KCNK10, a Tandem Pore Domain Potassium Channel, Is a Regulator of Mitotic Clonal Expansion during the Early Stage of Adipocyte Differentiation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
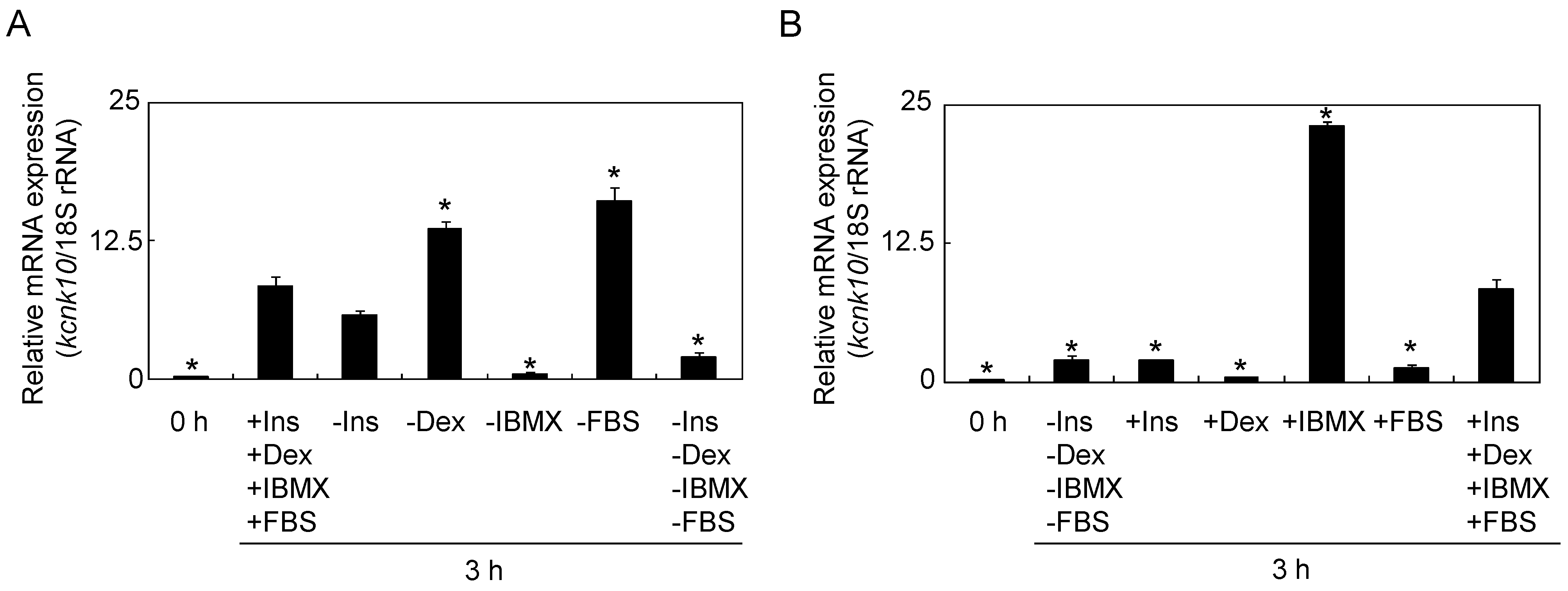
2.1.1. Effect of Adipogenic Inducers on the Expression of kcnk10
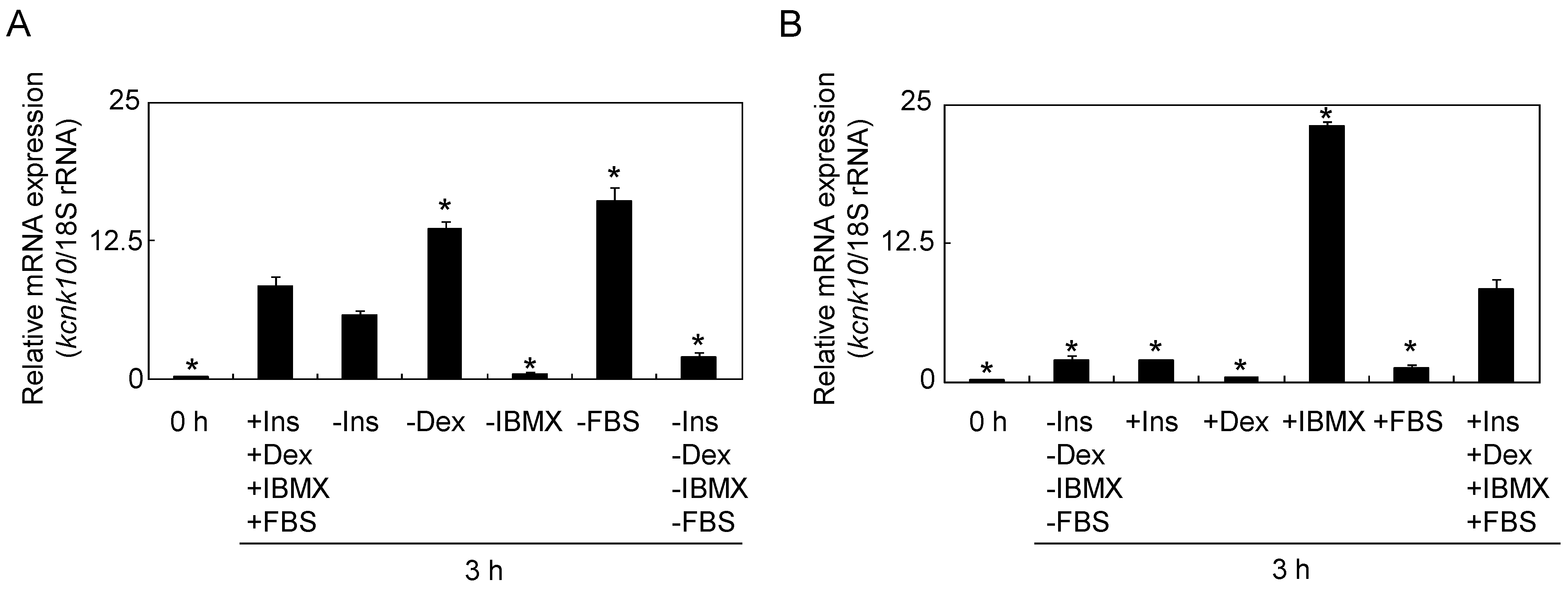
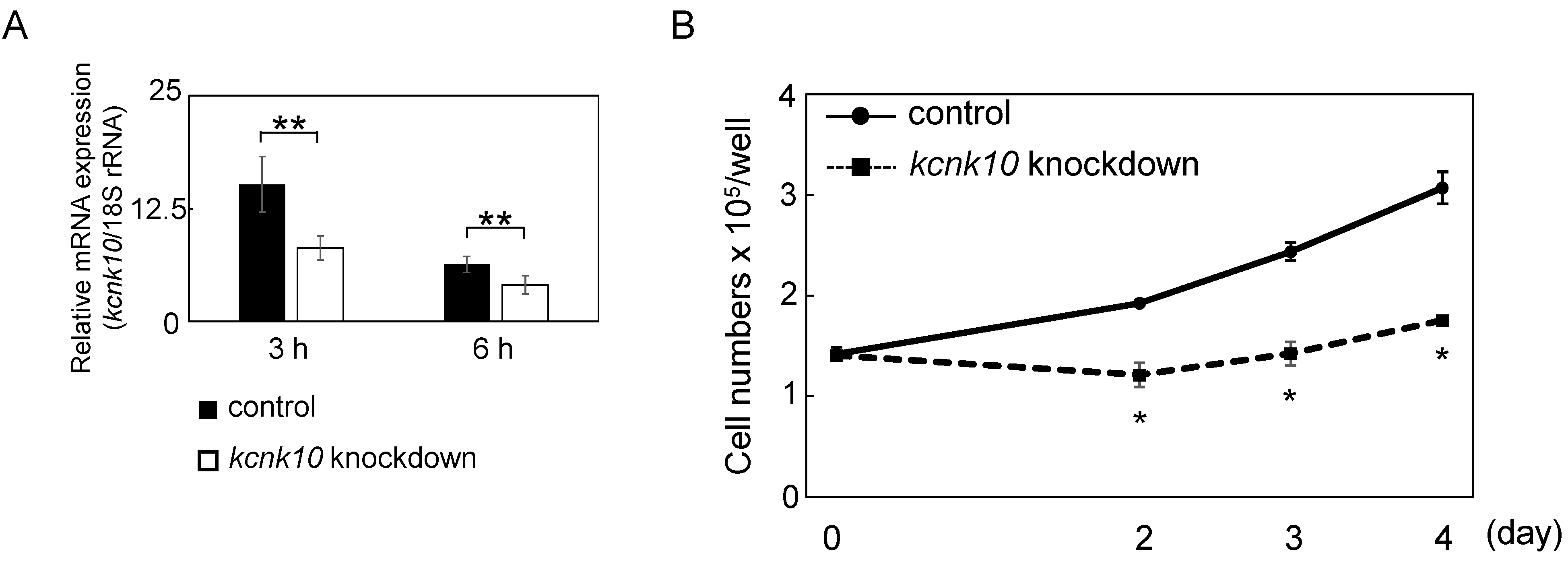
2.1.2. The Role of KCNK10 on MCE
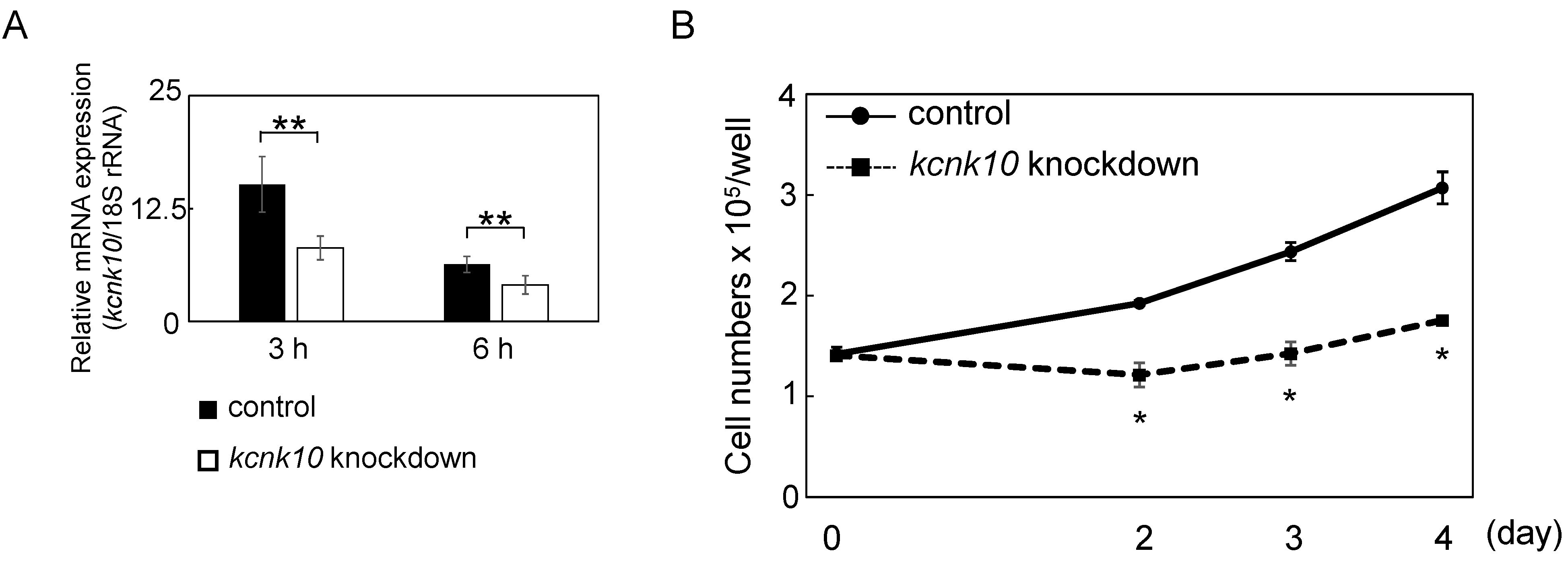
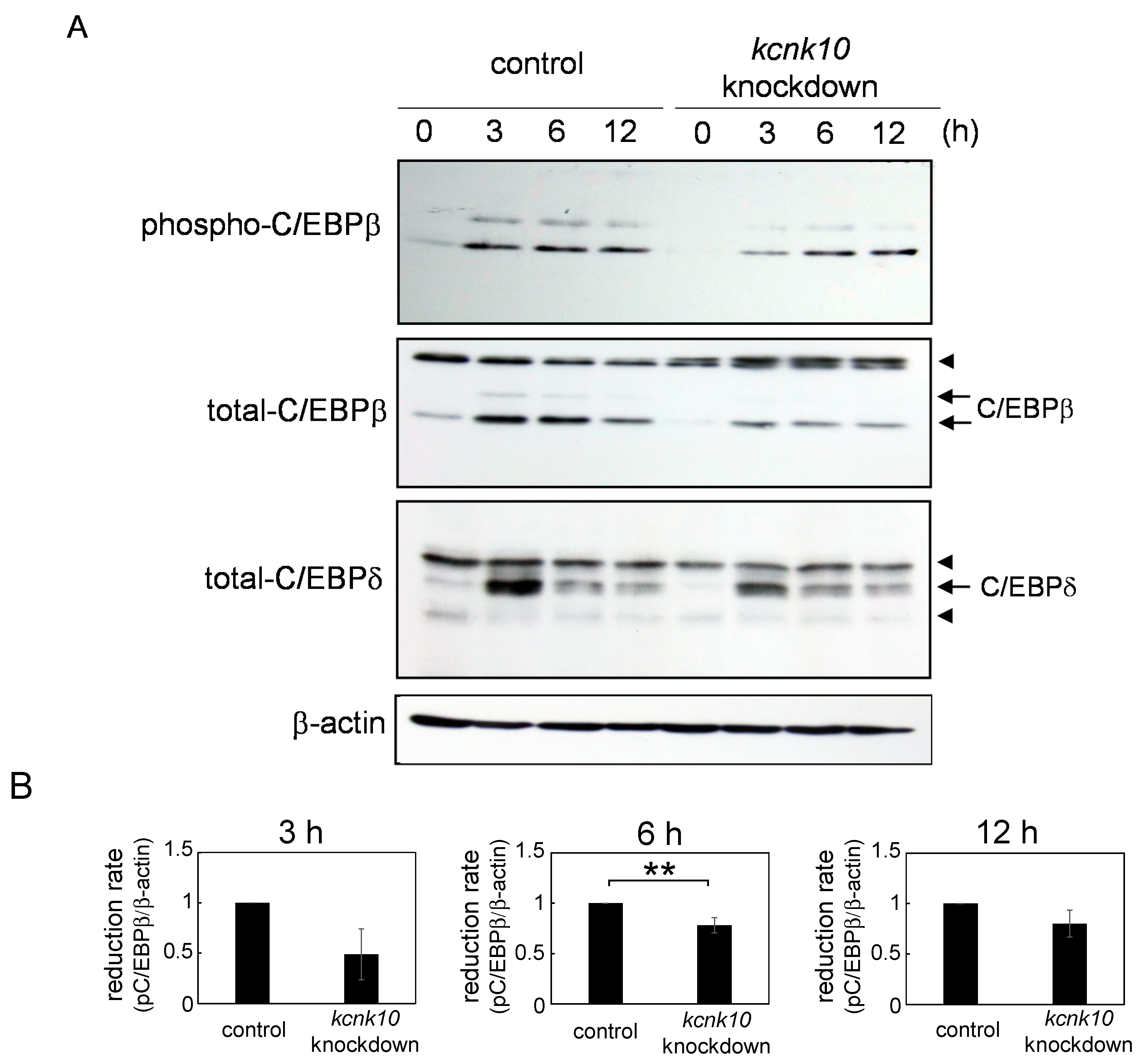
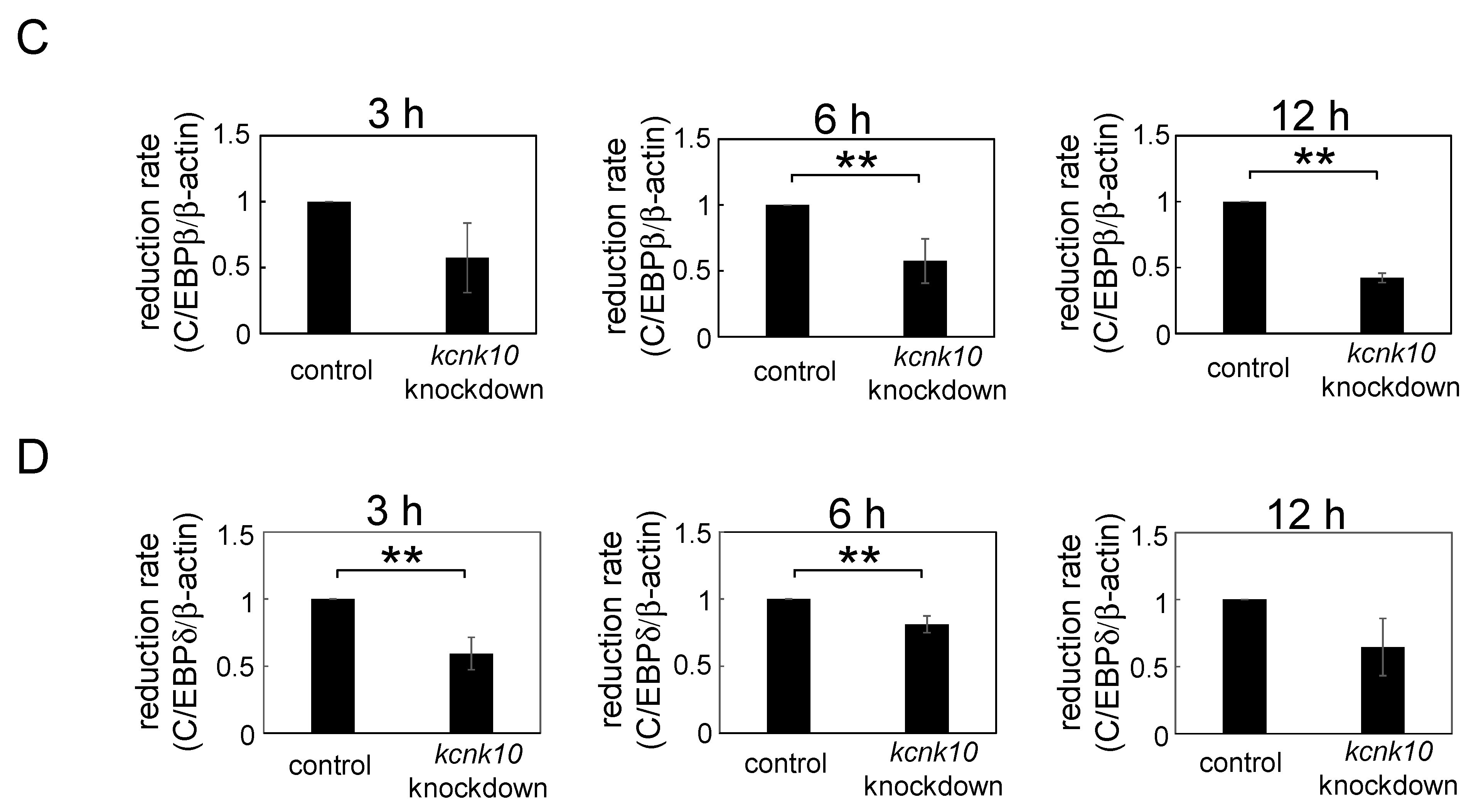
2.1.3. Effect of Knockdown of kcnk10 Expression on the Expression Level of C/EBPβ and C/EBPδ
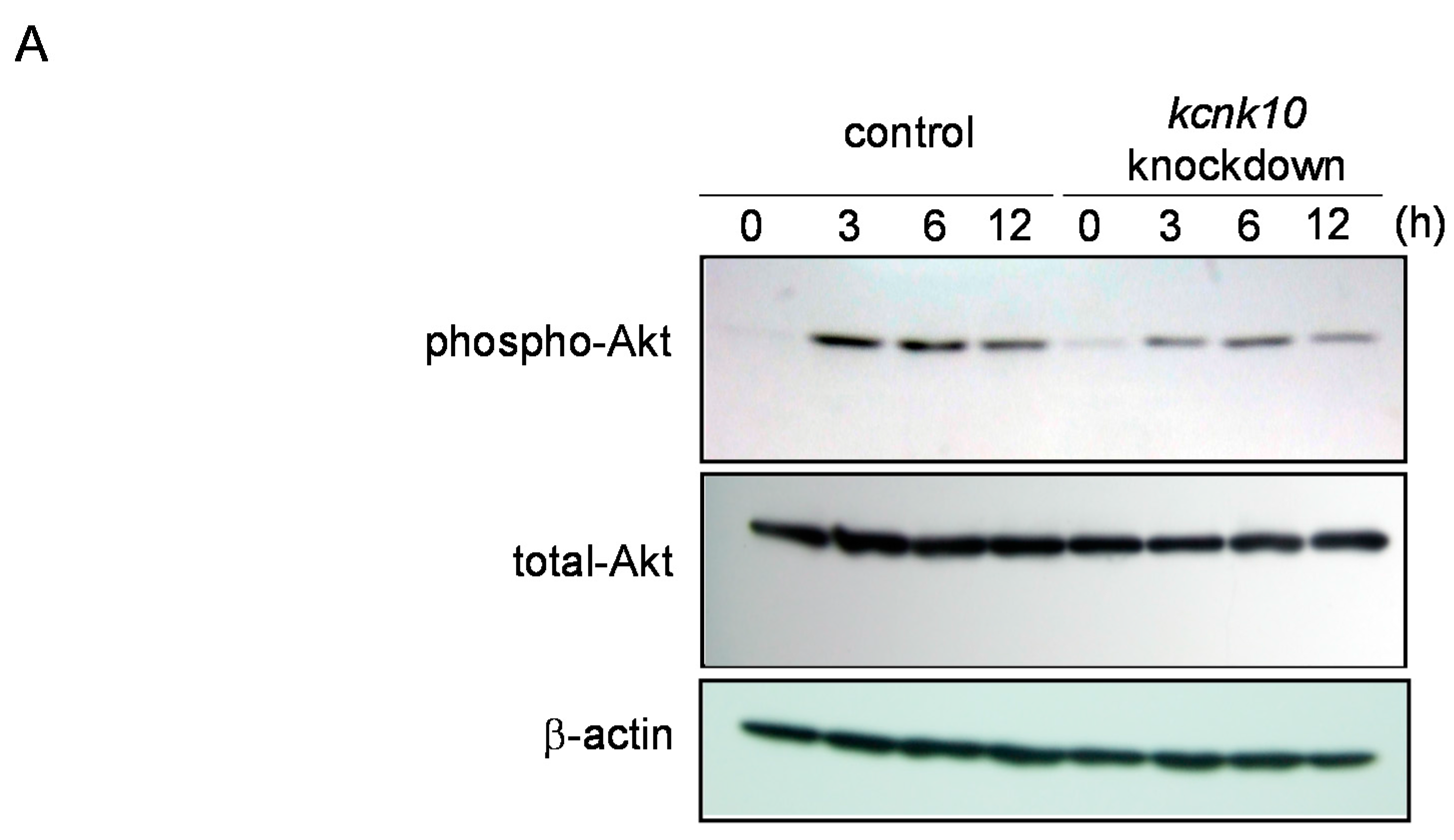
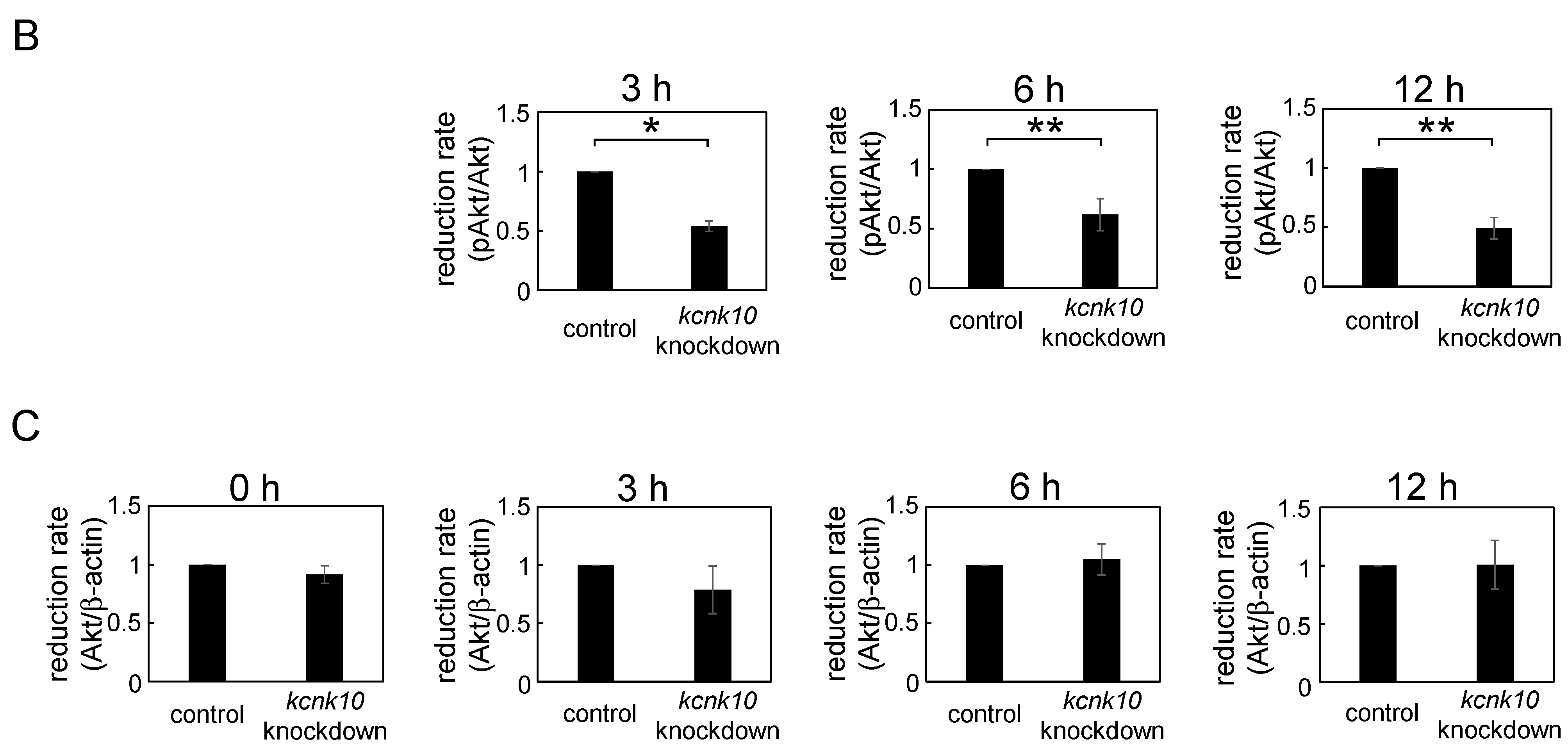
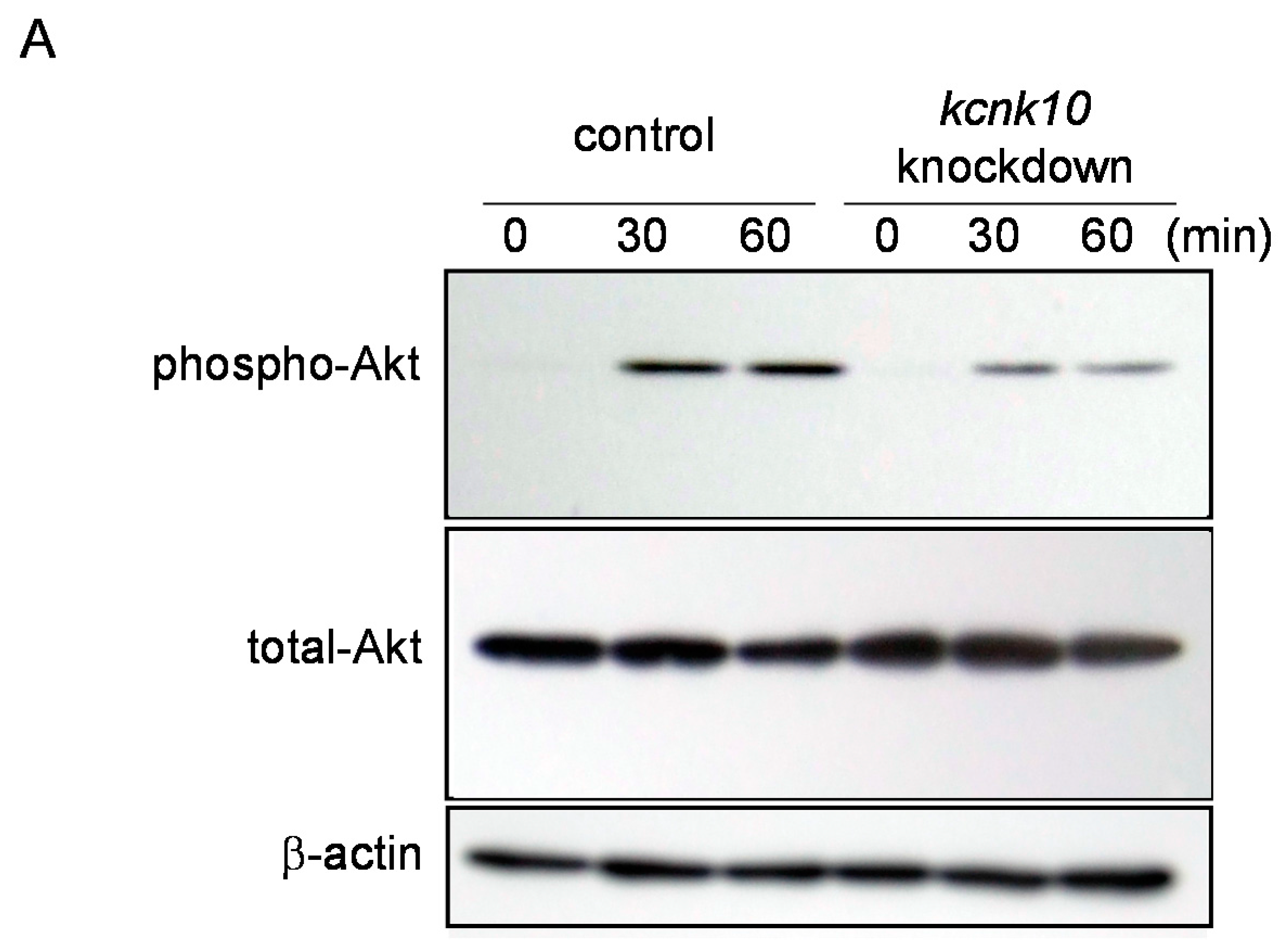
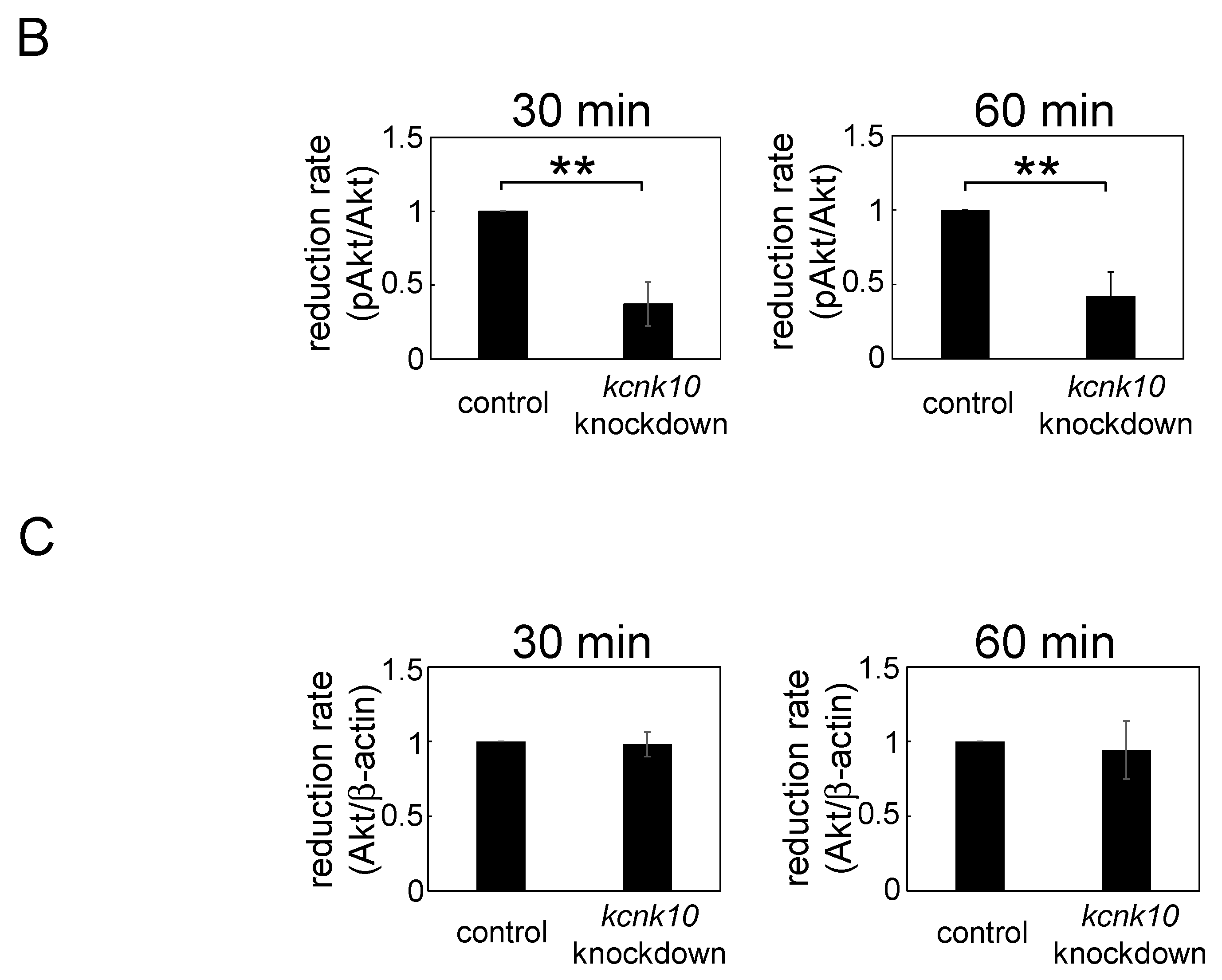
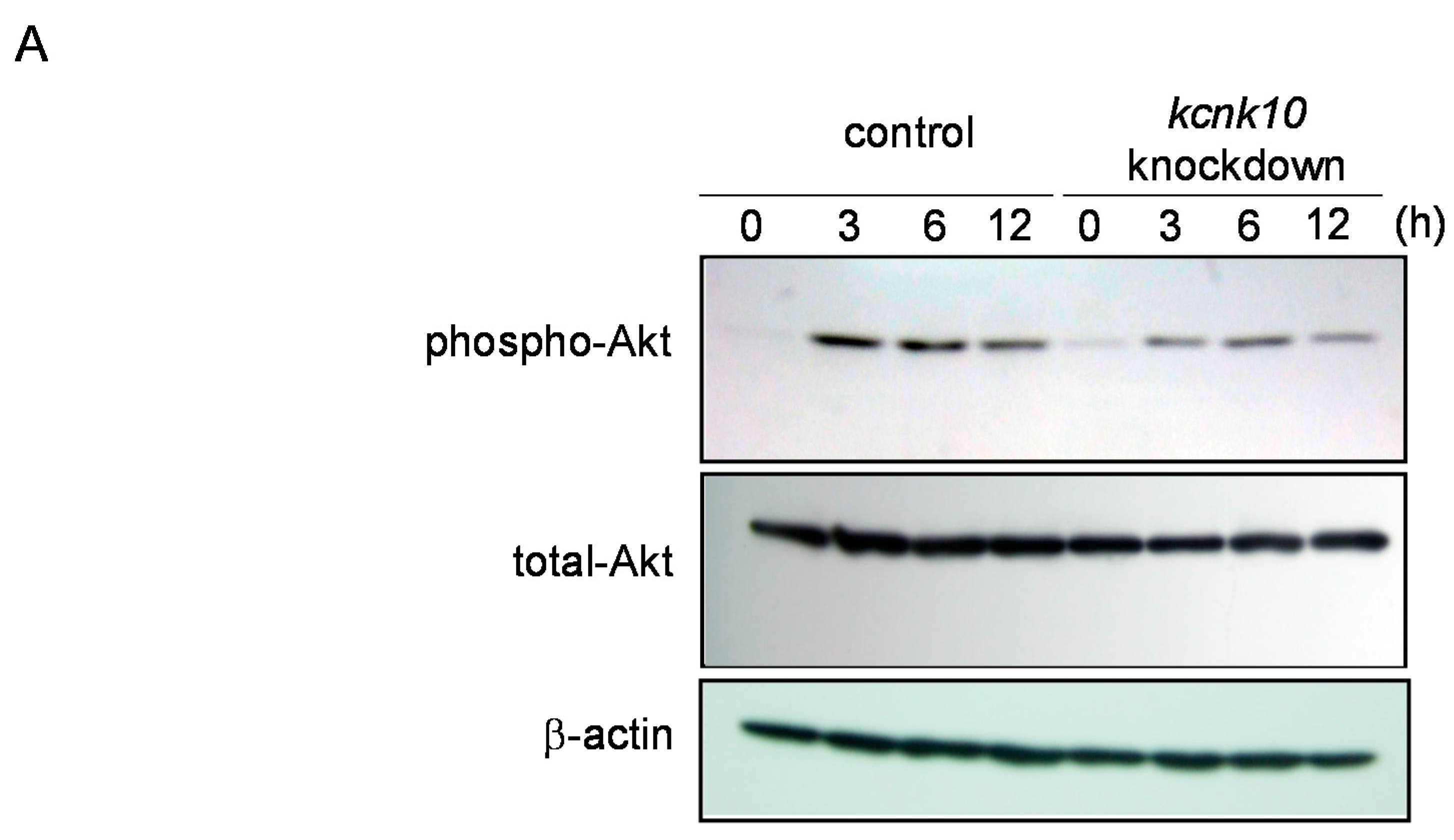
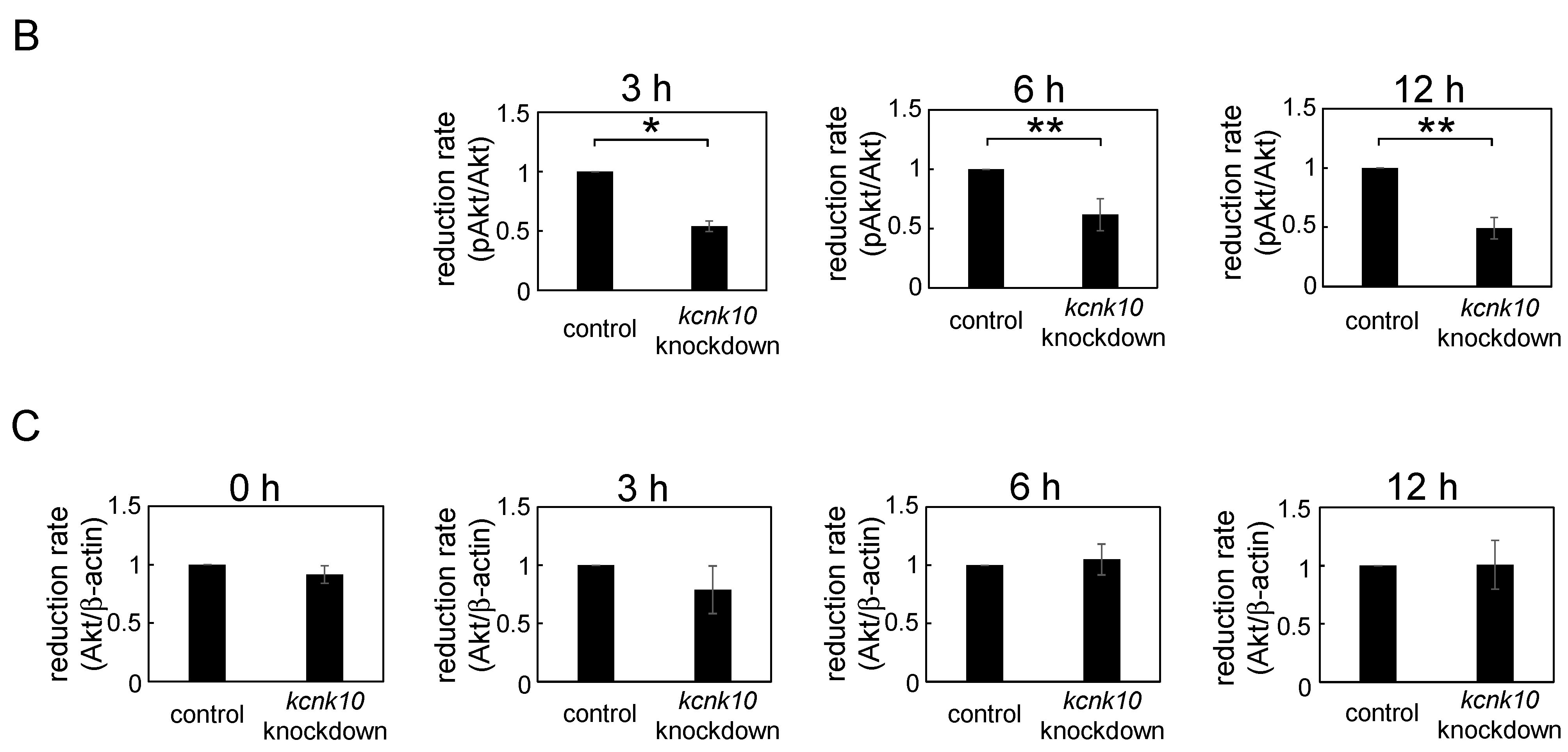
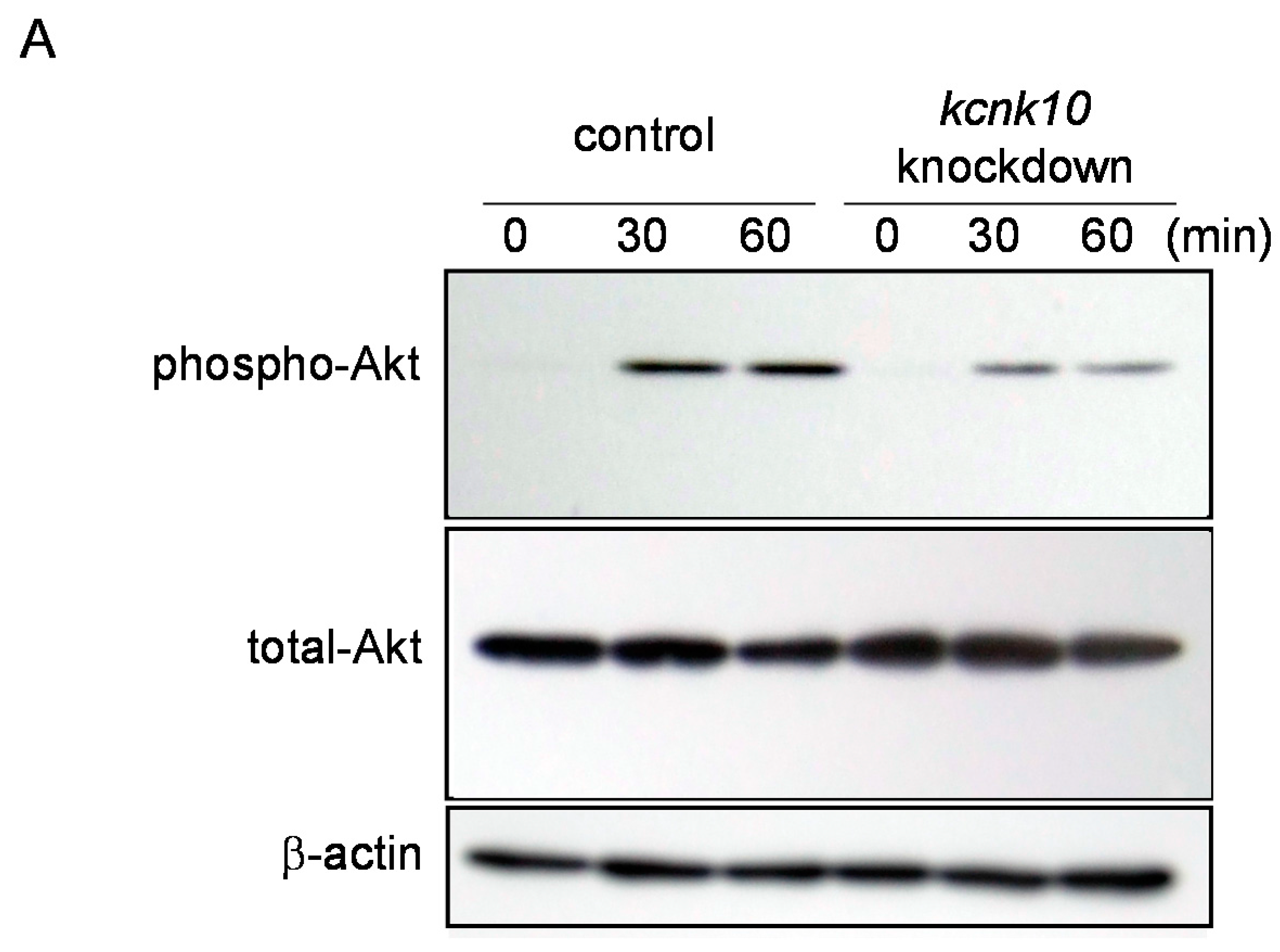
2.1.4. Effect of Knockdown of kcnk10 Expression on Insulin Signaling Pathway
2.2. Discussion
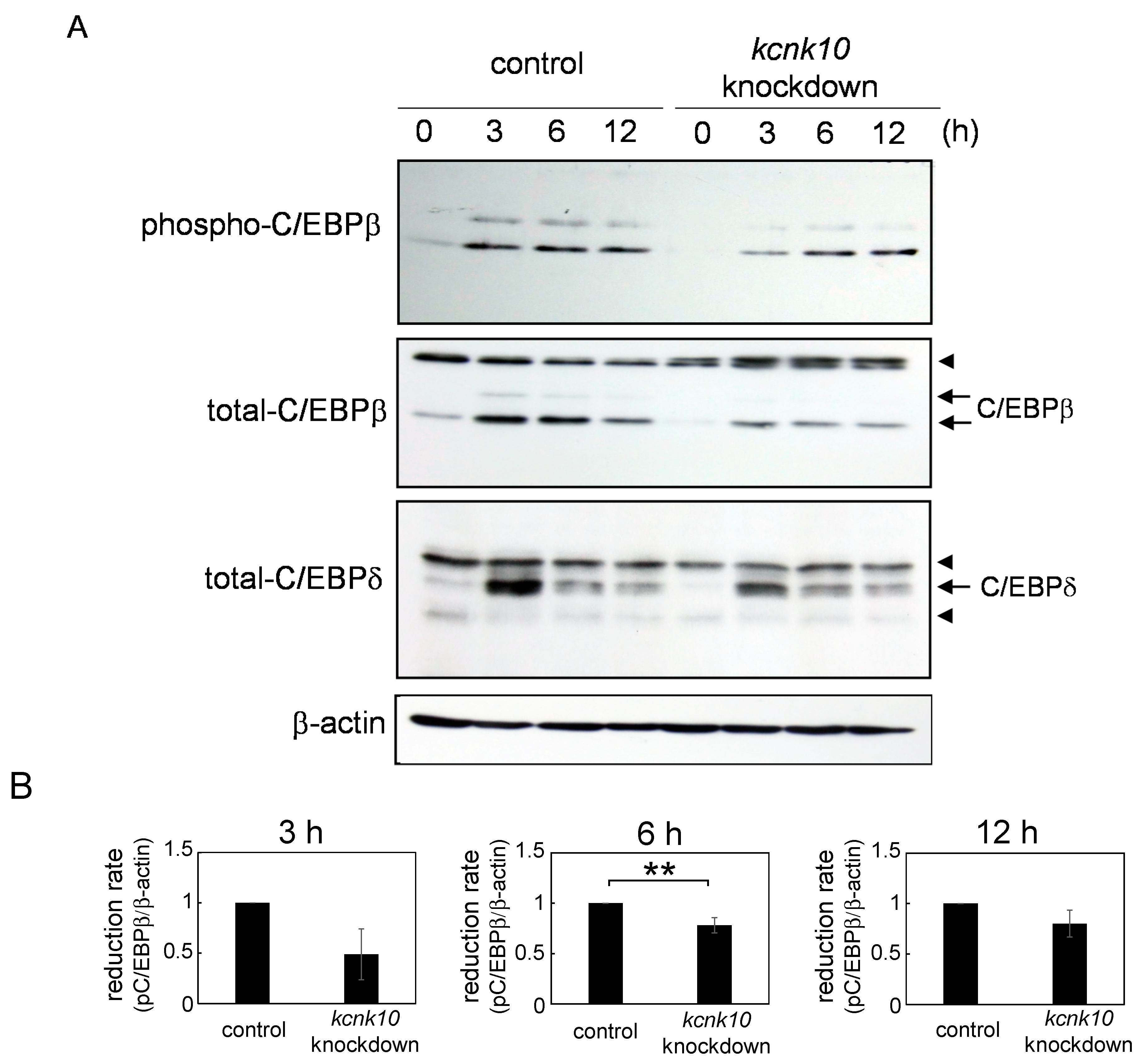
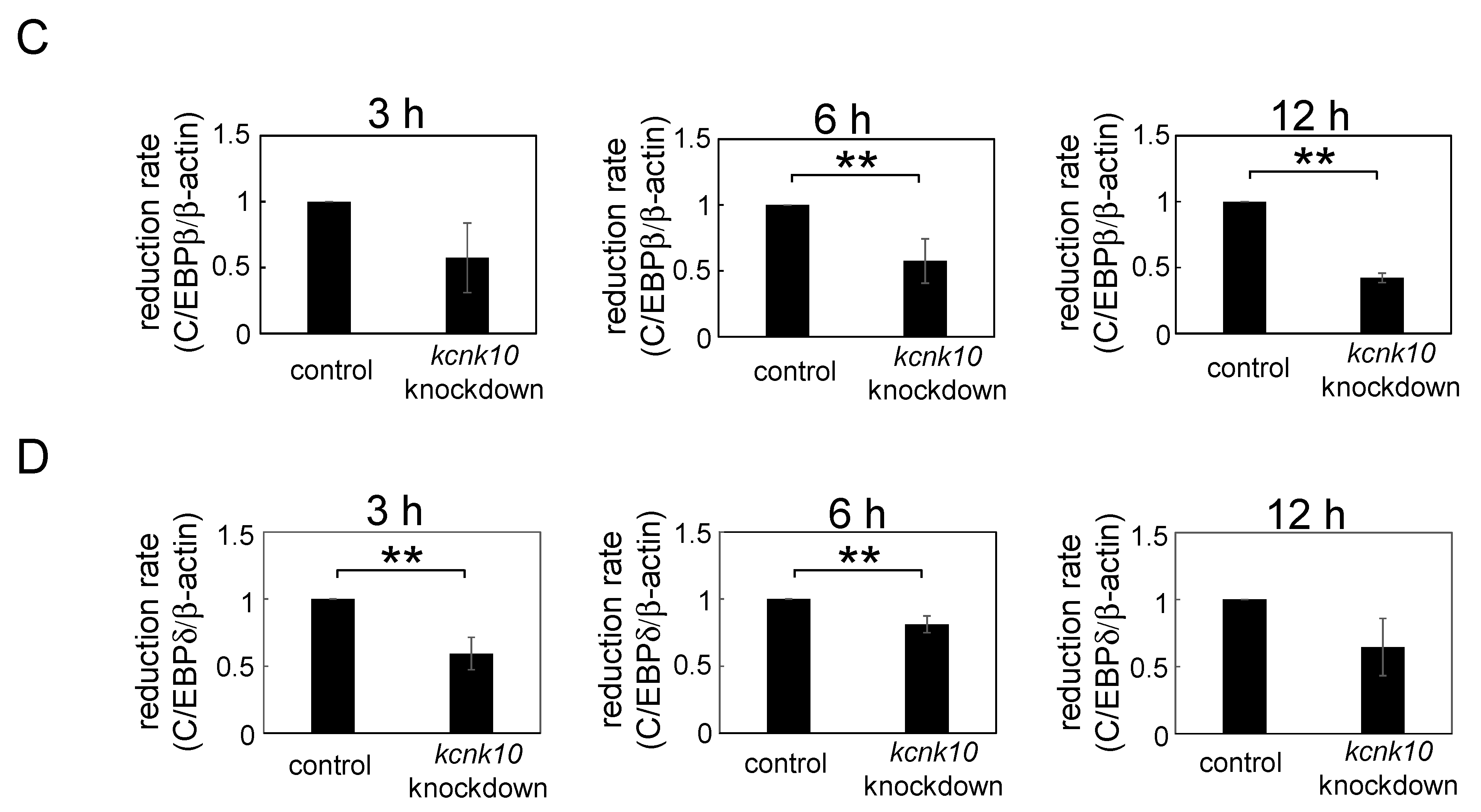

3. Experimental Section
3.1. Cell Culture and Differentiation
3.2. RNAi Experiments
3.3. Cell Counting
