Substrate | ZnL [a] | Oxidoreductase [b] (“Chiralscreen® OH”) | Product | Yield (%) [c] | Product ratio [d] | |||
|---|---|---|---|---|---|---|---|---|---|
| (1R,3R) | (1S,3R) | (1R,3S) | (1S,3S) | ||||||
| 1 | 6a | 36 (l-ZnL8) | E001 | 76a | 88 | <1 | <1 | 96 | 4 |
| 2 | 6a | 36 (l-ZnL8) | E039 | 76a | 88 | 95 | 5 | <1 | <1 |
| 3 | 6a | 37 (d-ZnL8) | E001 | 76a | 84 | <1 | <1 | 4 | 96 |
| 4 | 6a | 37 (d-ZnL8) | E039 | 76a | 92 | 5 | 95 | <1 | <1 |
| 5 | 6b | 36 (l-ZnL8) | E001 | 76b | 83 | <1 | <1 | 93 | 7 |
| 6 | 6b | 36 (l-ZnL8) | E039 | 76b | 87 | 94 | 6 | <1 | <1 |
| 7 | 6b | 37 (d-ZnL8) | E001 | 76b | 91 | <1 | <1 | 4 | 96 |
| 8 | 6b | 37 (d-ZnL8) | E039 | 76b | 80 | 4 | 96 | <1 | <1 |
| 9 | 6c | 36 (l-ZnL8) | E001 | 76c | 68 | <1 | <1 | 96 | 4 |
| 10 | 6c | 36 (l-ZnL8) | E039 | 76c | 60 | 96 | 4 | <1 | <1 |
| 11 | 6c | 37 (d-ZnL8) | E001 | 76c | 60 | <1 | <1 | 5 | 95 |
| 12 | 6c | 37 (d-ZnL8) | E039 | 76c | 48 | 4 | 96 | <1 | <1 |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Itoh, S.; Sonoike, S.; Kitamura, M.; Aoki, S. Design and Synthesis of Chiral Zn2+ Complexes Mimicking Natural Aldolases for Catalytic C–C Bond Forming Reactions in Aqueous Solution. Int. J. Mol. Sci. 2014, 15, 2087-2118. https://doi.org/10.3390/ijms15022087
Itoh S, Sonoike S, Kitamura M, Aoki S. Design and Synthesis of Chiral Zn2+ Complexes Mimicking Natural Aldolases for Catalytic C–C Bond Forming Reactions in Aqueous Solution. International Journal of Molecular Sciences. 2014; 15(2):2087-2118. https://doi.org/10.3390/ijms15022087
Chicago/Turabian StyleItoh, Susumu, Shotaro Sonoike, Masanori Kitamura, and Shin Aoki. 2014. "Design and Synthesis of Chiral Zn2+ Complexes Mimicking Natural Aldolases for Catalytic C–C Bond Forming Reactions in Aqueous Solution" International Journal of Molecular Sciences 15, no. 2: 2087-2118. https://doi.org/10.3390/ijms15022087