The Importance of Polarity in the Evolution of the K+ Binding Site of Pyruvate Kinase

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
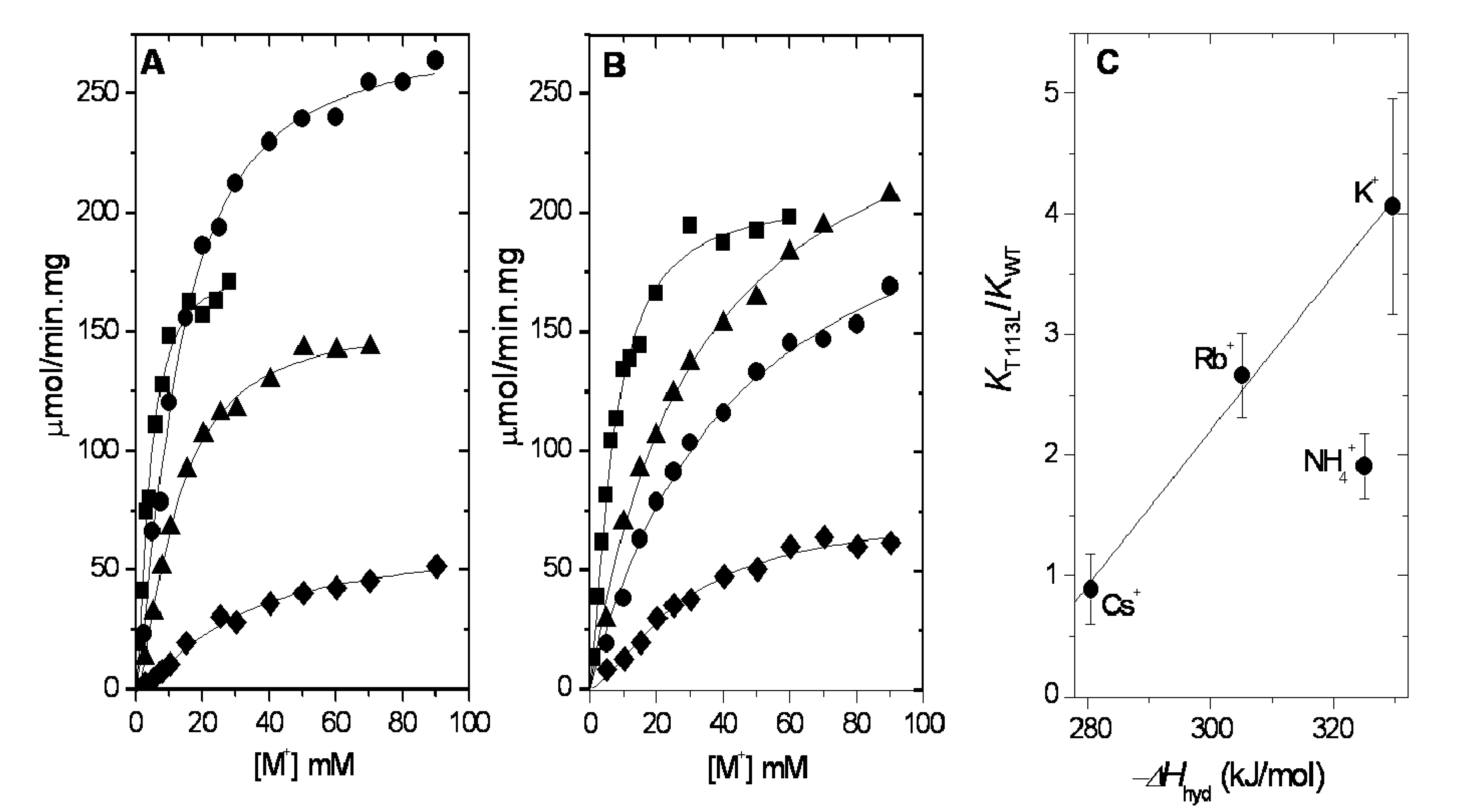
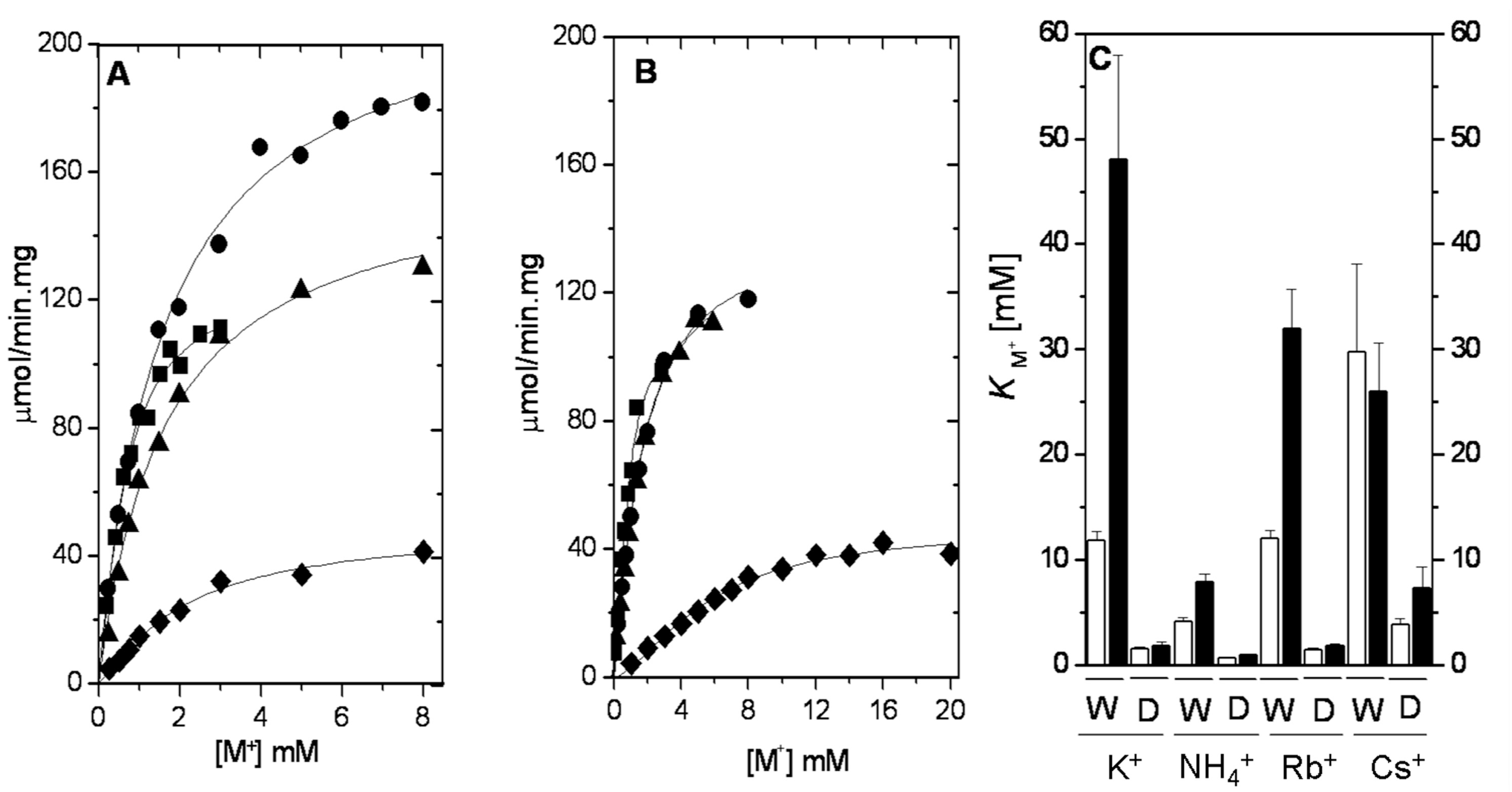
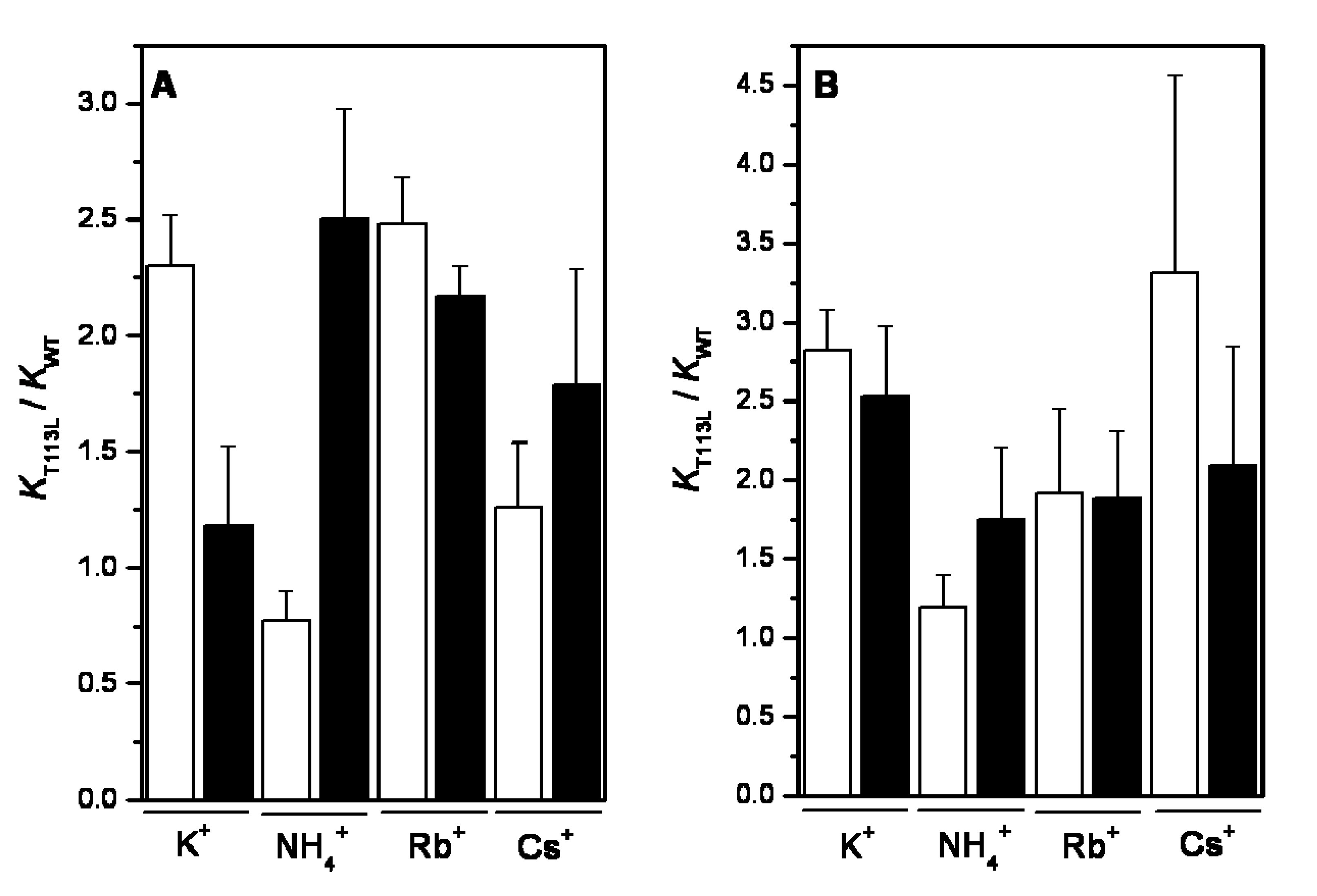
2.1. Ion Selectivity of WT-RMPK and T113L-RMPK in 100% Water
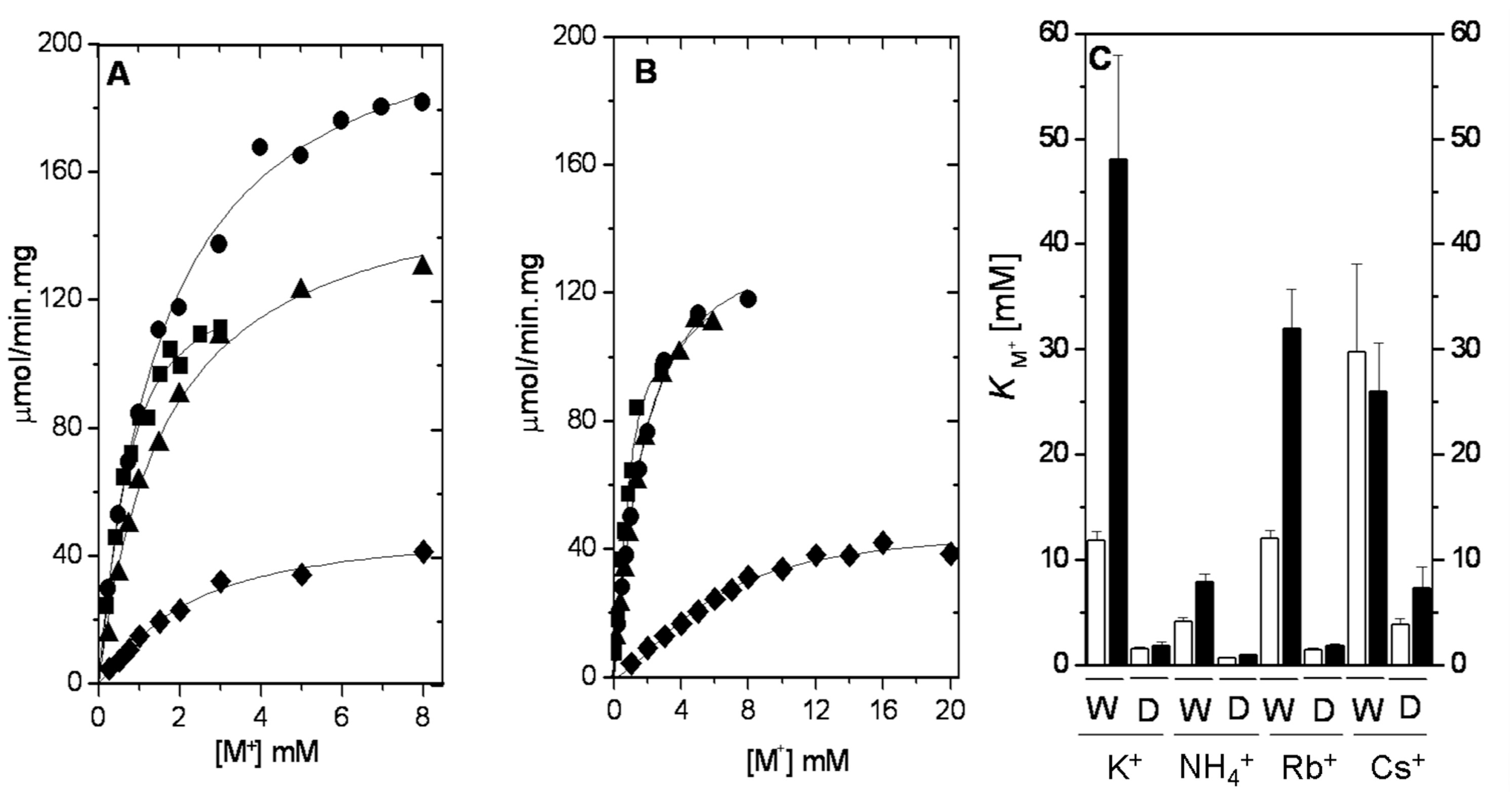
2.2. Ion Selectivity of WT-RMPK and T113L-RMPK with 20% DMSO
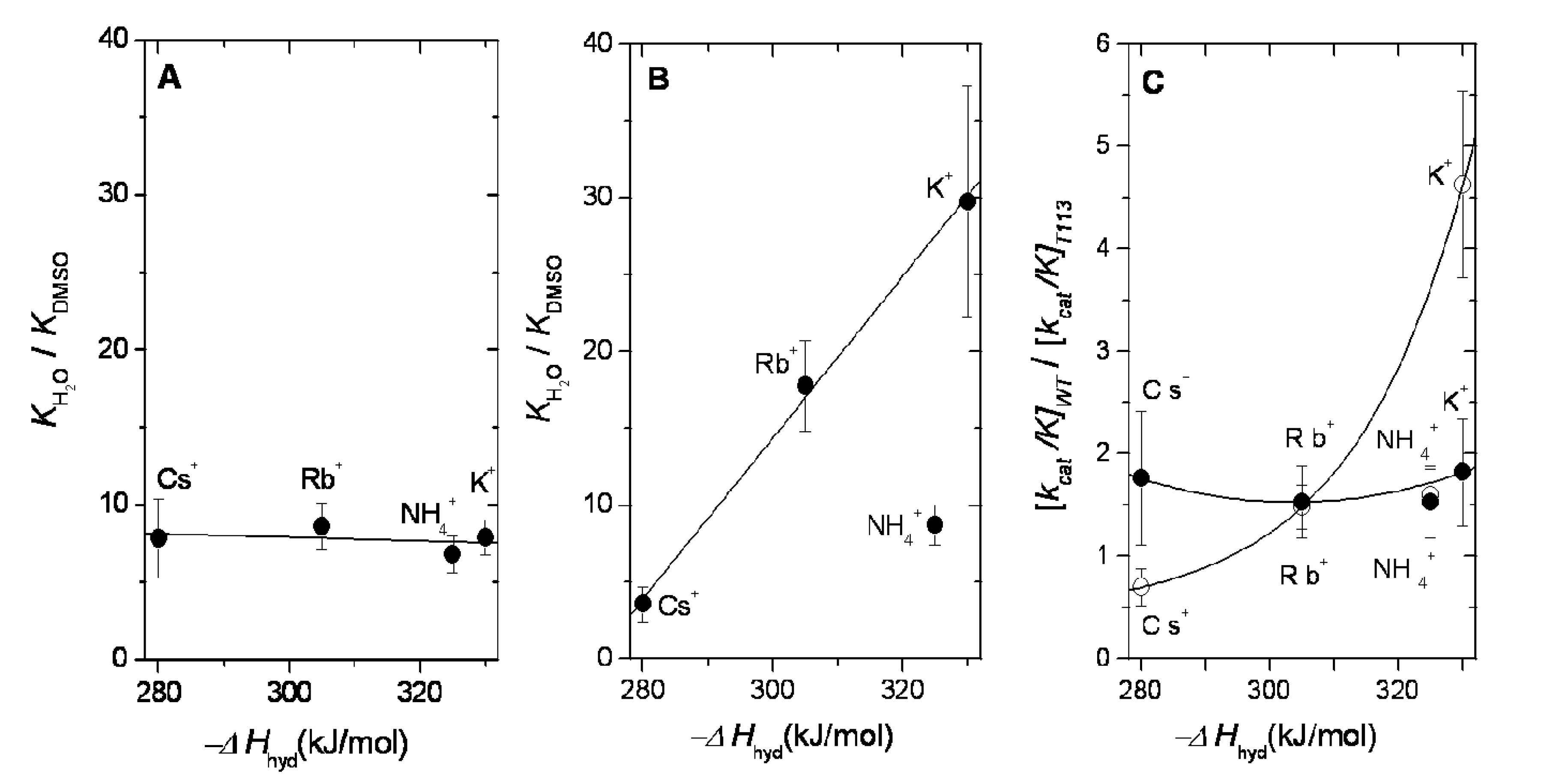
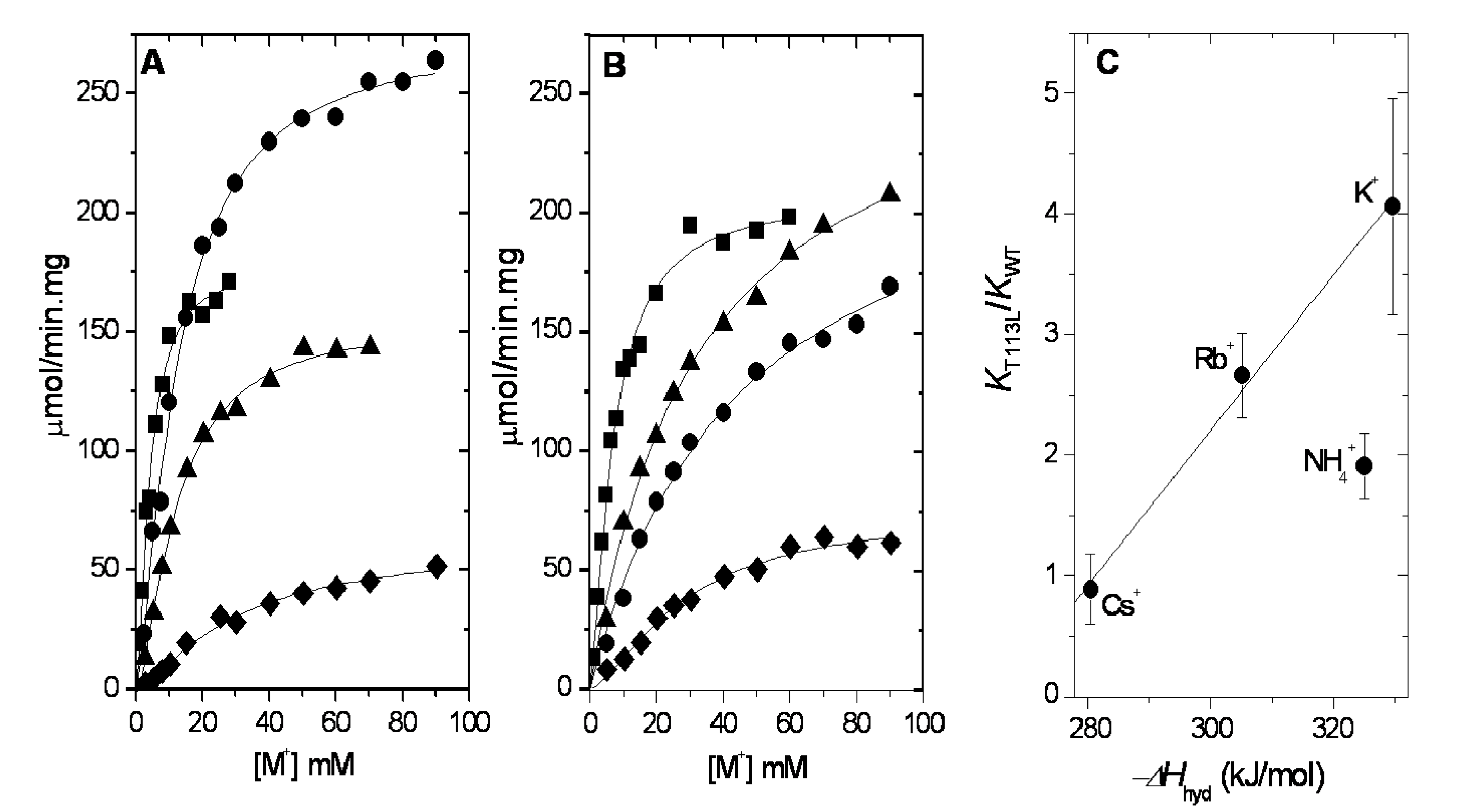
2.3. Dehydration Energy of K+, NH4+, Rb+ and Cs+ and Their Partition into the Catalytic Site of WT-RMPK and T113L-RMPK
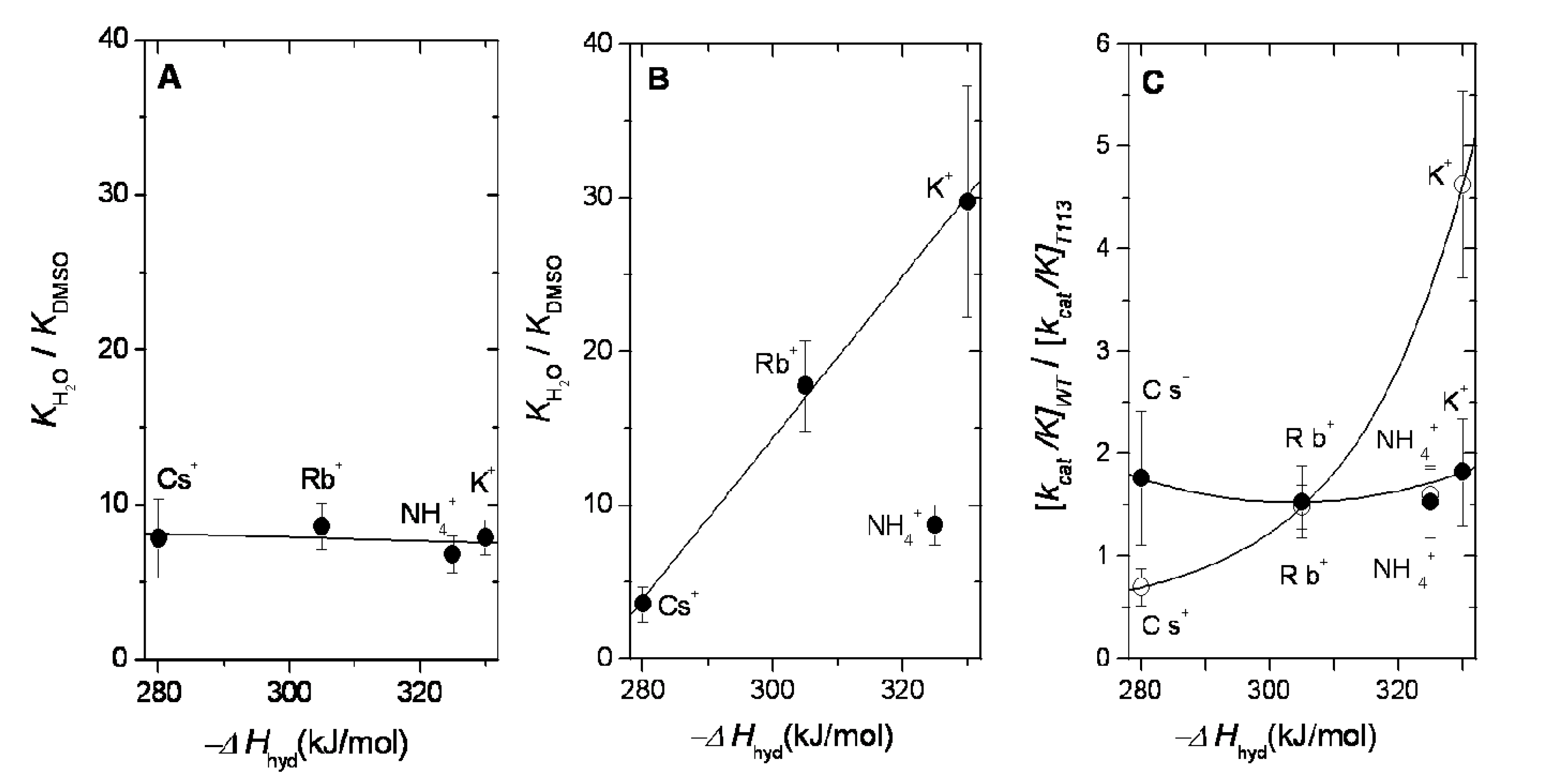
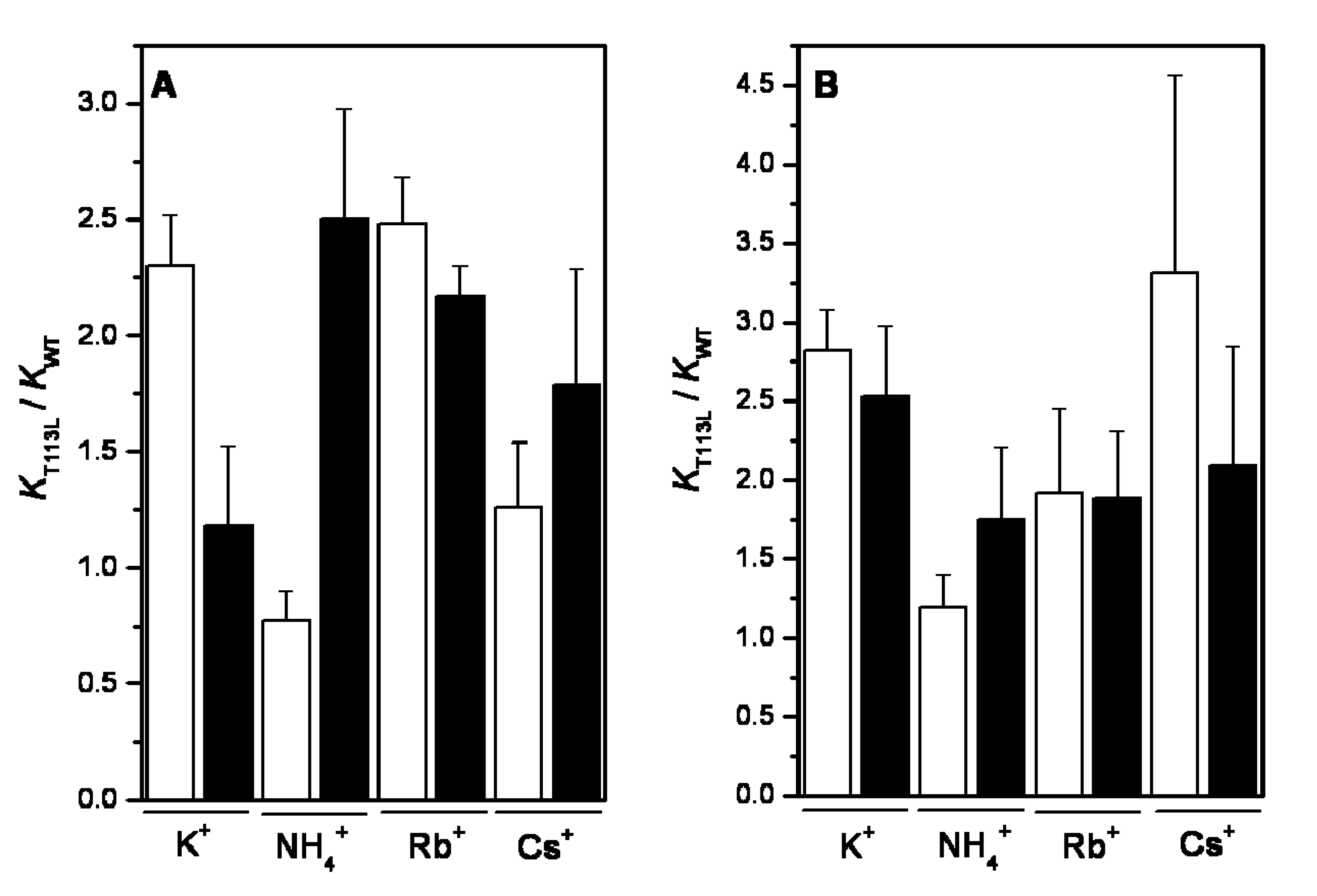
2.4. Dehydration Energy of K+, NH4+, Rb+ and Cs+ and Their Specificity for WT-RMPK and T113L-RMPK
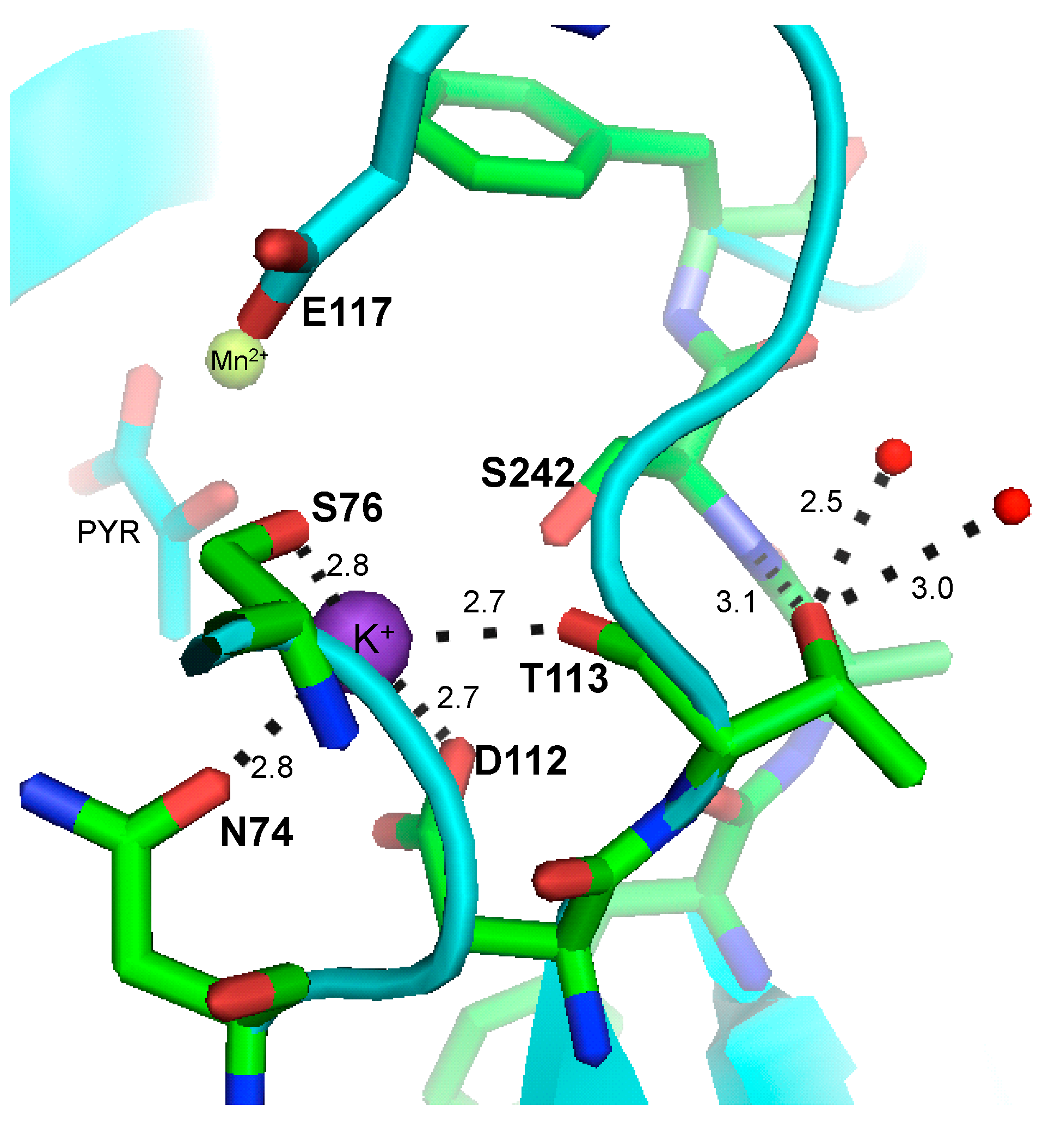
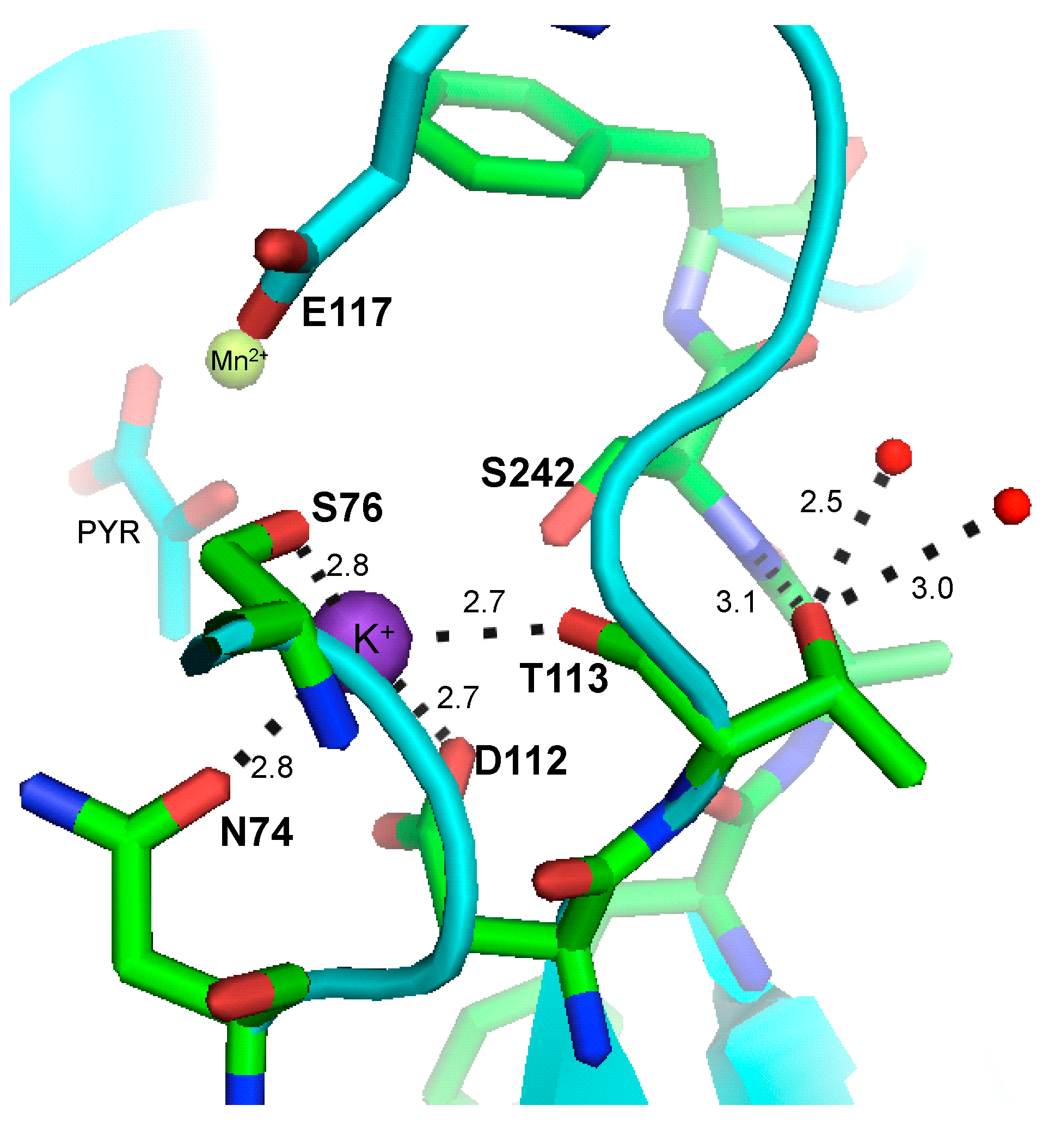
2.5. PEP and ADP-Mg Binding Sites
3. Materials and Methods
3.1. Mutagenesis of T113L Pyruvate Kinase (T113L-RMPK)
3.2. Cell Growth and Purification of the T113L Mutant
3.3. Assays of Pyruvate Kinase Activity