3.4. Q-PCR
3.5. Western Blotting
3.6. Statistical Analyses
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Enyedi, P.; Czirják, G. Molecular background of leak K+ currents: Two-pore domain potassium channels. Physiol. Rev. 2010, 90, 559–605. [Google Scholar]
- Noël, J.; Sandoz, G.; Lesage, F. Molecular regulations governing TREK and TRAAK channel functions. Channels (Austin) 2011, 5, 402–409. [Google Scholar]
- Bayliss, D.A.; Barrett, P.Q. Emerging roles for two-pore-domain potassium channels and their potential therapeutic impact. Trends Pharmacol. Sci. 2008, 29, 566–575. [Google Scholar] [CrossRef]
- Franks, N.P.; Honoré, E. The TREK K2P channels and their role in general anaesthesia and neuroprotection. Trends Pharmacol. Sci. 2004, 25, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Mathie, A.; Al-Moubarak, E.; Veale, E.L. Gating of two pore domain potassium channels. J. Physiol. 2010, 588, 3149–3156. [Google Scholar]
- Honoré, E. The neuronal background K2P channels: Focus on TREK1. Nat. Rev. Neurosci. 2007, 8, 251–261. [Google Scholar] [CrossRef]
- Bang, H.; Kim, Y.; Kim, D. TREK-2, a new member of the mechanosensitive tandem-pore K+ channel family. J. Biol. Chem. 2000, 275, 17412–17419. [Google Scholar] [CrossRef]
