Amelioration of Mitochondrial Dysfunction-Induced Insulin Resistance in Differentiated 3T3-L1 Adipocytes via Inhibition of NF-κB Pathways

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
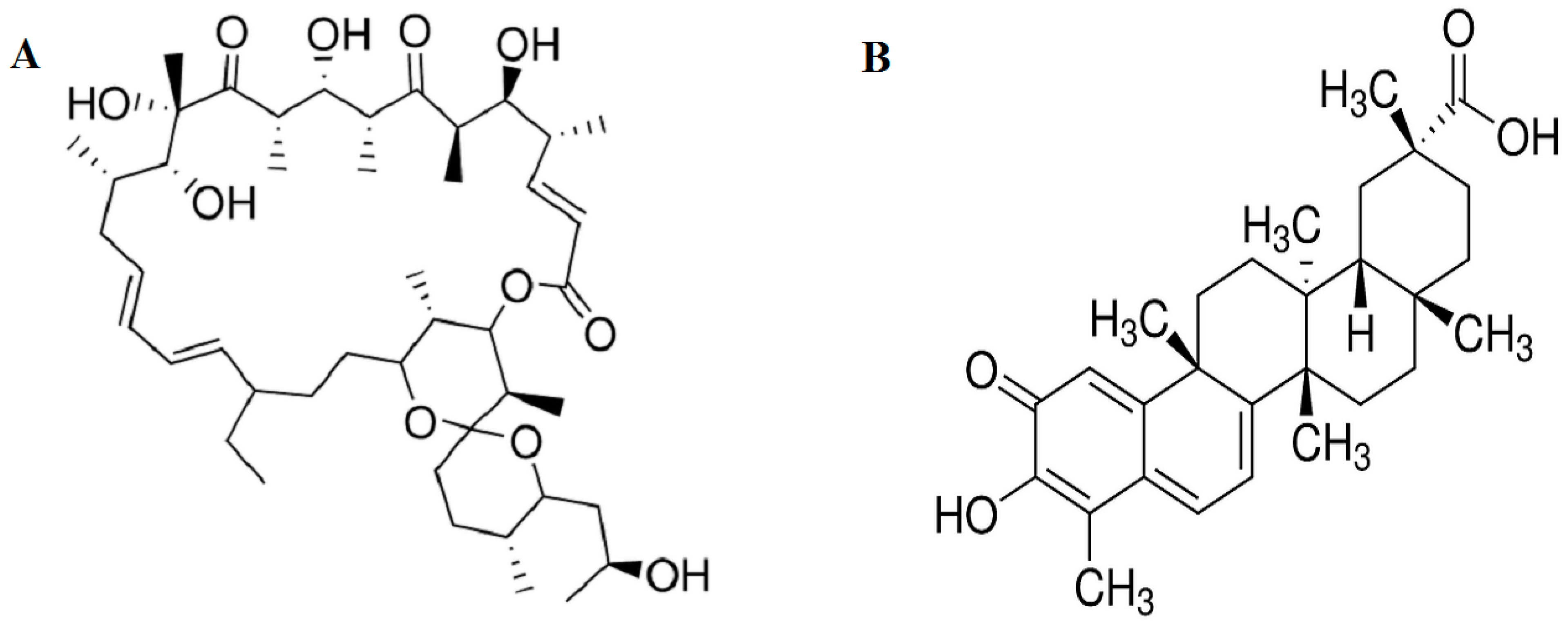
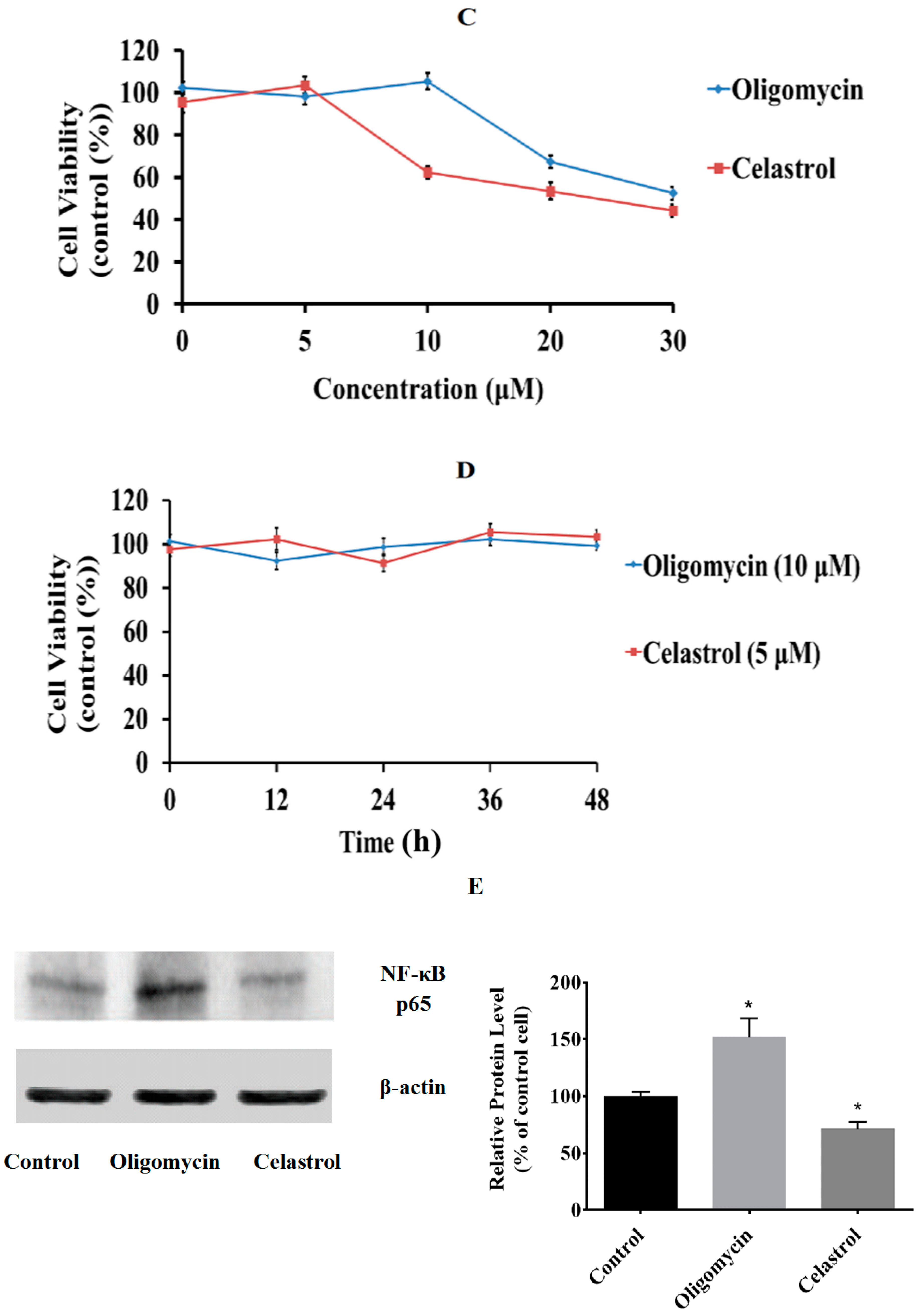
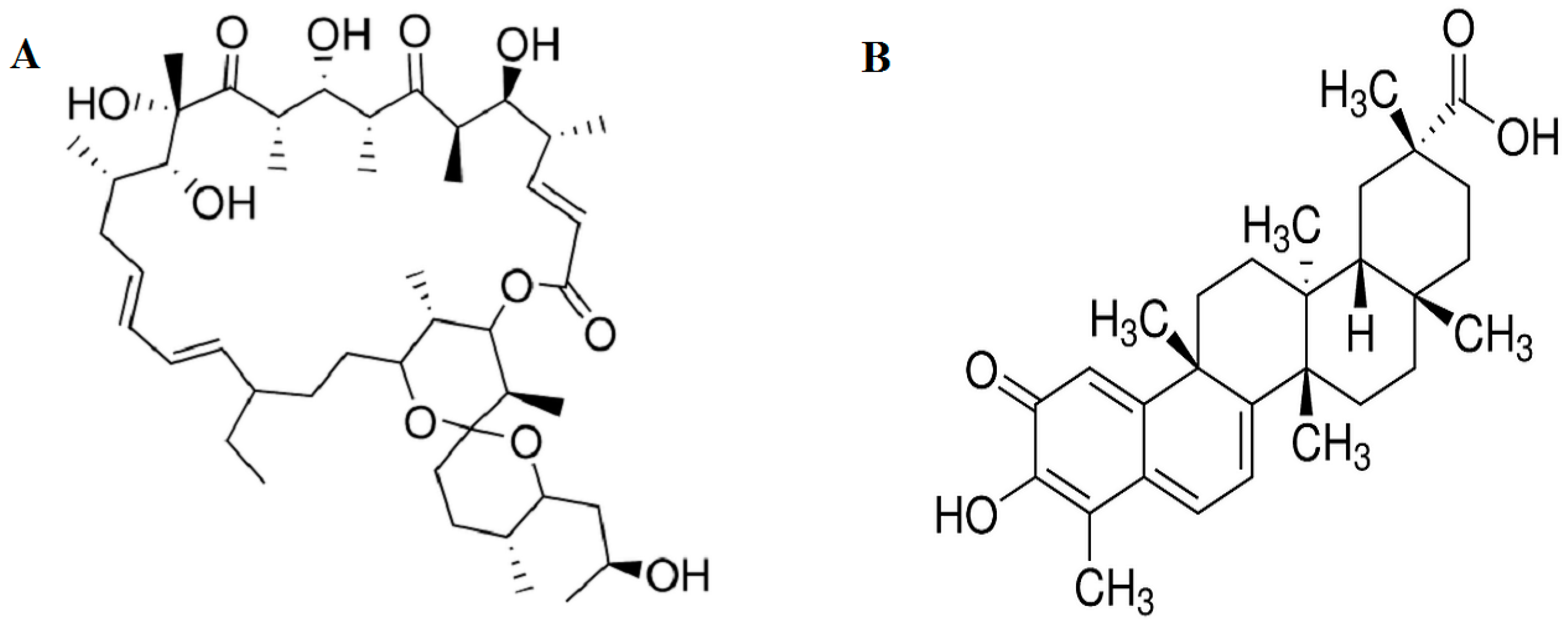
2.1. Effect of Oligomycin and Celastrol on the Cell Viability and NF-κB Expression of 3T3-L1 Adipocytes

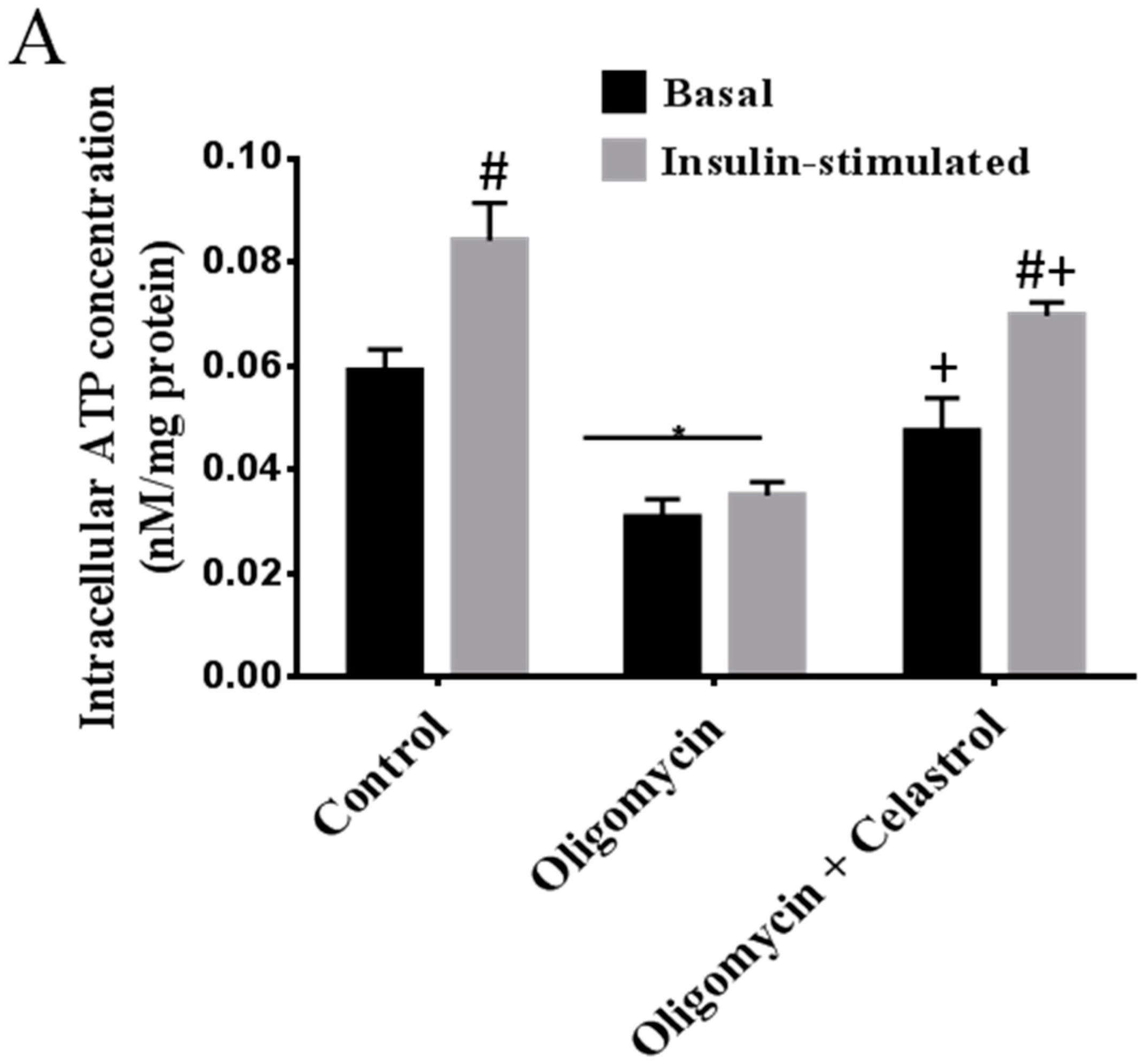
2.2. NF-κB Inhibitor Recues 3T3-L1 Adipocytes from Oligomycin Induced Mitochondrial Dysfunction

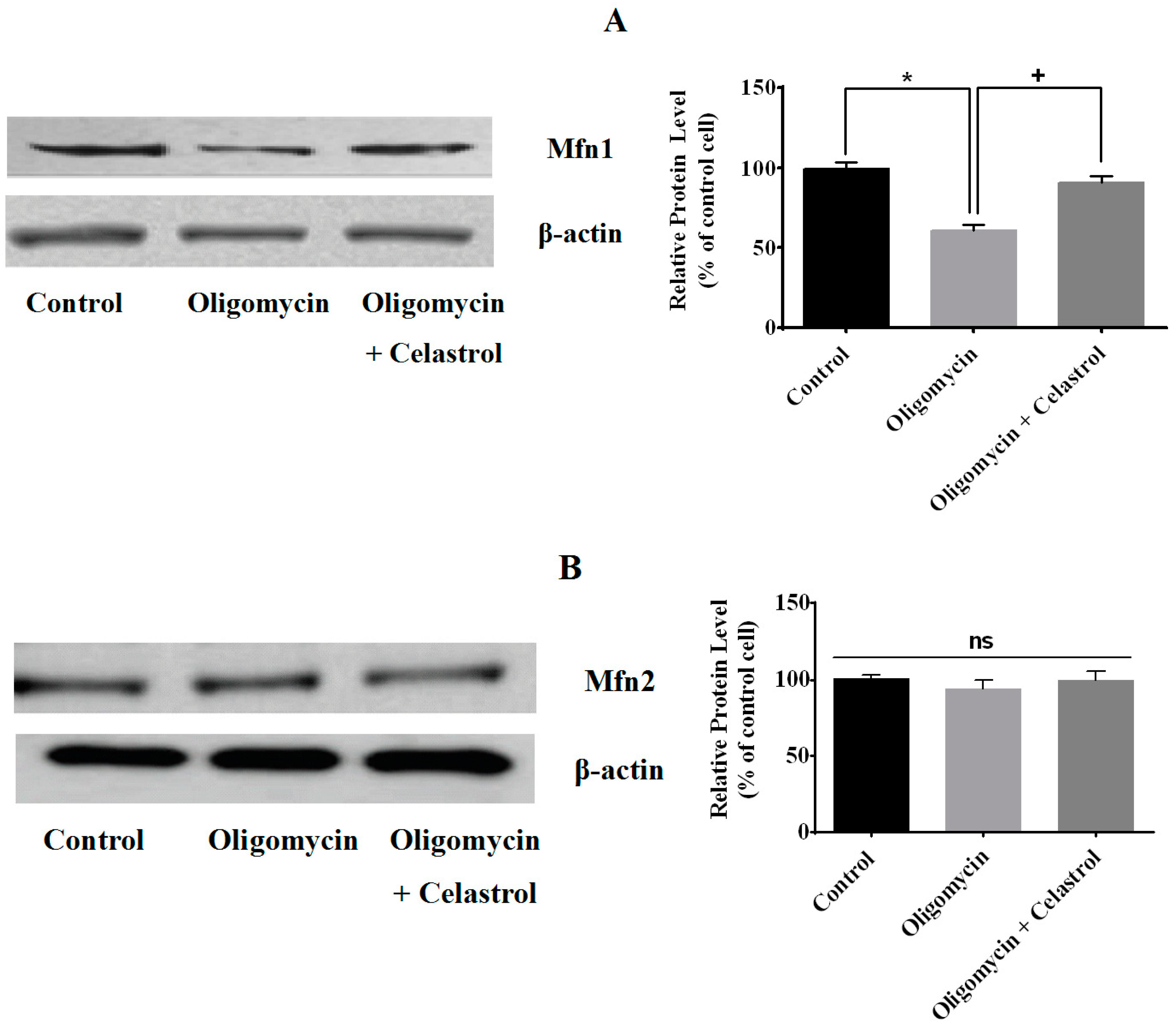
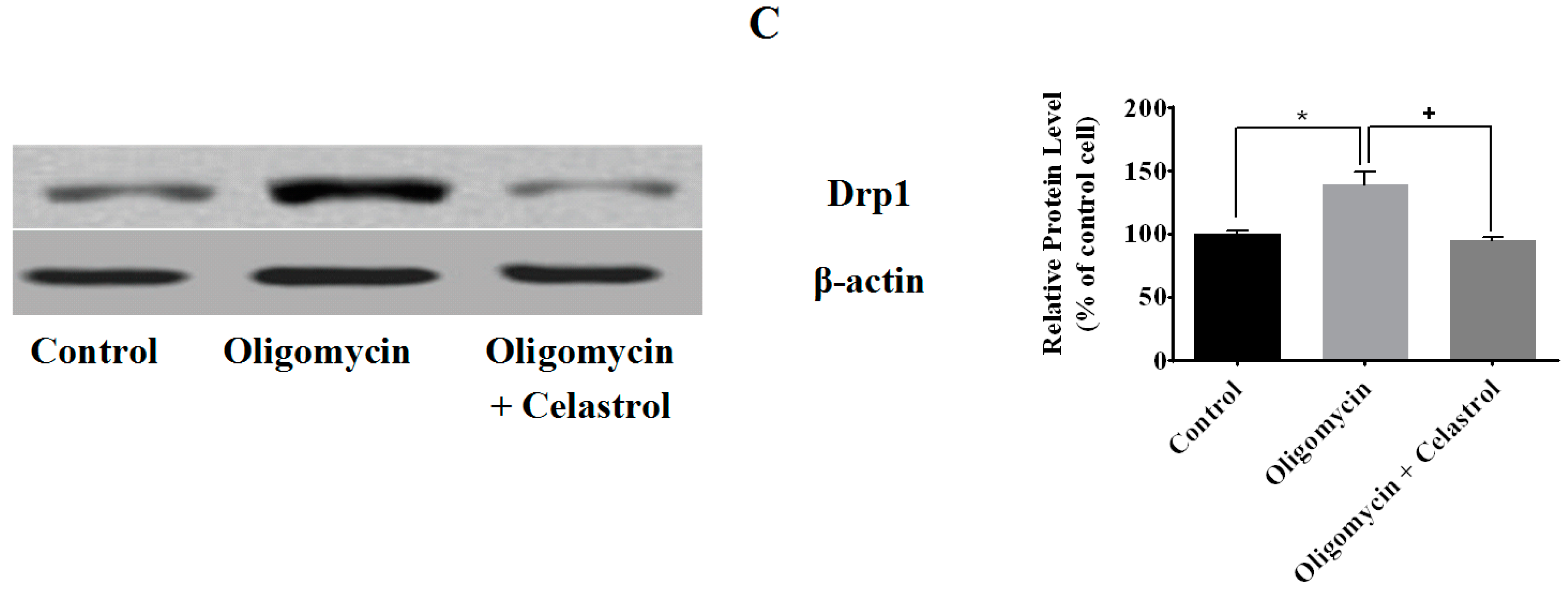
2.3. Effect of Celastrol on Mitochondrial Dynamics of 3T3-L1 Adipocytes with Mitochondrial Dysfunction
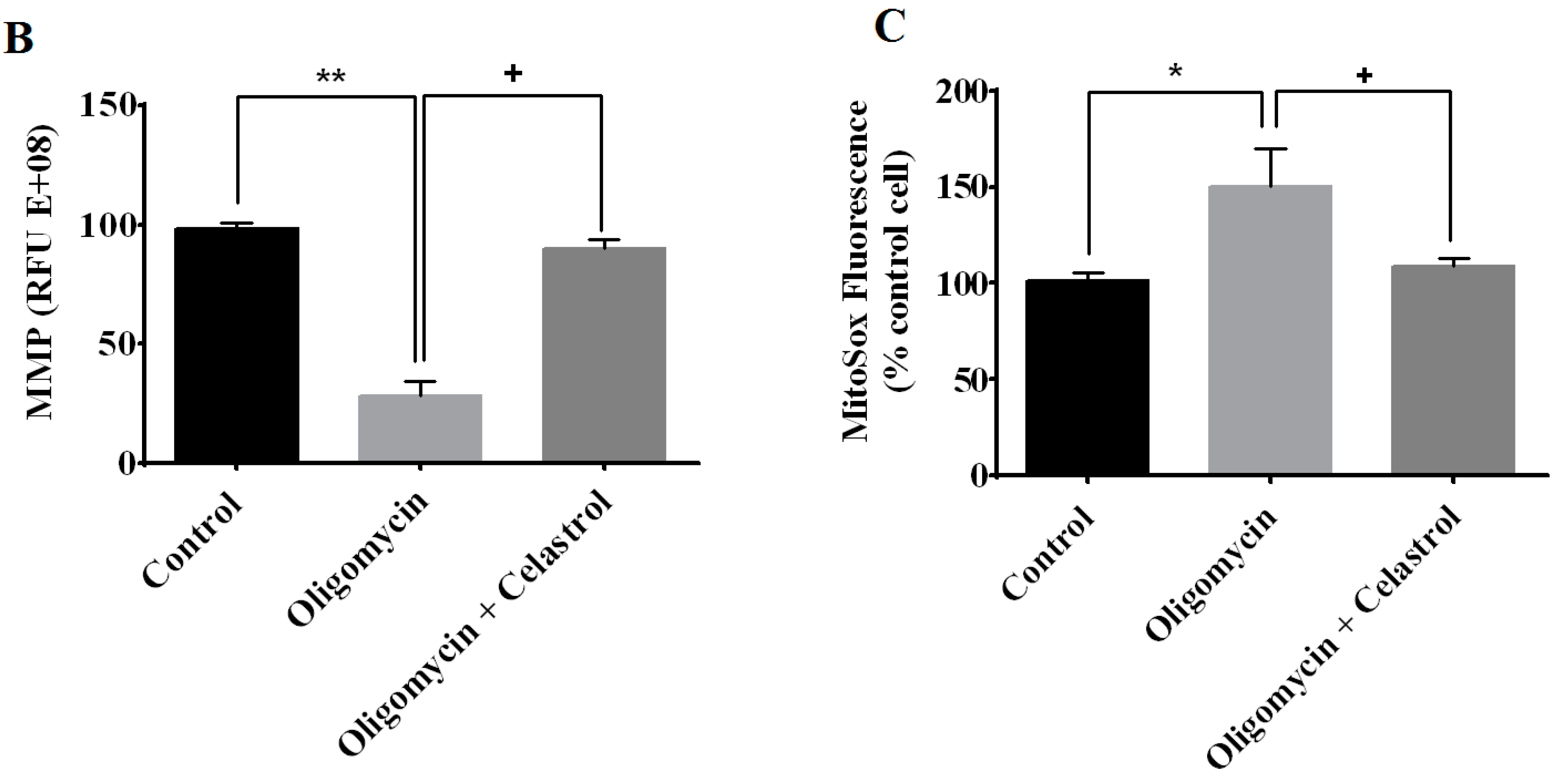
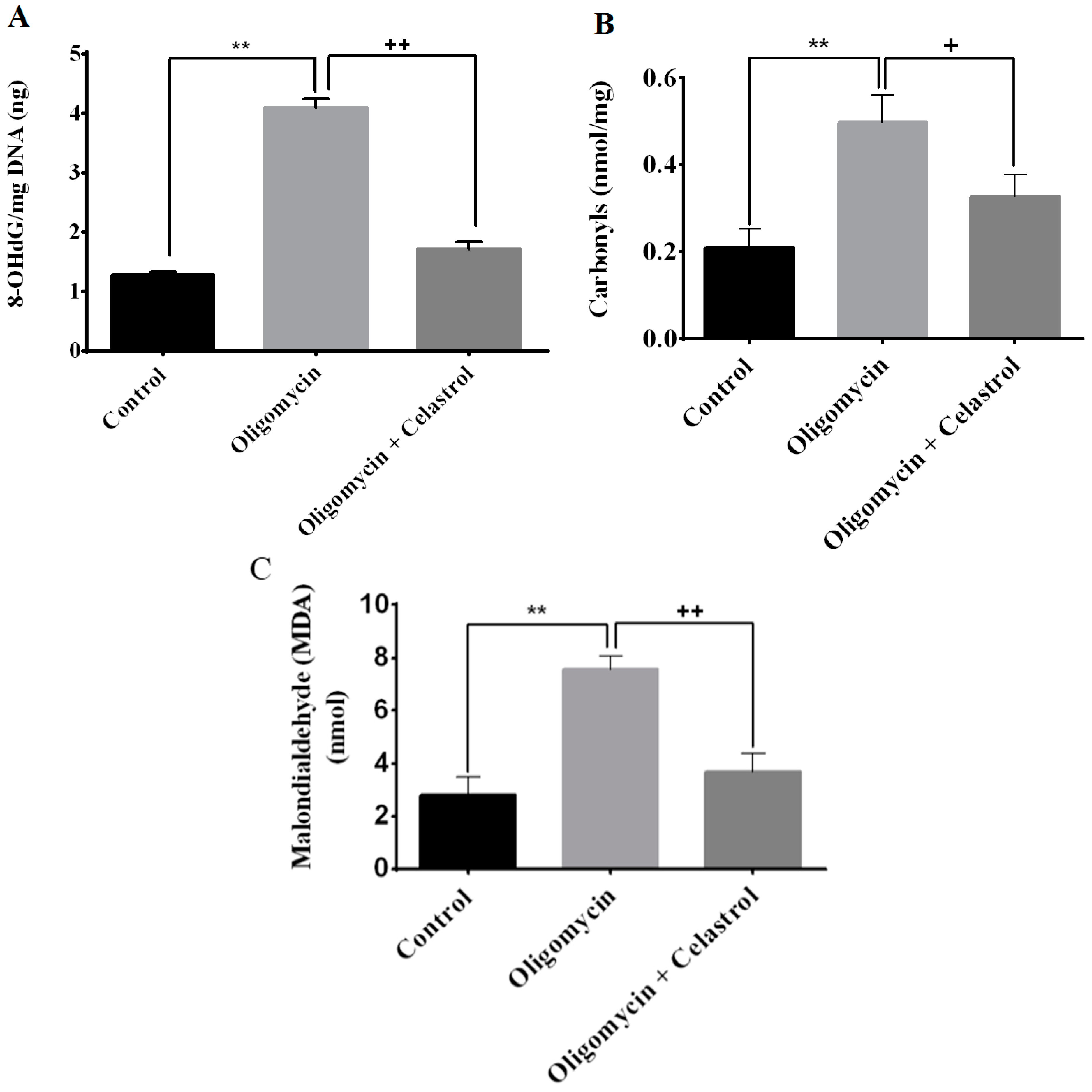
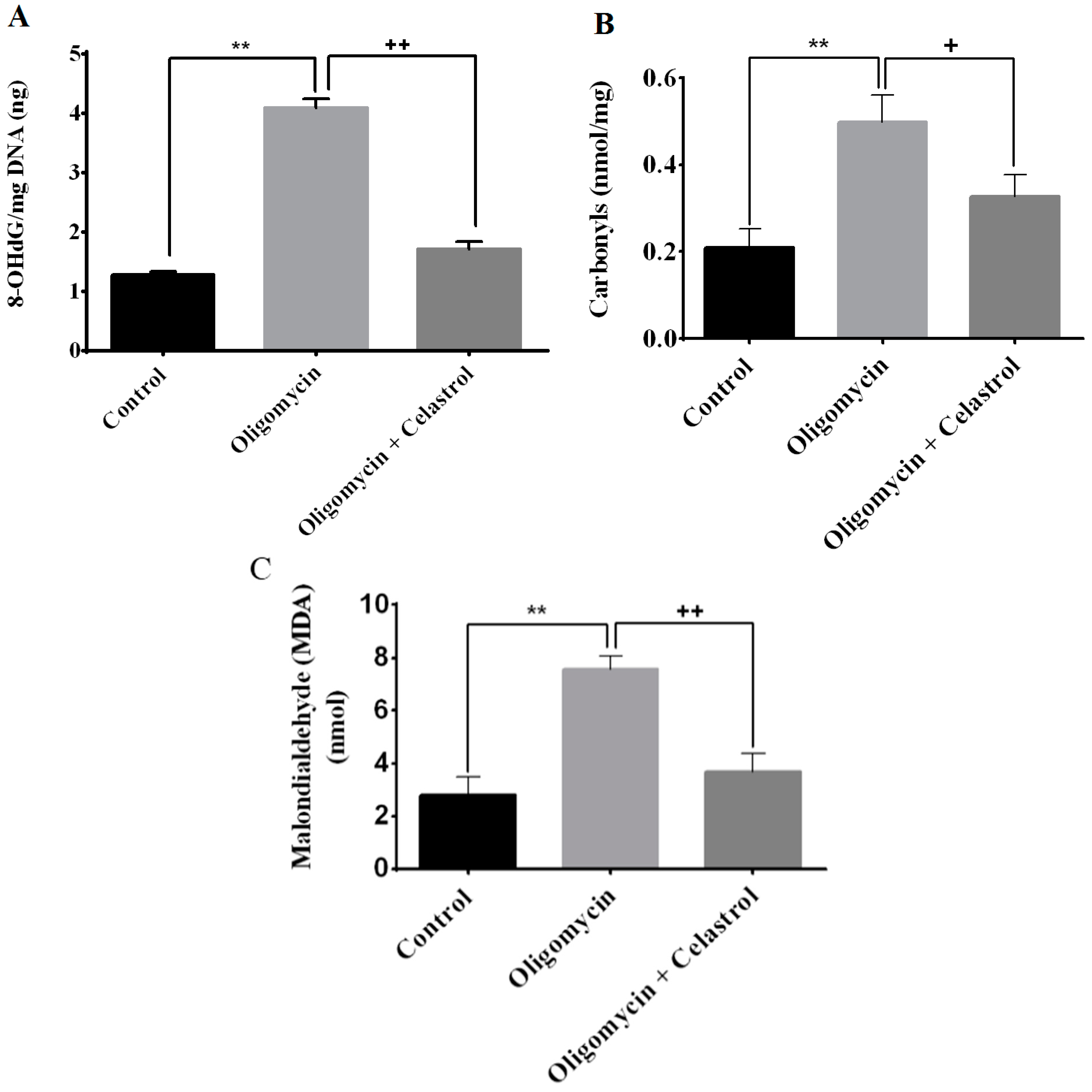
2.4. Effect of Celastrol on Impairment of Mitochondrial Respiratory Chain Elicited Oxidative Stress in 3T3-L1 Adipocytes


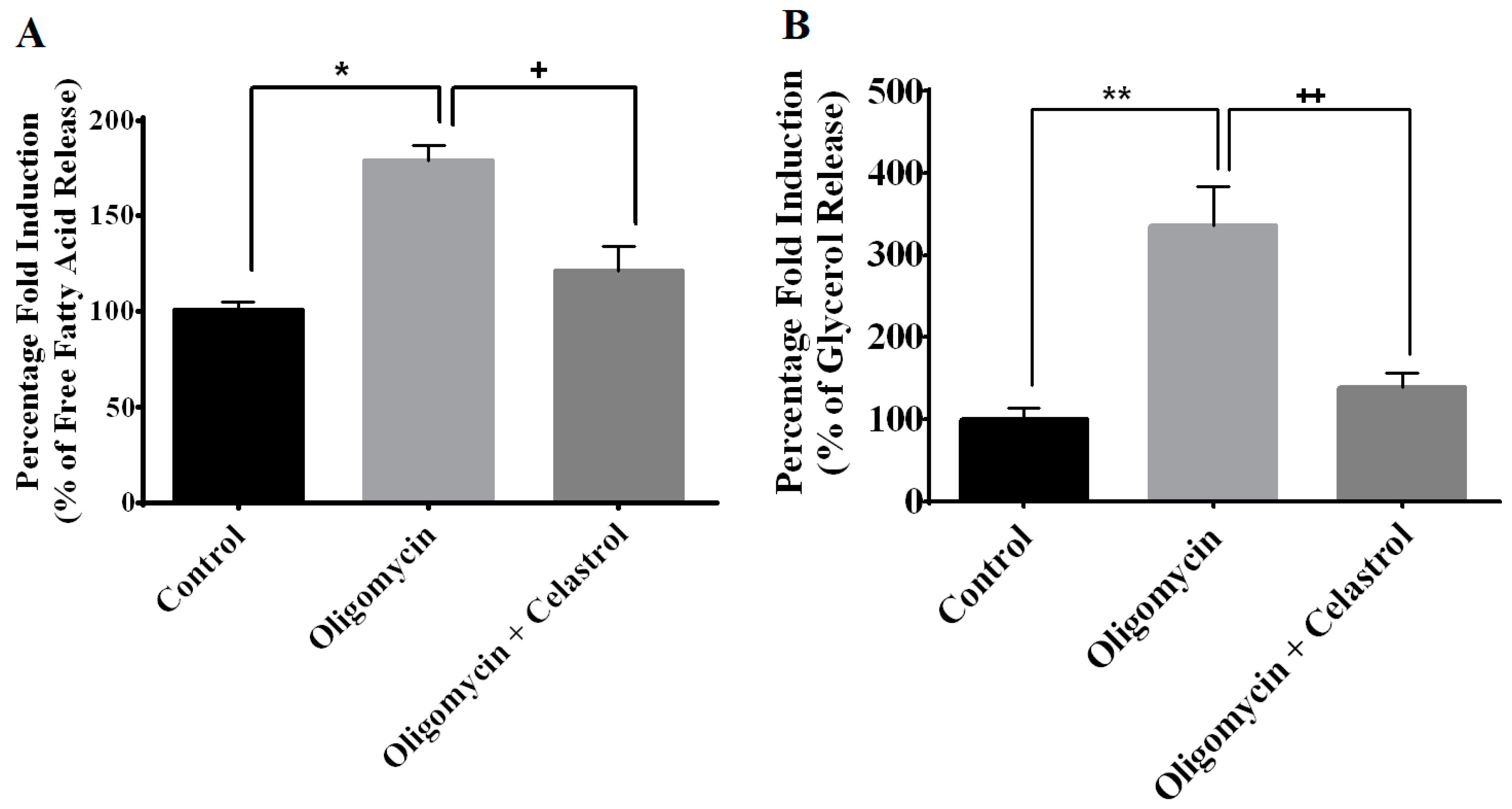
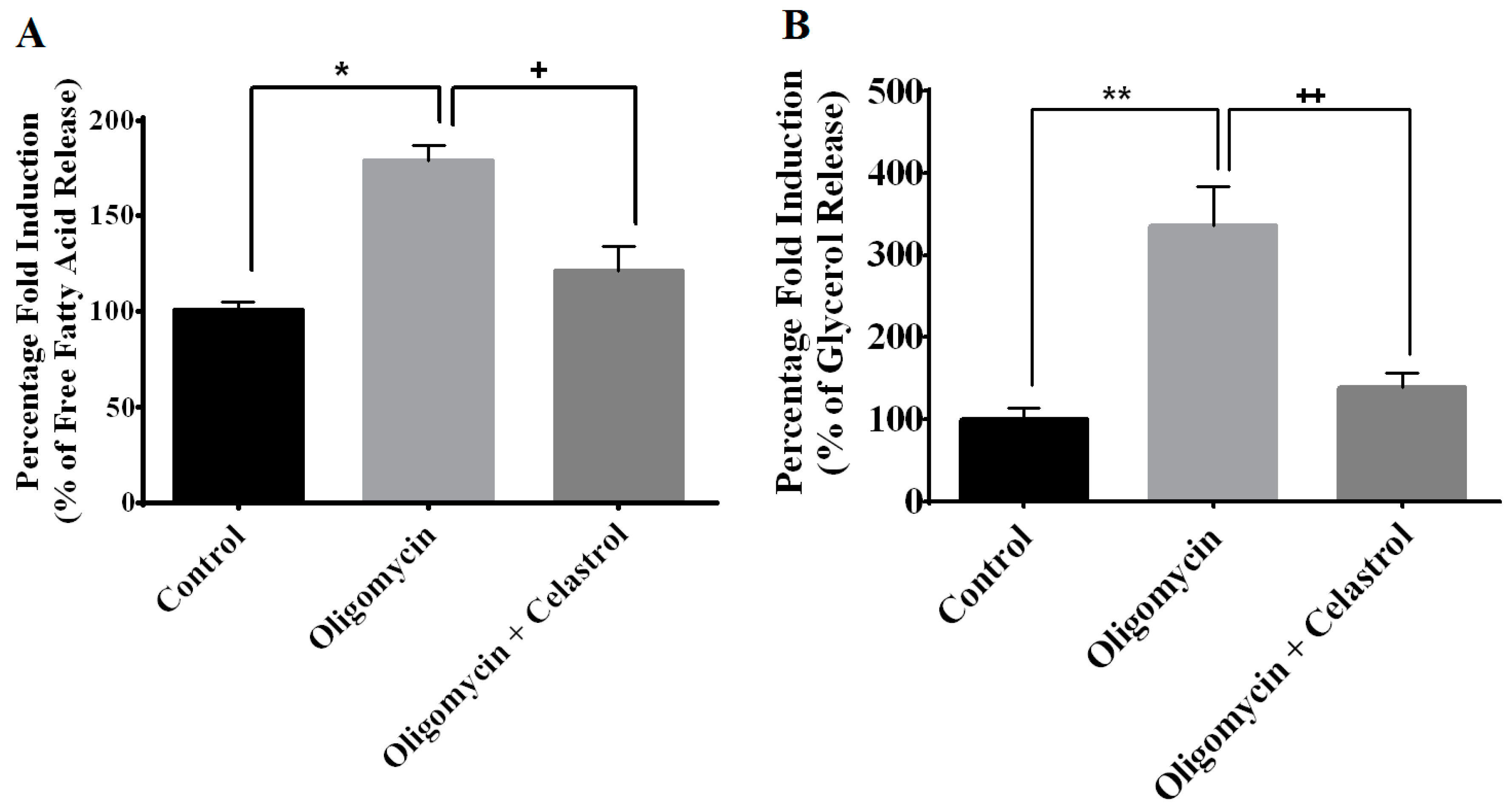
2.5. NF-κB Inhibitor Reduces Lipolysis and Alleviates Accelerated Intracellular Lpid Accumulation in 3T3-L1 Adipocytes with Mitochondria Dysfunction
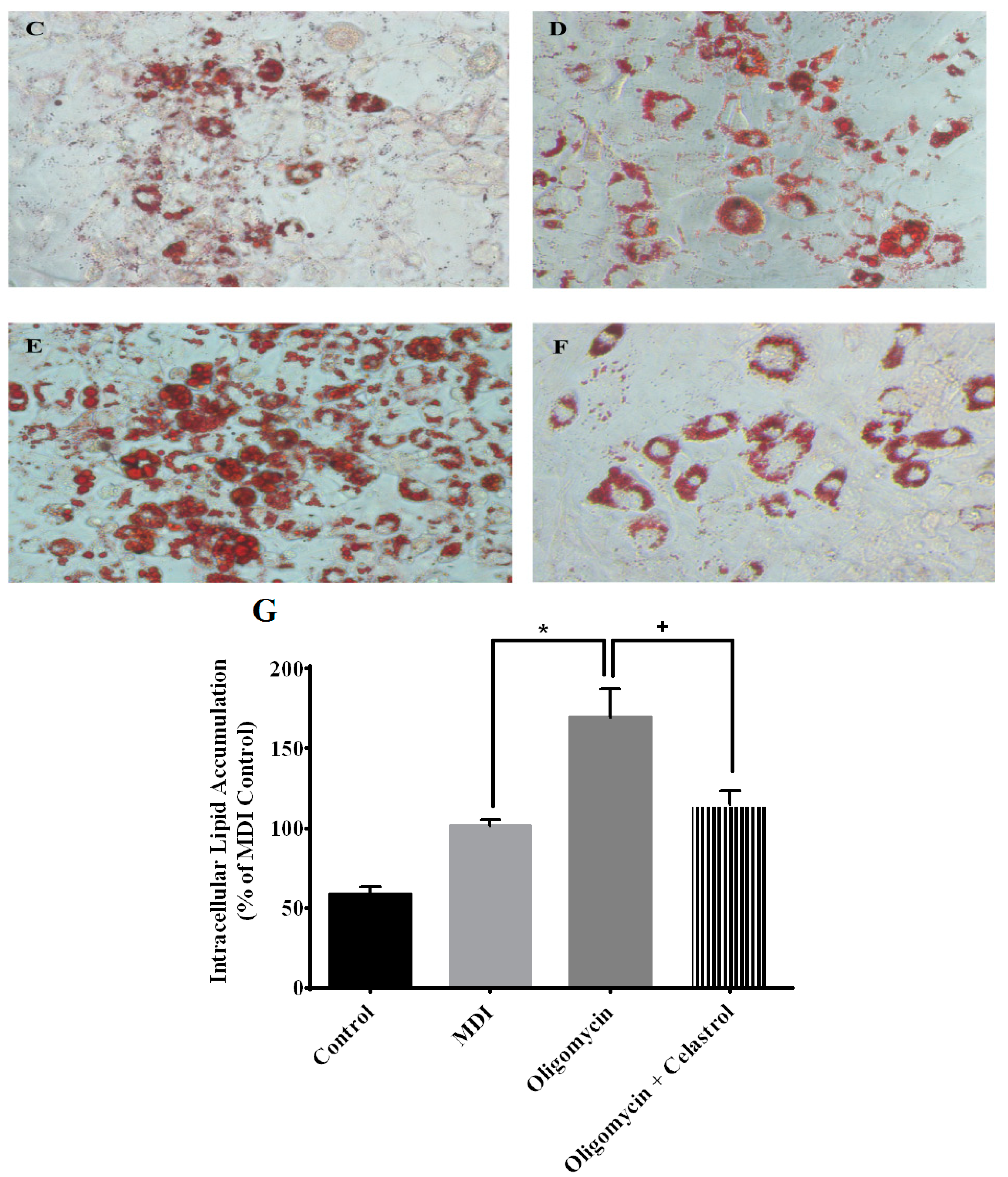
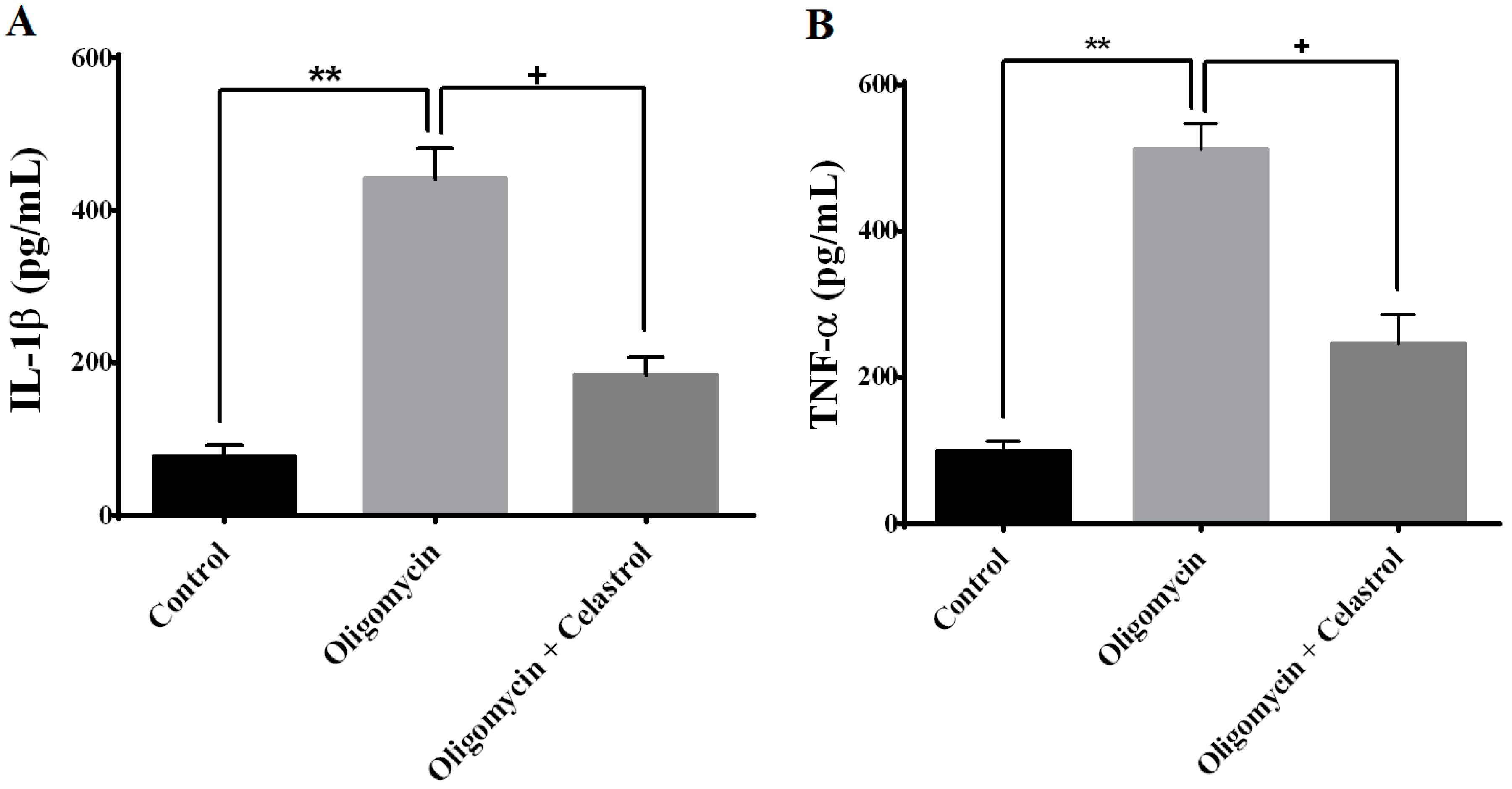
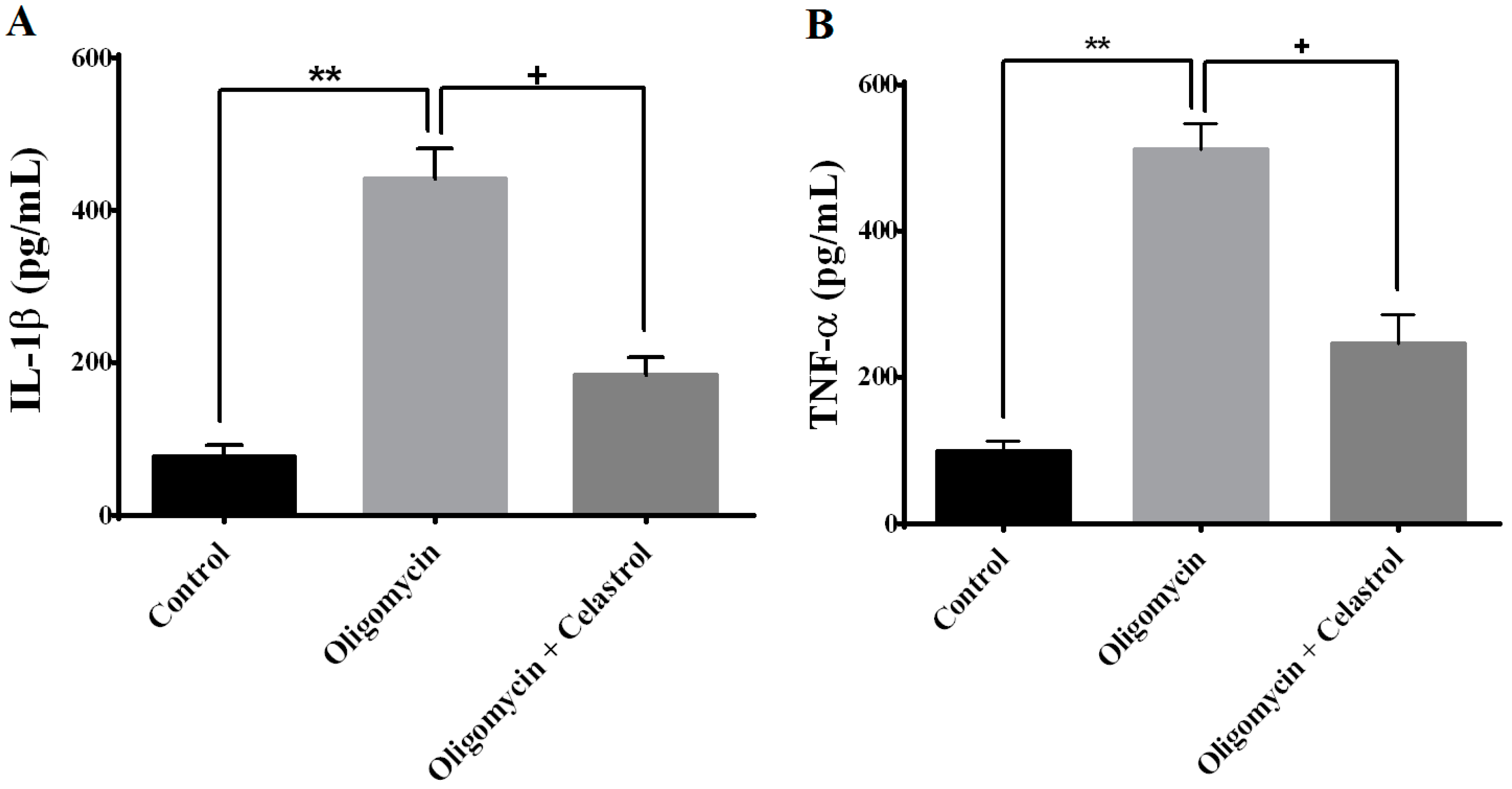
2.6. NF-κB Inhibitor Suppress Production of Tumor Necrosis Factor-α (TNF-α) and Interleukin-1β (IL-1β) in the Event of Mitochondrial Dysfunction of 3T3-L1 Adipocytes
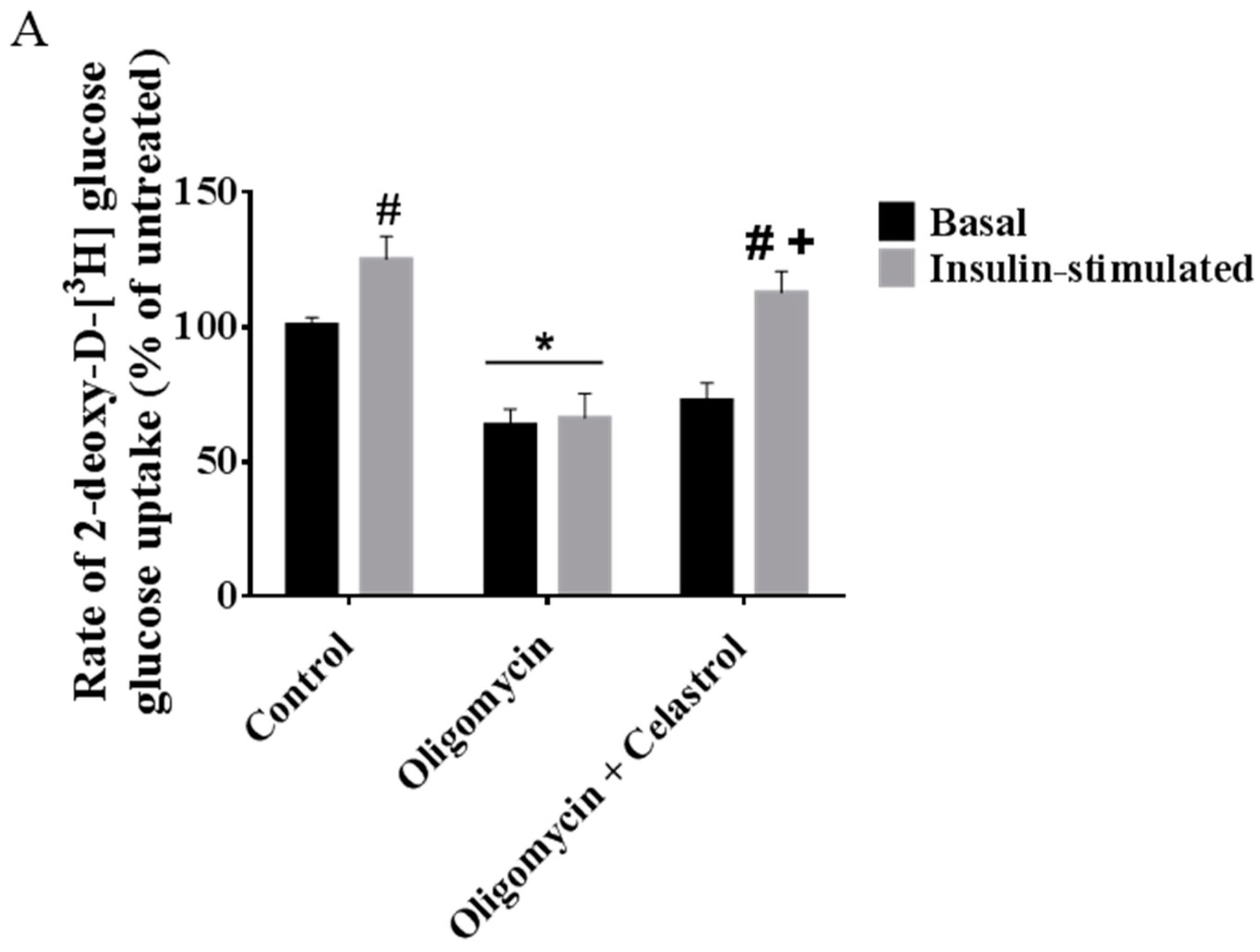
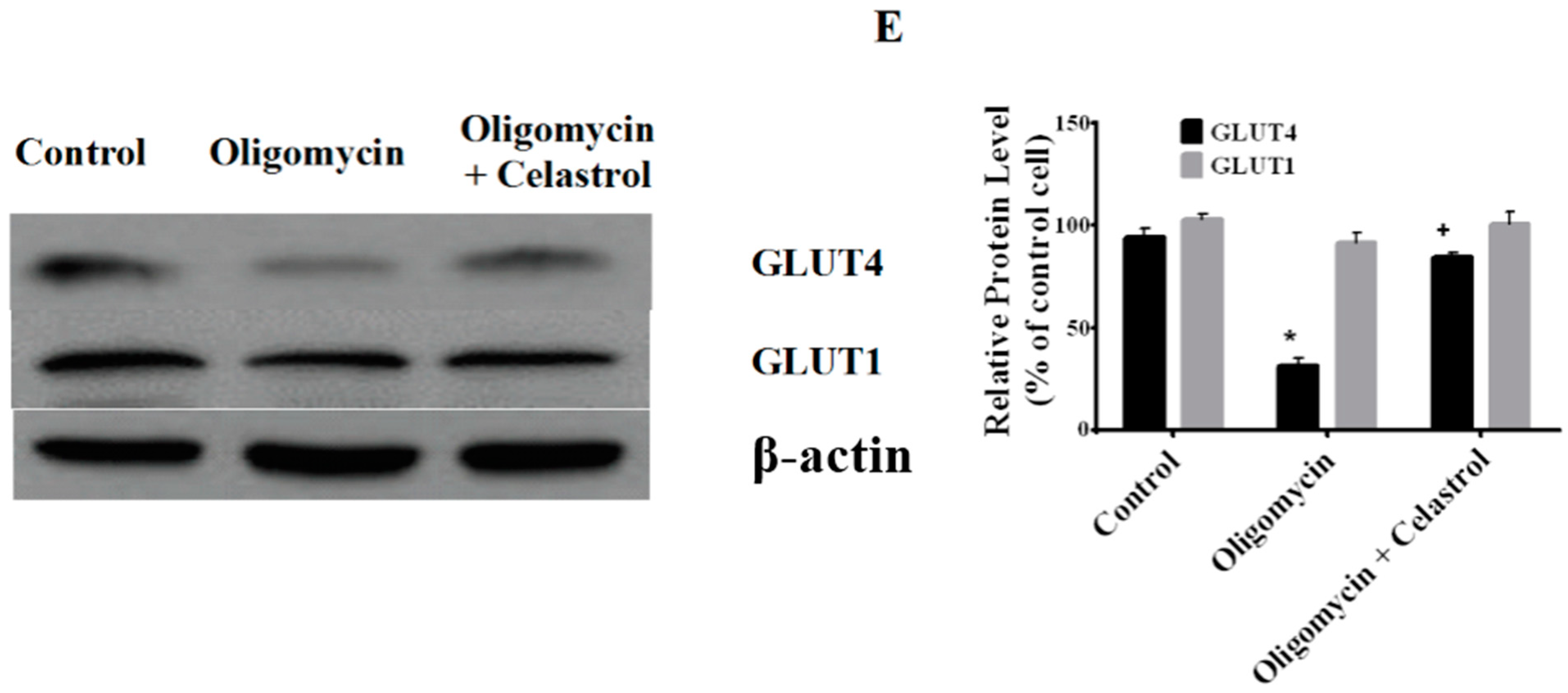
2.7. Co-Incubation of Celastrol Lead to the Restoration of Insulin-Induced Glucose Uptake Activity and Improved Insulin Signaling Cascades in Oligomycin-Treated 3T3-L1 Adipocytes
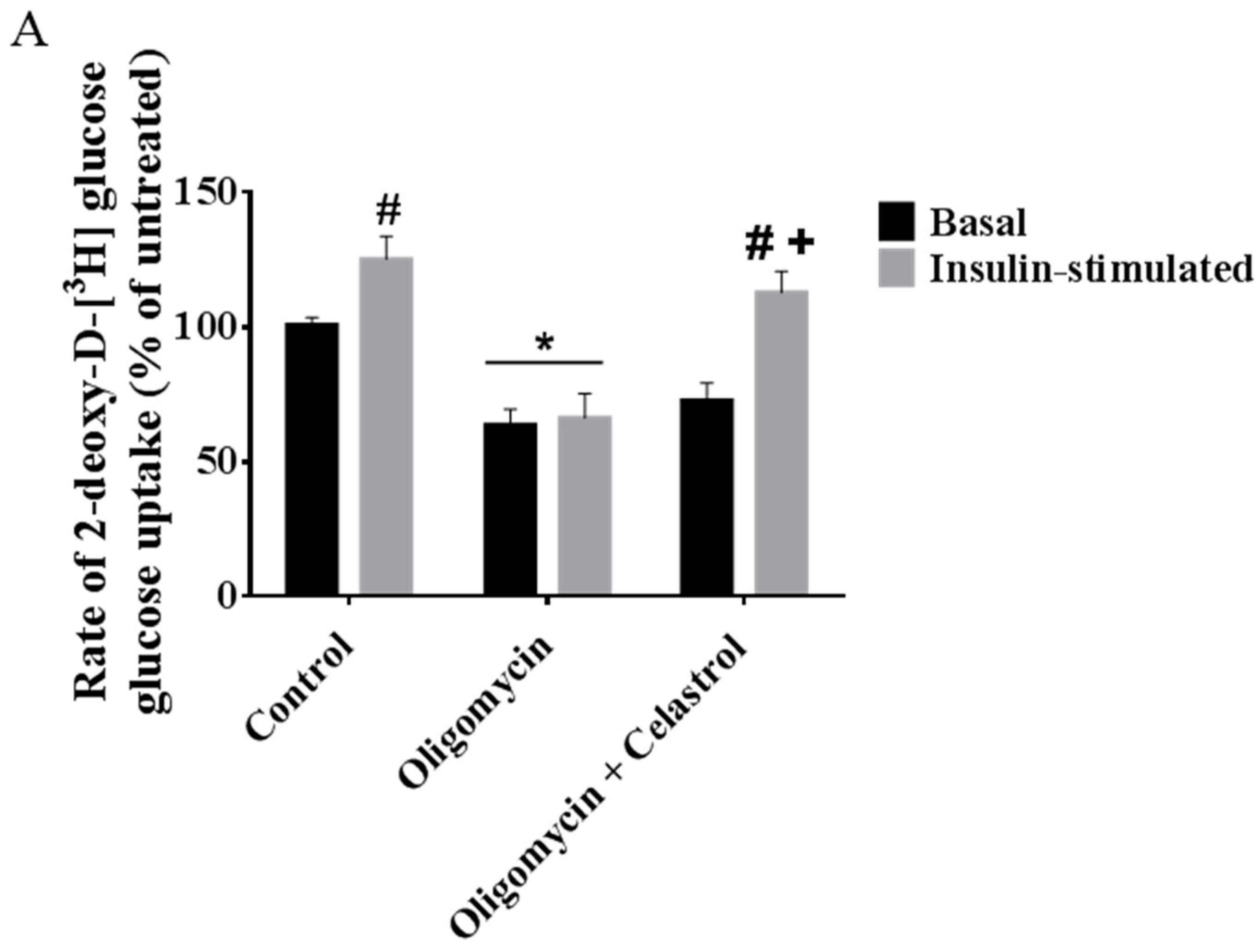
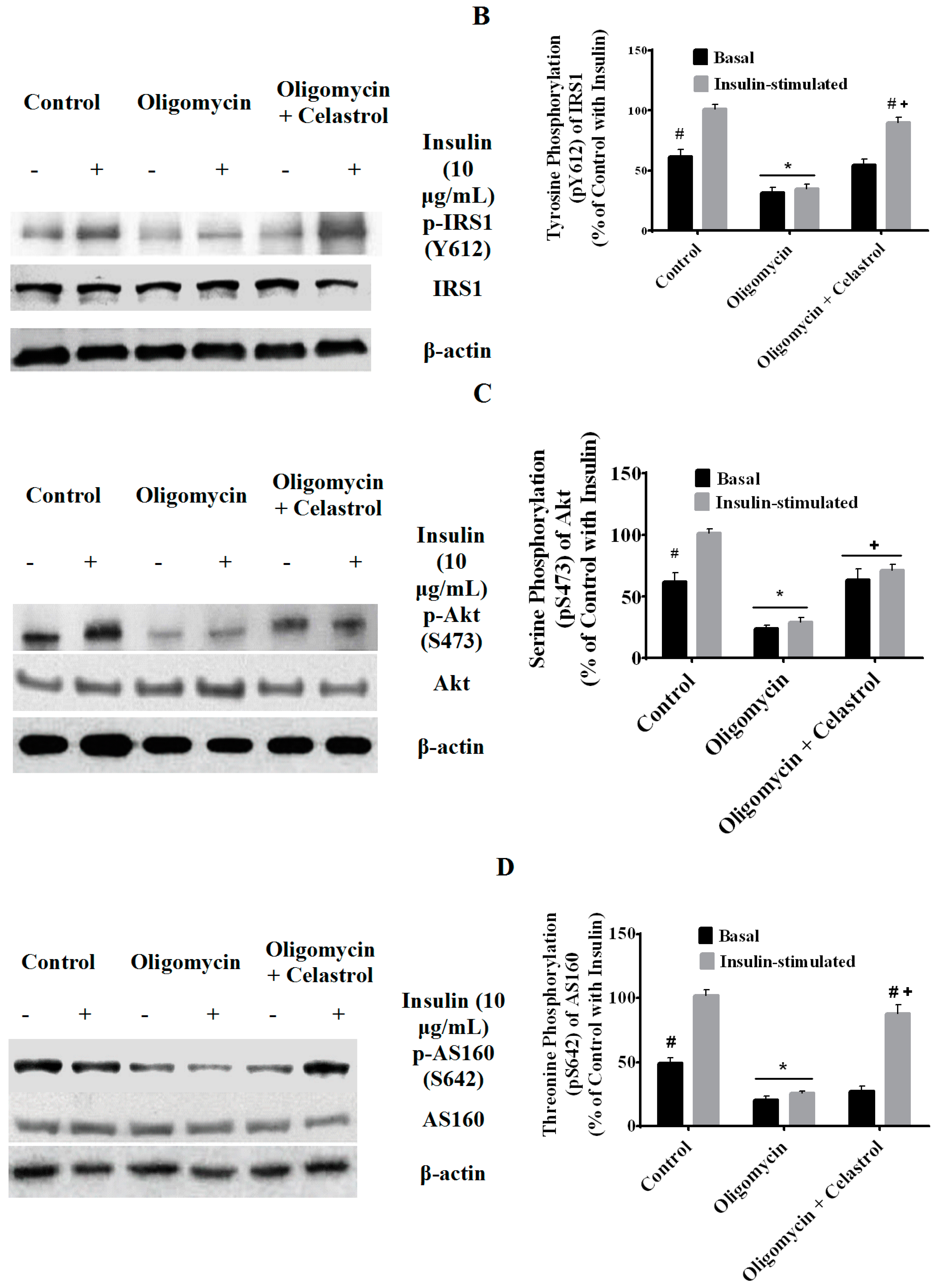
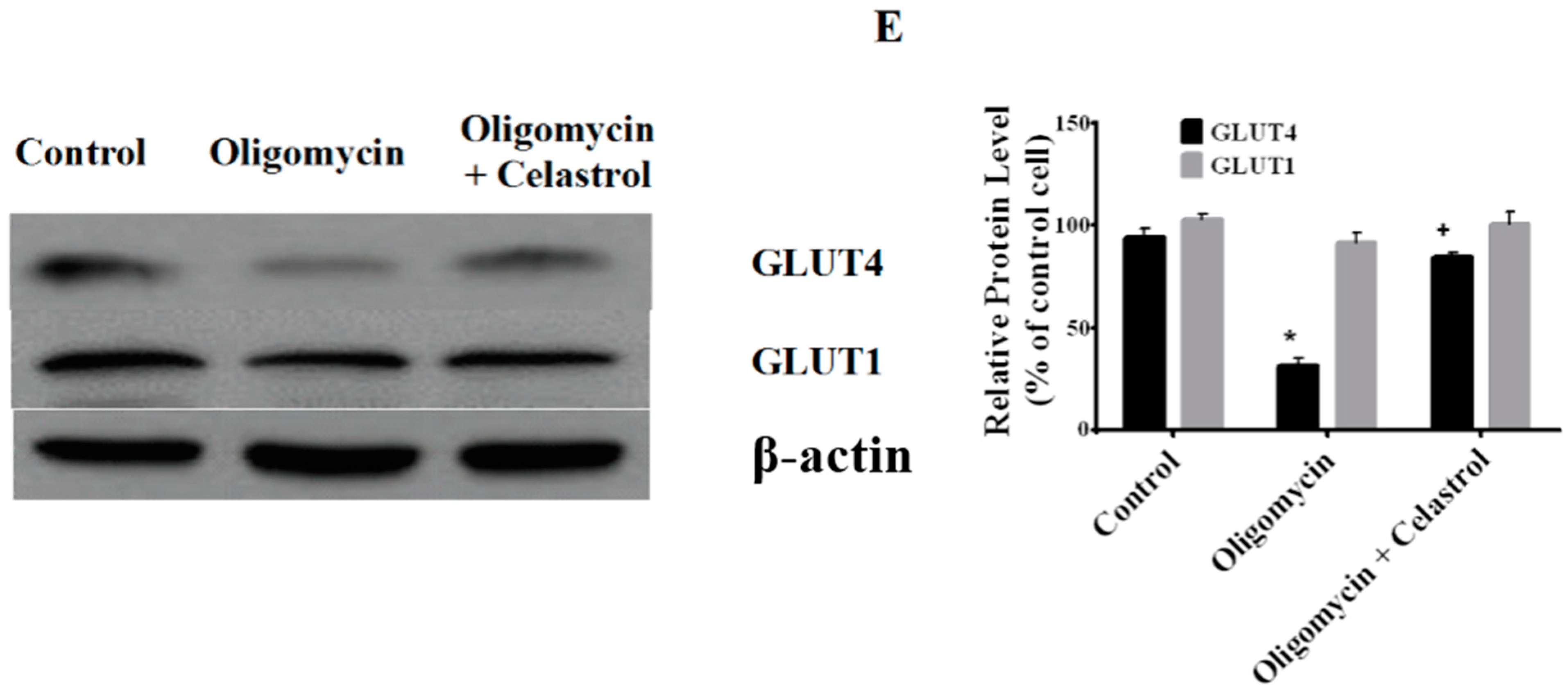
2.8. Discussion
3. Experimental Section
3.1. Cell Culture
3.2. Oligomycin and Celastrol Treatment
3.3. Measurement of Cell Viability
3.4. Intracellular ATP Concentration
3.5. Measurement of Mitochondrial Membrane Potentials (ΔΨm)
3.6. Mitochondrial Superoxide Measurement
3.7. Immunoblotting
3.8. Measurement of DNA Oxidative Damage and Lipid Peroxidation
3.9. Protein Carbonylation
3.10. Measurement of Lipolysis
3.11. Quantification of Lipid Contents by Oil Red O Assay
3.12. Cytokine ELISA for TNF-α and IL-1β Measurement