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kachmar, J.F.; Boyer, P.D. Kinetic analysis of enzyme reactions: II. The potassium activation and calcium inhibition of pyruvic phosphoferase. J. Biol. Chem. 1953, 200, 669–682. [Google Scholar]
- Kayne, F.J. Thallium (I) activation of pyruvate kinase. Arch. Biochem. Biophys. 1971, 143, 232–239. [Google Scholar]
- Mildvan, A.S.; Cohn, M. Kinetics and magnetic resonance studies of the pyruvate kinase reaction II. Complexes of enzyme, metal, and substrates. J. Biol. Chem. 1966, 241, 1178–1193. [Google Scholar]
- Kayne, F.J.; Reuben, J. Thallium (I) activation of pyruvate kinase. J. Am. Chem. Soc. 1970, 92, 220–222. [Google Scholar]
- Reuben, J.; Kayne, F.J. Thallium-205 nuclear magnetic resonance study of pyruvate kinase and its substrates. J. Biol. Chem. 1971, 246, 6227–6234. [Google Scholar]
- Nowak, T.; Mildvan, A.S. Nuclear magnetic resonance studies of the function of potassium in the mechanism of pyruvate kinase. Biochemistry 1972, 11, 2819–2828. [Google Scholar]
- Raushel, F.M.; Villafranca, J.J. A multinuclear nuclear magnetic resonance study of the monovalent-divalent cation sites of pyruvate kinase. Biochemistry 1980, 19, 5481–5485. [Google Scholar]
- Oria-Hernández, J.; Cabrera, N.; Pérez-Montfort, R.; Ramírez-Silva, L. Pyruvate kinase revisited. The activating effect of K+. J. Biol. Chem. 2005, 280, 37294–37929. [Google Scholar]
- Larsen, T.M.; Laughlin, T.; Holden, H.M.; Rayment, I.; Reed, G.H. Structure of rabbit muscle pyruvate kinase complexed with Mn2+, K+, and pyruvate. Biochemistry 1994, 33, 6301–6309. [Google Scholar]
- Williams, R.; Holyoak, T.; Mc Donald, G.; Gui, C.; Fenton, A.W. Differentiating a ligand’s chemical requirement for allosteric interactions from those for protein binding. Phenylalanine inhibition of pyruvate kinase. Biochemistry 2006, 45, 5421–5429. [Google Scholar]
- Larsen, T.M.; Benning, M.M.; Wesenberg, G.E.; Rayment, I.; Reed, G.H. Ligand-induced domain movement in pyruvate kinase: Structure of the enzyme from rabbit muscle with Mg2+, K+, and L-phospholactate at 2.7 Ǻ resolution. Arch. Biochem. Biophys. 1997, 345, 199–206. [Google Scholar]