- Cadaveira-Mosquera, A.; Ribeiro, S.J.; Reboreda, A.; Pérez, M.; Lamas, J.A. Activation of TREK currents by the neuroprotective agent riluzole in mouse sympathetic neurons. J. Neurosci. 2011, 31, 1375–1385. [Google Scholar] [CrossRef]
- Deng, P.Y.; Xiao, Z.; Yang, C.; Rojanathammanee, L.; Grisanti, L.; Watt, J.; Geiger, J.D.; Liu, R.; Porter, J.E.; Lei, S. GABAB receptor activation inhibits neuronal excitability and spatial learning in the entorhinal cortex by activating TREK-2 K+ channels. Neuron 2009, 63, 230–243. [Google Scholar] [CrossRef]
- Sato, S.; Hasebe, H.; Sato, S.; Asahi, Y.; Hayashi, T.; Kobayashi, E.; Sugimoto, Y. High-resolution physical mapping and construction of a porcine contig spanning the intramuscular fat content QTL. Anim. Genet. 2006, 37, 113–120. [Google Scholar] [CrossRef]
- Sato, S.; Nishizuka, M.; Asano, M.; Ohtake, T.; Imagawa, M.; Kobayashi, E. RNA interference-mediated knockdown of the mouse gene encoding potassium channel subfamily K member 10 inhibits hormone-induced differentiation of 3T3-L1 preadipocytes. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2010, 157, 46–53. [Google Scholar] [CrossRef]