3.13. Glucose Uptake Assay
3.14. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wei, Y.; Chen, K.; Whaley-Connell, A.T.; Stump, C.S.; Ibdah, J.A.; Sowers, J.R. Skeletal muscle insulin resistance: Role of inflammatory cytokines and reactive oxygen species. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 23, R673–R680. [Google Scholar]
- Hafizi Abu Bakar, M.; Kian Kai, C.; Wan Hassan, W.N.; Sarmidi, M.R.; Yaakob, H.; Zaman Huri, H. Mitochondrial dysfunction as a central event for mechanisms underlying insulin resistance: The roles of long chain fatty acids. Diabetes Metab. Res. Rev. 2014. [Google Scholar] [CrossRef]
- Martins, A.R.; Nachbar, R.T.; Gorjao, R.; Vinolo, M.A.; Festuccia, W.T.; Lambertucci, R.H.; Cury-Boaventura, M.F.; Silveira, L.R.; Curi, R.; Hirabara, S.M. Mechanisms underlying skeletal muscle insulin resistance induced by fatty acids: Importance of the mitochondrial function. Lipids Health Dis. 2012, 11, 30. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Sánchez, A.; Madrigal-Santillán, E.; Bautista, M.; Esquivel-Soto, J.; Morales-González, A.; Esquivel-Chirino, C.; Durante-Montiel, I.; Sánchez-Rivera, G.; Valadez-Vega, C.; Morales-González, J.A. Inflammation, oxidative stress, and obesity. Int. J. Mol. Sci. 2011, 12, 3117–3132. [Google Scholar] [CrossRef]
- Hernández-Aguilera, A.; Rull, A.; Rodríguez-Gallego, E.; Riera-Borrull, M.; Luciano-Mateo, F.; Camps, J.; Menéndez, J.A.; Joven, J. Mitochondrial dysfunction: A basic mechanism in inflammation-related non-communicable diseases and therapeutic opportunities. Mediators Inflamm. 2013. [Google Scholar] [CrossRef]
- Bondia-Pons, I.; Ryan, L.; Martinez, J.A. Oxidative stress and inflammation interactions in human obesity. J. Physiol. Biochem. 2012, 68, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Naik, E.; Dixit, V.M. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J. Exp. Med. 2011, 208, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Halberg, N.; Wernstedt-Asterholm, I.; Scherer, P.E. The adipocyte as an endocrine cell. Endocrinol. Metab. Clin. N. Am. 2008, 37, 753–768. [Google Scholar] [CrossRef]
- Cinti, S.; Mitchell, G.; Barbatelli, G.; Murano, I.; Ceresi, E.; Faloia, E.; Wang, S.; Fortier, M.; Greenberg, A.S.; Obin, M.S. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J. Lipid Res. 2005, 46, 2347–2355. [Google Scholar] [CrossRef] [PubMed]
