Validation of Bmi1 as a Therapeutic Target of Hepatocellular Carcinoma in Mice

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
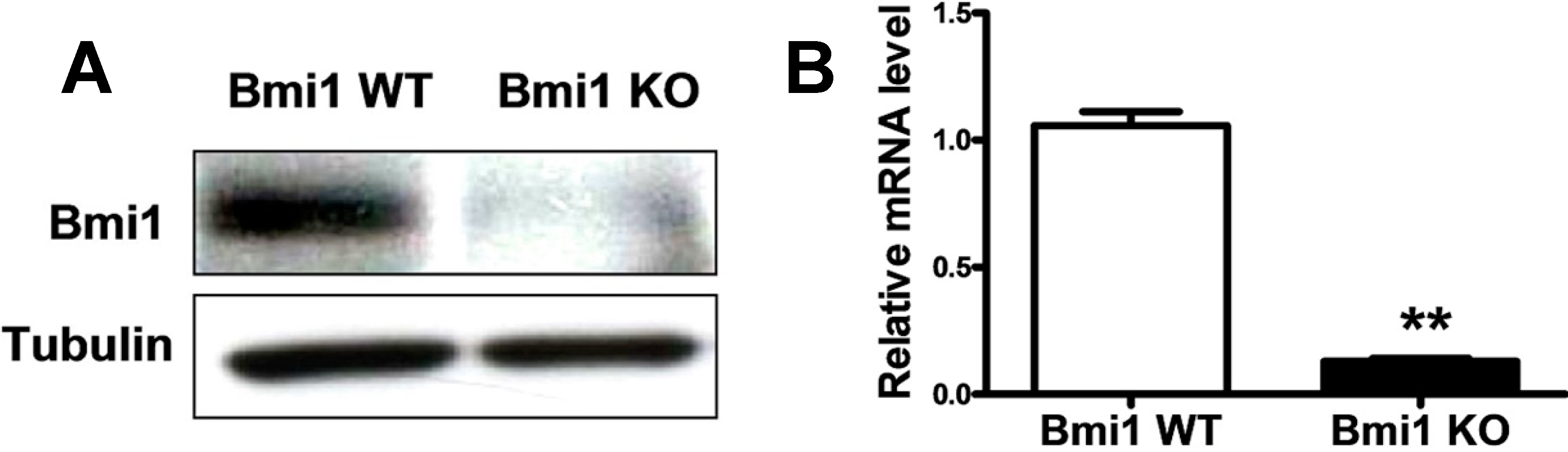
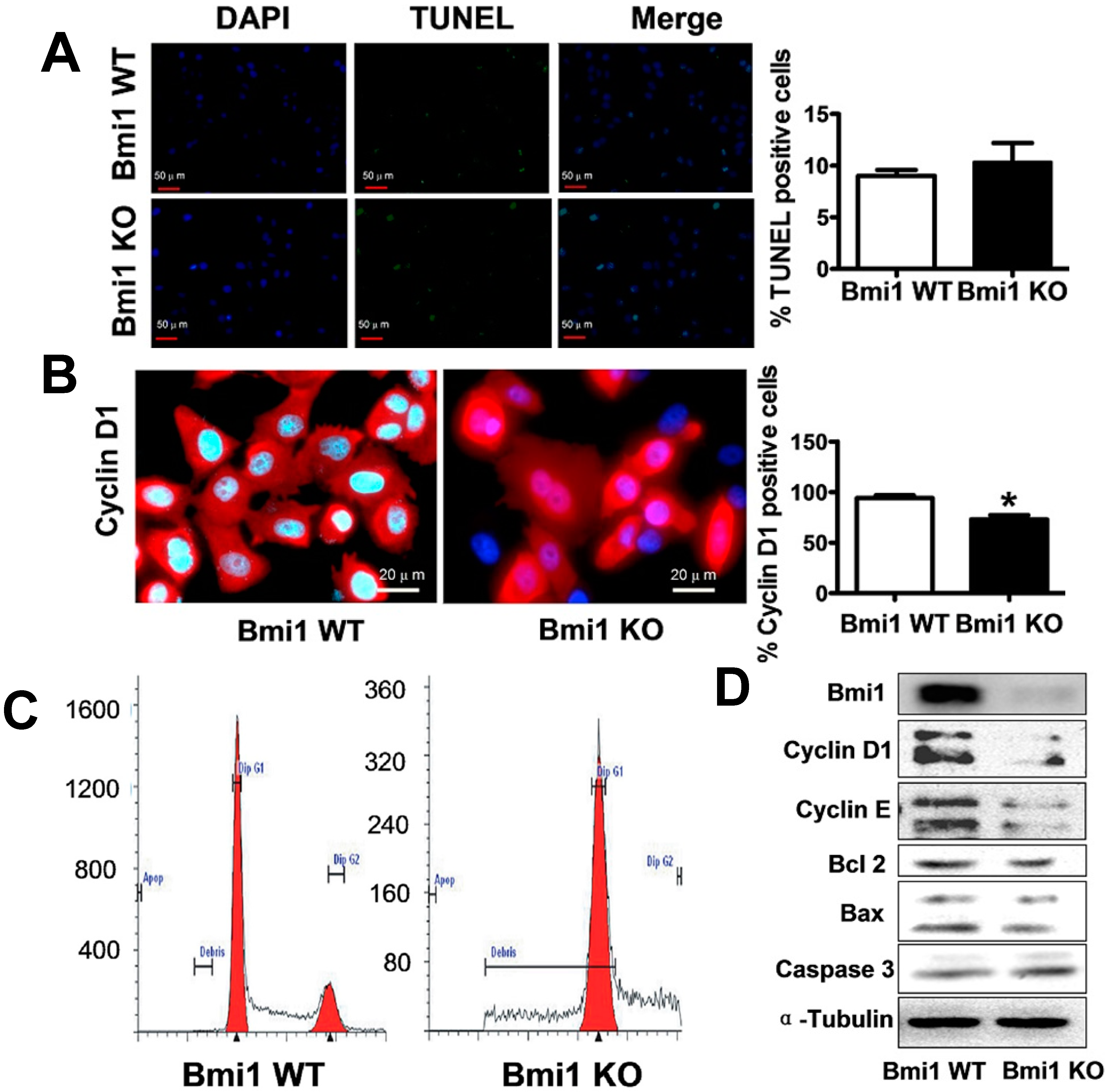
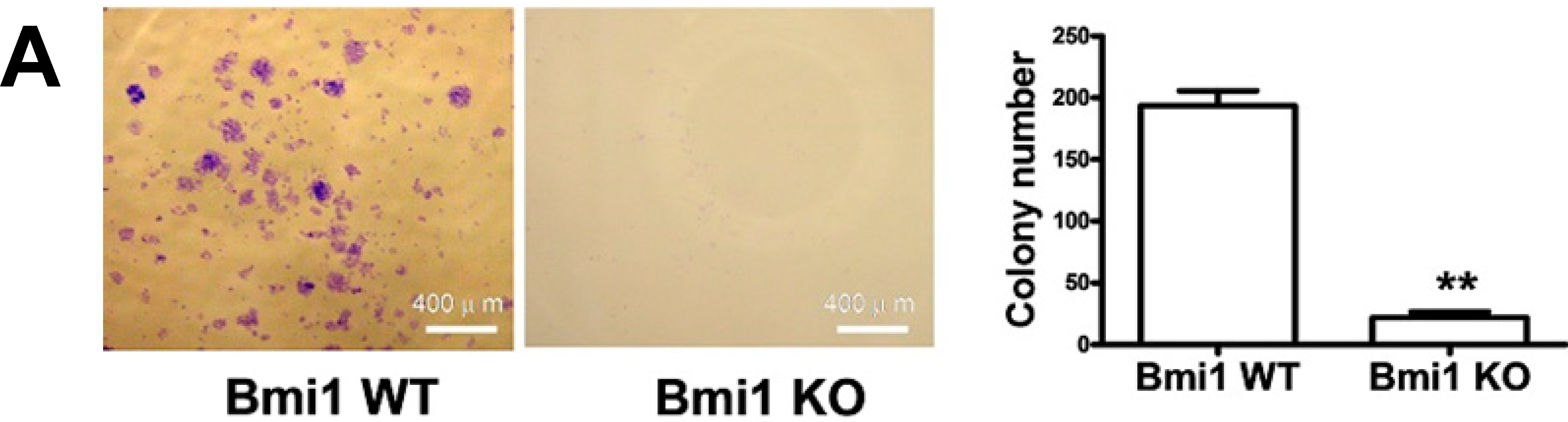