- Tang, Q.Q.; Otto, T.C.; Lane, M.D. CCAAT/enhancer-binding protein β is required for mitotic clonal expansion during adipogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 850–855. [Google Scholar] [CrossRef]
- Hishida, T.; Nishizuka, M.; Osada, S.; Imagawa, M. The role of C/EBPΔ in the early stages of adipogenesis. Biochimie 2009, 91, 654–657. [Google Scholar] [CrossRef]
- Descombes, P.; Schibler, U. A liver-enriched transcriptional activator protein, LAP, and a transcriptional inhibitory protein, LIP, are translated from the same mRNA. Cell 1991, 67, 569–579. [Google Scholar] [CrossRef]
- Nakae, J.; Kitamura, T.; Kitamura, Y.; Biggs, W.H., III; Arden, K.C.; Accili, D. The forkhead transcription factor Foxo1 regulates adipocyte differentiation. Dev. Cell 2003, 4, 119–129. [Google Scholar] [CrossRef]
- Auld, C.A.; Caccia, C.D.; Morrison, R.F. Hormonal induction of adipogenesis induces Skp2 expression through PI3K and MAPK pathways. J. Cell. Biochem. 2007, 100, 204–216. [Google Scholar] [CrossRef]
- Gagnon, A.; Chen, C.S.; Sorisky, A. Activation of protein kinase B and induction of adipogenesis by insulin in 3T3-L1 preadipocytes: Contribution of phosphoinositide-3,4,5-trisphosphate versus phosphoinositide-3,4-bisphosphate. Diabetes 1999, 48, 691–698. [Google Scholar] [CrossRef]
- Tang, Q.Q.; Lane, M.D. Adipogenesis: From stem cell to adipocyte. Annu. Rev. Biochem. 2012, 81, 715–736. [Google Scholar] [CrossRef]
- Auld, C.A.; Fernandes, K.M.; Morrison, R.F. Skp2-mediated p27Kip1 degradation during S/G2 phase progression of adipocyte hyperplasia. J. Cell. Physiol. 2007, 211, 101–111. [Google Scholar] [CrossRef]
- Kawaji, A.; Nishizuka, M.; Osada, S.; Imagawa, M. TC10-like/TC10βLong regulates adipogenesis by controlling mitotic clonal expansion. Biol. Pharm. Bull. 2010, 33, 404–409. [Google Scholar] [CrossRef]
- Johmura, Y.; Osada, S.; Nishizuka, M.; Imagawa, M. FAD24 acts in concert with histone acetyltransferase HBO1 to promote adipogenesis by controlling DNA replication. J. Biol. Chem. 2008, 283, 2265–2274. [Google Scholar] [CrossRef]
- Hishida, T.; Naito, K.; Osada, S.; Nishizuka, M.; Imagawa, M. Peg10, an imprinted gene, plays a crucial role in adipocyte differentiation. FEBS Lett. 2007, 581, 4272–4278. [Google Scholar] [CrossRef]