- Trayhurn, P.; Wood, I.S. Adipokines: Inflammation and the pleiotropic role of white adipose tissue. Br. J. Nutr. 2004, 92, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.C.; Lee, J. Cellular and molecular players in adipose tissue inflammation in the development of obesity-induced insulin resistance. Biochim. Biophys. Acta 2014, 1842, 446–462. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Cildir, G.; Akıncılar, S.C.; Tergaonkar, V. Chronic adipose tissue inflammation: All immune cells on the stage. Trends Mol. Med. 2013, 19, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.H.; Zhao, Y.P.; Xue, M.; Ji, C.B.; Gao, C.L.; Zhu, J.G.; Qin, D.N.; Kou, C.Z.; Qin, X.H.; Tong, M.L.; et al. TNF-α induces mitochondrial dysfunction in 3T3-L1 adipocytes. Mol. Cell Endocrinol. 2010, 328, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.L.; Zhu, C.; Zhao, Y.P.; Chen, X.H.; Ji, C.B.; Zhang, C.M.; Zhu, J.G.; Xia, Z.K.; Tong, M.L.; Guo, X.R. Mitochondrial dysfunction is induced by high levels of glucose and free fatty acids in 3T3-L1 adipocytes. Mol. Cell Endocrinol. 2010, 320, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Youssef-Elabd, E.M.; McGee, K.C.; Tripathi, G.; Aldaghri, N.; Abdalla, M.S.; Sharada, H.M.; Ash, E.; Amin, A.I.; Ceriello, A.; O’Hare, J.P.; et al. Acute and chronic saturated fatty acid treatment as a key instigator of the TLR-mediated inflammatory response in human adipose tissue, in vitro. J. Nutr. Biochem. 2012, 23, 39–50. [Google Scholar] [CrossRef]
- Reynolds, C.M.; McGillicuddy, F.C.; Harford, K.A.; Finucane, O.M.; Mills, K.H.G.; Roche, H.M. Dietary saturated fatty acids prime the NLRP3 inflammasome via TLR4 in dendritic cells—Implications for diet-induced insulin resistance. Mol. Nutr. Food Res. 2012, 56, 1212–1222. [Google Scholar] [CrossRef] [PubMed]
- Escames, G.; López, L.; García, J.; García-Corzo, L.; Ortiz, F.; Acuña-Castroviejo, D. Mitochondrial DNA and inflammatory diseases. Hum. Genet. 2012, 131, 161–173. [Google Scholar] [CrossRef]
- López-Armada, M.J.; Riveiro-Naveira, R.R.; Vaamonde-García, C.; Valcárcel-Ares, M.N. Mitochondrial dysfunction and the inflammatory response. Mitochondrion 2013, 13, 106–118. [Google Scholar]
- Patel, S.; Santani, D. Role of NF-κB in the pathogenesis of diabetes and its associated complications. Pharmacol. Rep. 2009, 61, 595–603. [Google Scholar] [PubMed]