- Larsen, T.M.; Benning, M.M.; Rayment, I.; Reed, G.H. Structure of the bis(Mg2+)-ATP-oxalate complex of the rabbit muscle pyruvate kinase at 2.1 Å resolution: ATP binding over a barrel. Biochemistry 1998, 37, 6247–6255. [Google Scholar]
- Laughlin, I.T.; Reed, G.H. The monovalent cation requirement of rabbit muscle pyruvate kinase is eliminated by substitution of lysine for glutamate 117. Arch. Biochem. Biophys. 1997, 348, 262–267. [Google Scholar]
- Oria-Hernández, J.; Riveros-Rosas, H.; Ramírez-Silva, L. Dichotomic phylogenetic tree of the pyruvate kinase family. K+-dependent and -indepependent enzymes. J. Biol. Chem. 2006, 281, 30717–30724. [Google Scholar]
- Ramírez-Silva, L.; Oria-Hernández, J. Selectivity of pyruvate kinase for Na+ and K+ in water/dimethylsulfoxide mixtures. Eur. J. Biochem. 2003, 270, 2377–2385. [Google Scholar]
- Marcus, Y.A. Simple empirical model describing the thermodynamics of hydration of ions of widely varying charges, sizes, and shapes. Biophys. Chem. 1994, 51, 111–127. [Google Scholar]
- Collins, K.D. Sticky ions in biological systems. Proc. Natl. Acad. Sci. USA 1995, 92, 5553–5557. [Google Scholar]