- Fox, K.E.; Fankell, D.M.; Erickson, P.F.; Majka, S.M.; Crossno, J.T., Jr.; Klemm, D.J. Depletion of cAMP-response element-binding protein/ATF1 inhibits adipogenic conversion of 3T3-L1 cells ectopically expressing CCAAT/enhancer-binding protein (C/EBP) α, C/EBPβ, or PPARγ2. J. Biol. Chem. 2006, 281, 40341–4035. [Google Scholar] [CrossRef]
- Zhang, J.W.; Klemm, D.J.; Vinson, C.; Lane, M.D. Role of CREB in transcriptional regulation of CCAAT/enhancer-binding protein β gene during adipogenesis. J. Biol. Chem. 2004, 279, 4471–4478. [Google Scholar] [CrossRef]
- MacDougald, O.A.; Cornelius, P.; Liu, R.; Lane, M.D. Insulin regulates transcription of the CCAAT/enhancer binding protein (C/EBP) α, β and δ genes in fully-differentiated 3T3-L1 adipocytes. J. Biol. Chem. 1995, 270, 647–654. [Google Scholar] [CrossRef]
- Sundelacruz, S.; Levin, M.; Kaplan, D.L. Membrane potential controls adipogenic and osteogenic differentiation of mesenchymal stem cells. PLoS One 2008, 3, e3737. [Google Scholar] [CrossRef]
- Chatterjee, S.; Browning, E.A.; Hong, N.; de Bolt, K.; Sorokina, E.M.; Liu, W.; Birnbaum, M.J.; Fisher, A.B. Membrane depolarization is the trigger for PI3K/Akt activation and leads to the generation of ROS. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H105–H114. [Google Scholar] [CrossRef]