- Vaamonde-García, C.; Riveiro-Naveira, R.R.; Valcárcel-Ares, M.N.; Hermida-Carballo, L.; Blanco, F.J.; López-Armada, M.J. Mitochondrial dysfunction increases inflammatory responsiveness to cytokines in normal human chondrocytes. Arthritis Rheumatol. 2012, 64, 2927–2936. [Google Scholar] [CrossRef]
- Yang, H.; Chen, D.; Cui, Q.C.; Yuan, X.; Dou, Q.P. Celastrol, a triterpene extracted from the Chinese “Thunder of God Vine”, is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res. 2006, 66, 4758–4765. [Google Scholar] [CrossRef] [PubMed]
- Allison, A.C.; Cacabelos, R.; Lombardi, V.R.M.; Álvarez, X.A.; Vigo, C. Celastrol, a potent antioxidant and anti-inflammatory drug, as a possible treatment for Alzheimer’s disease. Prog. Neuro-Psychoph. Biol. Psychiatry 2001, 25, 1341–1357. [Google Scholar] [CrossRef]
- Lee, J.H.; Koo, T.H.; Yoon, H.; Jung, H.S.; Jin, H.Z.; Lee, K.; Hong, Y.S.; Lee, J.J. Inhibition of NF-κB activation through targeting IκB kinase by celastrol, a quinone methide triterpenoid. Biochem. Pharmacol. 2006, 72, 1311–1321. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Lee, M.H.; Nam, D.H.; Song, H.K.; Kang, Y.S.; Lee, J.E.; Kim, H.W.; Cha, J.J.; Hyun, Y.Y.; Han, S.Y.; et al. Celastrol, an NF-κB inhibitor, improves insulin resistance and attenuates renal injury in db/db mice. PLoS One 2013, 8, e62068. [Google Scholar] [CrossRef] [PubMed]
- Frohnert, B.I.; Bernlohr, D.A. Protein carbonylation, mitochondrial dysfunction, and insulin resistance. Adv. Nutr. 2013, 4, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Suzawa, M.; Takada, I.; Yanagisawa, J.; Ohtake, F.; Ogawa, S.; Yamauchi, T.; Kadowaki, T.; Takeuchi, Y.; Shibuya, H.; Gotoh, Y.; et al. Cytokines suppress adipogenesis and PPARγfunction through the TAK1/TAB1/NIK cascade. Nat. Cell Biol. 2003, 5, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.; Hacohen, N.; Golub, T.R.; van Parijs, L.; Lodish, H.F. Tumor necrosis factor-α suppresses adipocyte-specific genes and activates expression of preadipocyte genes in 3T3-L1 adipocytes: Nuclear factor-κB activation by TNF-α is obligatory. Diabetes 2002, 51, 1319–1336. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.H.; Wang, C.C.; Wei, Y.H. Mitochondrial dysfunction in insulin insensitivity: Implication of mitochondrial role in type 2 diabetes. Ann. N. Y. Acad. Sci. 2010, 1201, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.H.; Wang, C.C.; Huang, H.C.; Wei, Y.H. Mitochondrial dysfunction leads to impairment of insulin sensitivity and adiponectin secretion in adipocytes. FEBS J. 2013, 280, 1039–1050. [Google Scholar] [CrossRef] [PubMed]