- Ben-Naim, A.; Marcus, Y. Solvation thermodynamics of nonionic solutes. J. Chem. Phys. 1984, 81, 2016–2027. [Google Scholar]
- Ramírez-Silva, L.; Oria-Hernández, J.; Uribe, S. Function and Structure of Proteins in Water-Cosolvent Binary Systems. In Encyclopedia of Surface and Colloid Science; Somasundaran, P., Ed.; Marcel Dekker, Inc.: New York, NY, USA, 2004. [Google Scholar]
- Tuena de Gómez-Puyou, M.; García, J.J.; Gómez-Puyou, A. Synthesis of pyrophosphate and ATP by soluble mitochondrial F1. Biochemistry 1993, 32, 2213–2218. [Google Scholar]
- Robinson, J.D. Modification of ligand binding to the Na+/K+-activated ATPase. Biochim. Biophys. Acta 1989, 997, 41–48. [Google Scholar]
- De Meis, L.; Martins, O.M.; Alves, E.W. Role of water, hydrogen ion, and temperature on the synthesis of adenosine triphosphate by the sarcoplasmic reticulum adenosine triphosphatase in the absence of a calcium ion gradient. Biochemistry 1980, 19, 4252–4261. [Google Scholar]
- Suzuki, H.; Kanazawa, T. Reduction in water activity greatly retards the phosphoryl transfer from ATP to enzyme protein in the catalytic cycle of sarcoplasmic reticulum Ca2+-ATPase. J. Biol. Chem. 1996, 271, 5481–5486. [Google Scholar]
- Carvalho, P.C.; Scofano, H.M. The hydrolytic cycle of sarcoplasmic reticulum Ca2+-ATPase in the absence of calcium. J. Biol. Chem. 1987, 262, 6610–6614. [Google Scholar]
- Ramírez-Silva, L.; Oria, J.; Gómez-Puyou, A.; Tuena de Gómez-Puyou, M. The contribution of water to the selectivity of pyruvate kinase for Na+ and K+. Eur. J. Biochem. 1997, 250, 583–589. [Google Scholar]
- Suelter, C.H.; Singleton, R., Jr.; Kayne, F.J.; Arrington, S.; Glass, J.; Mildvan, A.S. Studies on the interaction of substrate and monovalent and divalent cations with pyruvate kinase. Biochemistry 1966, 5, 131–139. [Google Scholar]
- Nairn, J.; Duncan, D.; Gray, L.M.; Urquhart, G.; Binnie, M.; Byron, O.; Fothergill-Gilmore, I.A.; Price, N.C. Purification and characterization of pyruvate kinase from Schizosaccharomyces pombe: Evidence for an unusual quaternary structure. Protein Expr. Purif. 1998, 14, 247–253. [Google Scholar]