- Lauritzen, I.; Chemin, J.; Honoré, E.; Jodar, M.; Guy, N.; Lazdunski, M.; Patel, A.J. Cross-talk between the mechano-gated K2P channel TREK-1 and the actin cytoskeleton. EMBO Rep. 2005, 6, 642–648. [Google Scholar] [CrossRef]
- Nobusue, H.; Onishi, N.; Shimizu, T.; Sugihara, E.; Oki, Y.; Sumikawa, Y.; Chiyoda, T.; Akashi, K.; Saya, H.; Kano, K. Regulation of MKL1 via actin cytoskeleton dynamics drives adipocyte differentiation. Nat. Commun. 2014, 5, 3368. [Google Scholar] [CrossRef]
- Petersen, R.K.; Madsen, L.; Pedersen, L.M.; Hallenborg, P.; Hagland, H.; Viste, K.S.; Døskeland, O.; Kristiansen, K. Cyclic AMP (cAMP)-mediated stimulation of adipocyte differentiation requires the synergistic action of Epac- and cAMP-dependent protein kinase-dependent processes. Mol. Cell. Biol. 2008, 28, 3804–3816. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Yue, J.; Che, H.; Sun, H.Y.; Tse, H.F.; Li, G.R. BKCa and hEag1 channels regulate cell proliferation and differentiation in human bone marrow-derived mesenchymal stem cells. J. Cell. Physiol. 2014, 229, 202–212. [Google Scholar] [CrossRef]