- Choo, H.J.; Kim, J.H.; Kwon, O.B.; Lee, C.; Mun, J.; Han, S.; Yoon, Y.S.; Yoon, G.; Choi, K.M.; Ko, Y.G. Mitochondria are impaired in the adipocytes of type 2 diabetic mice. Diabetologia 2006, 49, 784–791. [Google Scholar] [CrossRef] [PubMed]
- Kusminski, C.M.; Scherer, P.E. Mitochondrial dysfunction in white adipose tissue. Trends Endocrinol. Metab. 2012, 23, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Vaamonde-García, C.; Valcarcel-Ares, N.; Riveiro-Naveira, R.; Lema, B.; Blanco, F.J.; López-Armada, M.J. Inflammatory response is modulated by mitochondrial dysfunction in cultured normal human chondrocytes. Ann. Rheum. Dis. 2010, 69, A15–A16. [Google Scholar] [CrossRef]
- Bulua, A.C.; Simon, A.; Maddipati, R.; Pelletier, M.; Park, H.; Kim, K.Y.; Sack, M.N.; Kastner, D.L.; Siegel, R.M. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med. 2011, 208, 519–533. [Google Scholar] [CrossRef] [PubMed]
- Ungvari, Z.; Orosz, Z.; Labinskyy, N.; Rivera, A.; Zhao, X.M.; Smith, K.; Csiszar, A. Increased mitochondrial H2O2 production promotes endothelial NF-κB activation in aged rat arteries. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H37–H47. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, N.; Eura, Y.; Mihara, K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J. Cell Sci. 2004, 117, 6535–6546. [Google Scholar] [CrossRef] [PubMed]
- Suen, D.F.; Norris, K.L.; Youle, R.J. Mitochondrial dynamics and apoptosis. Genes Dev. 2008, 22, 1577–1590. [Google Scholar] [CrossRef]
- Zemirli, N.; Pourcelot, M.; Ambroise, G.; Hatchi, E.; Vazquez, A.; Arnoult, D. Mitochondrial hyperfusion promotes NF-κB activation via the mitochondrial E3 ligase MULAN. FEBS J. 2014, 281, 3095–3112. [Google Scholar] [CrossRef] [PubMed]
- Mariappan, N.; Elks, C.M.; Sriramula, S.; Guggilam, A.; Liu, Z.; Borkhsenious, O.; Francis, J. NF-κB-induced oxidative stress contributes to mitochondrial and cardiac dysfunction in type II diabetes. Cardiovasc. Res. 2010, 85, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Inflammaging: Disturbed interplay between autophagy and inflammasomes. Aging (Albany NY) 2012, 4, 166–175. [Google Scholar]