- Ponce, E.; Flores, N.; Martínez, A.; Valle, F.; Bolívar, F. Cloning of the two pyruvate kinase isoenzyme structural genes from Escherichia coli: The relative roles of these enzymes in pyruvate biosynthesis. J. Bacteriol. 1995, 177, 5719–5722. [Google Scholar]
- Ramírez-Silva, L.; Tuena de Gómez-Puyou, M.; Gómez-Puyou, A. Water induced transitions in the K+ requirements for the activity of pyruvate kinase entrapped in reverse micelles. Biochemistry 1993, 32, 5332–5338. [Google Scholar]
- Büchner, T.; Pleiderer, G. Pyruvate kinase from muscle. In Methods in Enzymology; Colowick, S., Kaplan, N., Eds.; Academic Press: New York, NY, USA, 1955; Volume 1, pp. 435–440. [Google Scholar]
- Schoemakers, T.J.M.; Visser, G.J.; Flik, G.; Theuvenet, P.R. CHELATOR: An improved method for computing metal ion concentrations in physiological solutions. Biotechniques 1992, 12, 870–879. [Google Scholar]
- Dougherty, T.M.; Cleland, W.W. pH Studies of the chemical mechanism of rabbit muscle pyruvate kinase. 2. Physiological substrates and phosphoenol-αketobutyrate. Biochemistry 1985, 24, 5875–5880. [Google Scholar]