- You, M.H.; Song, M.S.; Lee, S.K.; Ryu, P.D.; Lee, S.Y.; Kim, D.Y. Voltage-gated K+ channels in adipogenic differentiation of bone marrow-derived human mesenchymal stem cells. Acta Pharmacol. Sin. 2013, 34, 129–136. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nishizuka, M.; Hayashi, T.; Asano, M.; Osada, S.; Imagawa, M. KCNK10, a Tandem Pore Domain Potassium Channel, Is a Regulator of Mitotic Clonal Expansion during the Early Stage of Adipocyte Differentiation. Int. J. Mol. Sci. 2014, 15, 22743-22756. https://doi.org/10.3390/ijms151222743
Nishizuka M, Hayashi T, Asano M, Osada S, Imagawa M. KCNK10, a Tandem Pore Domain Potassium Channel, Is a Regulator of Mitotic Clonal Expansion during the Early Stage of Adipocyte Differentiation. International Journal of Molecular Sciences. 2014; 15(12):22743-22756. https://doi.org/10.3390/ijms151222743
Chicago/Turabian StyleNishizuka, Makoto, Takahiro Hayashi, Mami Asano, Shigehiro Osada, and Masayoshi Imagawa. 2014. "KCNK10, a Tandem Pore Domain Potassium Channel, Is a Regulator of Mitotic Clonal Expansion during the Early Stage of Adipocyte Differentiation" International Journal of Molecular Sciences 15, no. 12: 22743-22756. https://doi.org/10.3390/ijms151222743