- Lee, H.K.; Song, J.H.; Shin, C.S.; Park, D.J.; Park, K.S.; Lee, K.U.; Koh, C.S. Decreased mitochondrial DNA content in peripheral blood precedes the development of non-insulin-dependent diabetes mellitus. Diabetes Res. Clin. Pract. 1998, 42, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Morino, K.; Petersen, K.F.; Dufour, S.; Befroy, D.; Frattini, J.; Shatzkes, N.; Neschen, S.; White, M.F.; Bilz, S.; Sono, S.; et al. Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J. Clin. Investig. 2005, 115, 3587–3593. [Google Scholar] [CrossRef] [PubMed]
- Stienstra, R.; Joosten, L.A.B.; Koenen, T.; van Tits, B.; van Diepen, J.A.; van den Berg, S.A.A.; Rensen, P.C.N.; Voshol, P.J.; Fantuzzi, G.; Hijmans, A.; et al. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab. 2010, 12, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Yano, S.; Yamaguchi, T.; Sugimoto, T. Advanced glycation end products-induced reactive oxygen species generation is partly through NF-κB activation in human aortic endothelial cells. J. Diabetes Complicat. 2013, 27, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Curtis, J.M.; Hahn, W.S.; Stone, M.D.; Inda, J.J.; Droullard, D.J.; Kuzmicic, J.P.; Donoghue, M.A.; Long, E.K.; Armien, A.G.; Lavandero, S.; et al. Protein carbonylation and adipocyte mitochondrial function. J. Biol. Chem. 2012, 287, 32967–32980. [Google Scholar] [CrossRef] [PubMed]
- Frohnert, B.I.; Sinaiko, A.R.; Serrot, F.J.; Foncea, R.E.; Moran, A.; Ikramuddin, S.; Choudry, U.; Bernlohr, D.A. Increased adipose protein carbonylation in human obesity. Obesity 2011, 19, 1735–1741. [Google Scholar]
- Curtis, J.M.; Grimsrud, P.A.; Wright, W.S.; Xu, X.; Foncea, R.E.; Graham, D.W.; Brestoff, J.R.; Wiczer, B.M.; Ilkayeva, O.; Cianflone, K.; et al. Downregulation of adipose glutathione S-transferase A4 leads to increased protein carbonylation, oxidative stress, and mitochondrial dysfunction. Diabetes 2010, 59, 1132–1142. [Google Scholar]
- Fang, J.; Holmgren, A. Inhibition of thioredoxin and thioredoxin reductase by 4-Hydroxy-2-nonenal in vitro and in vivo. J. Am. Chem. Soc. 2006, 128, 1879–1885. [Google Scholar]
- Yadav, U.C.S.; Ramana, K.V. Regulation of NF-κB-induced inflammatory signaling by lipid peroxidation-derived aldehydes. Oxid. Med. Cell Longev. 2013. [Google Scholar] [CrossRef]