- Kaatze, U.; Pottel, R.; Schäfer, M. Dielectric spectrum of dimethyl sulfoxide/water mixtures as a function of composition. J. Phys. Chem. 1989, 93, 5623–5627. [Google Scholar]
- Ku, H.H. Notes on the use of propagation of error formulas. J. Res. NBS. 1966, 70C, 262–273. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramírez-Silva, L.; Guerrero-Mendiola, C.; Cabrera, N. The Importance of Polarity in the Evolution of the K+ Binding Site of Pyruvate Kinase. Int. J. Mol. Sci. 2014, 15, 22214-22226. https://doi.org/10.3390/ijms151222214
Ramírez-Silva L, Guerrero-Mendiola C, Cabrera N. The Importance of Polarity in the Evolution of the K+ Binding Site of Pyruvate Kinase. International Journal of Molecular Sciences. 2014; 15(12):22214-22226. https://doi.org/10.3390/ijms151222214
Chicago/Turabian StyleRamírez-Silva, Leticia, Carlos Guerrero-Mendiola, and Nallely Cabrera. 2014. "The Importance of Polarity in the Evolution of the K+ Binding Site of Pyruvate Kinase" International Journal of Molecular Sciences 15, no. 12: 22214-22226. https://doi.org/10.3390/ijms151222214