- Altavilla, D.; Famulari, C.; Passaniti, M.; Campo, G.M.; MacrÌ, A.; Seminara, P.; Marini, H.; Calò, M.; Santamaria, L.B.; Bono, D.; et al. Lipid peroxidation inhibition reduces NF-κB activation and attenuates cerulein-induced pancreatitis. Free Radic. Res. 2003, 37, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.; Chen, X.; Gao, C.; Jiao, L.; Wang, J.; Xu, G.; Fu, H.; Guo, X.; Zhao, Y. IL-6 induces lipolysis and mitochondrial dysfunction, but does not affect insulin-mediated glucose transport in 3T3-L1 adipocytes. J. Bioenerg. Biomembr. 2011, 43, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Souza, S.C.; Palmer, H.J.; Kang, Y.H.; Yamamoto, M.T.; Muliro, K.V.; Paulson, K.E.; Greenberg, A.S. TNF-α induction of lipolysis is mediated through activation of the extracellular signal related kinase pathway in 3T3-L1 adipocytes. J. Cell Biochem. 2003, 89, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Laurencikiene, J.; van Harmelen, V.; Arvidsson Nordström, E.; Dicker, A.; Blomqvist, L.; Näslund, E.; Langin, D.; Arner, P.; Rydén, M. NF-κB is important for TNF-α-induced lipolysis in human adipocytes. J. Lipid Res. 2007, 48, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Zuany-Amorim, C.; Hastewell, J.; Walker, C. Toll-like receptors as potential therapeutic targets for multiple diseases. Nat. Rev. Drug Discov. 2002, 1, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Ortega, M.T.; Mora, S.; Chapes, S.K. Interactive changes between macrophages and adipocytes. Clin. Vaccine Immunol. 2010, 17, 651–659. [Google Scholar] [CrossRef]
- Li, N.; Karin, M. Is NF-κB the sensor of oxidative stress? FASEB J. 1999, 13, 1137–1143. [Google Scholar] [PubMed]
- Emanuela, F.; Grazia, M.; de Marco, R.; Paola, L.M.; Giorgio, F.; Marco, B. Inflammation as a link between obesity and metabolic syndrome. J. Nutr. Metab. 2012. [Google Scholar] [CrossRef]
- Mauro, C.; Leow, S.C.; Anso, E.; Rocha, S.; Thotakura, A.K.; Tornatore, L.; Moretti, M.; de Smaele, E.; Beg, A.A.; Tergaonkar, V.; et al. NF-κB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat. Cell Biol. 2011, 13, 1272–1279. [Google Scholar] [CrossRef] [PubMed]
- Bogan, J.S. Regulation of glucose transporter translocation in health and diabetes. Annu. Rev. Biochem. 2012, 81, 507–532. [Google Scholar] [PubMed]
- Nandi, A.; Kitamura, Y.; Kahn, C.R.; Accili, D. Mouse models of insulin resistance. Physiol. Rev. 2004, 84, 623–647. [Google Scholar] [CrossRef] [PubMed]
- Wei, B.; Ta, N.; Li, J.; Zhang, J.; Chen, B. Relationship between the inhibition of NF-κB and insulin resistance in uterrus of rats with gestational diabetes mellitus. Chin. J. Cell Mol. Immunol. 2010, 26, 235–237, 241. [Google Scholar]
- Wu, N.; Zheng, B.; Shaywitz, A.; Dagon, Y.; Tower, C.; Bellinger, G.; Shen, C.H.; Wen, J.; Asara, J.; McGraw, T.E.; et al. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol. Cell 2013, 49, 1167–1175. [Google Scholar] [CrossRef] [PubMed]
- Schubert, D. Glucose metabolism and Alzheimer’s disease. Ageing Res. Rev. 2005, 4, 240–257. [Google Scholar] [CrossRef] [PubMed]
- Kussmann, M.; Morine, M.; Hager, J.; Sonderegger, B.; Kaput, J. Perspective: A system approach to diabetes research. Front. Genet. 2013, 4, 205. [Google Scholar]
- Miki, H.; Yamauchi, T.; Suzuki, R.; Komeda, K.; Tsuchida, A.; Kubota, N.; Terauchi, Y.; Kamon, J.; Kaburagi, Y.; Matsui, J.; et al. Essential role of insulin receptor substrate 1 (IRS-1) and IRS-2 in adipocyte differentiation. Mol. Cell. Biol. 2001, 21, 2521–2532. [Google Scholar] [CrossRef]
- Hossner, K.L. Hormonal Regulation of Farm Animal Growth; CABI: Cambrige, MA, USA, 2005; p. 174. [Google Scholar]
- Huang, C.; Zhang, Y.; Gong, Z.; Sheng, X.; Li, Z.; Zhang, W.; Qin, Y. Berberine inhibits 3T3-L1 adipocyte differentiation through the PPARγ pathway. Biochem. Biophys. Res. Commun. 2006, 348, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Bhat, H.K. Superoxide dismutase 3 is induced by antioxidants, inhibits oxidative DNA damage and is associated with inhibition of estrogen-induced breast cancer. Carcinogenesis 2012, 33, 2601–2610. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bakar, M.H.A.; Sarmidi, M.R.; Kai, C.K.; Huri, H.Z.; Yaakob, H. Amelioration of Mitochondrial Dysfunction-Induced Insulin Resistance in Differentiated 3T3-L1 Adipocytes via Inhibition of NF-κB Pathways. Int. J. Mol. Sci. 2014, 15, 22227-22257. https://doi.org/10.3390/ijms151222227
Bakar MHA, Sarmidi MR, Kai CK, Huri HZ, Yaakob H. Amelioration of Mitochondrial Dysfunction-Induced Insulin Resistance in Differentiated 3T3-L1 Adipocytes via Inhibition of NF-κB Pathways. International Journal of Molecular Sciences. 2014; 15(12):22227-22257. https://doi.org/10.3390/ijms151222227
Chicago/Turabian StyleBakar, Mohamad Hafizi Abu, Mohamad Roji Sarmidi, Cheng Kian Kai, Hasniza Zaman Huri, and Harisun Yaakob. 2014. "Amelioration of Mitochondrial Dysfunction-Induced Insulin Resistance in Differentiated 3T3-L1 Adipocytes via Inhibition of NF-κB Pathways" International Journal of Molecular Sciences 15, no. 12: 22227-22257. https://doi.org/10.3390/ijms151222227