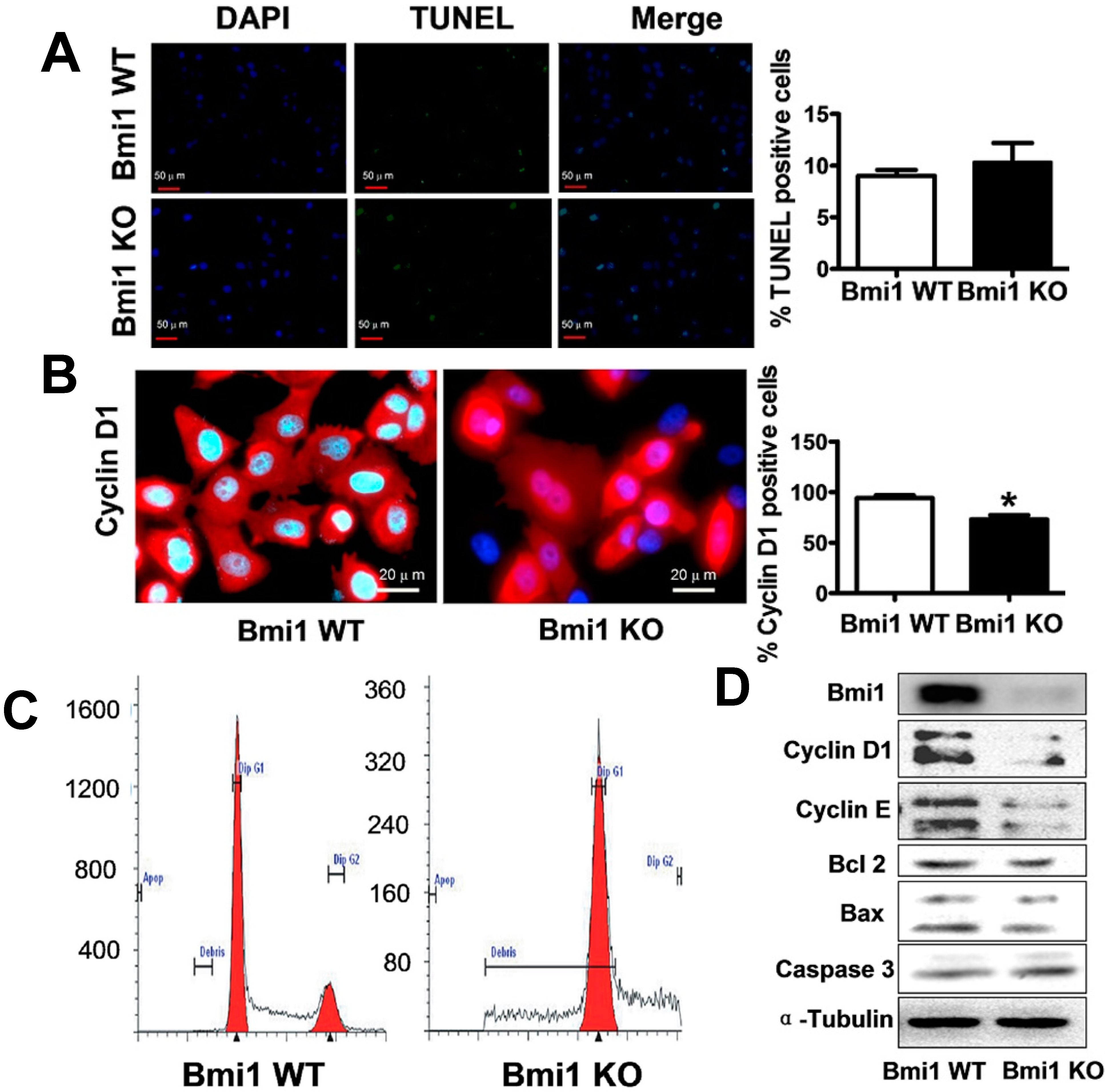
2.1. Bmi1 Knockdown Inhibits the Growth of Hep3B Cells by Inducing Cell Cycle Arrest
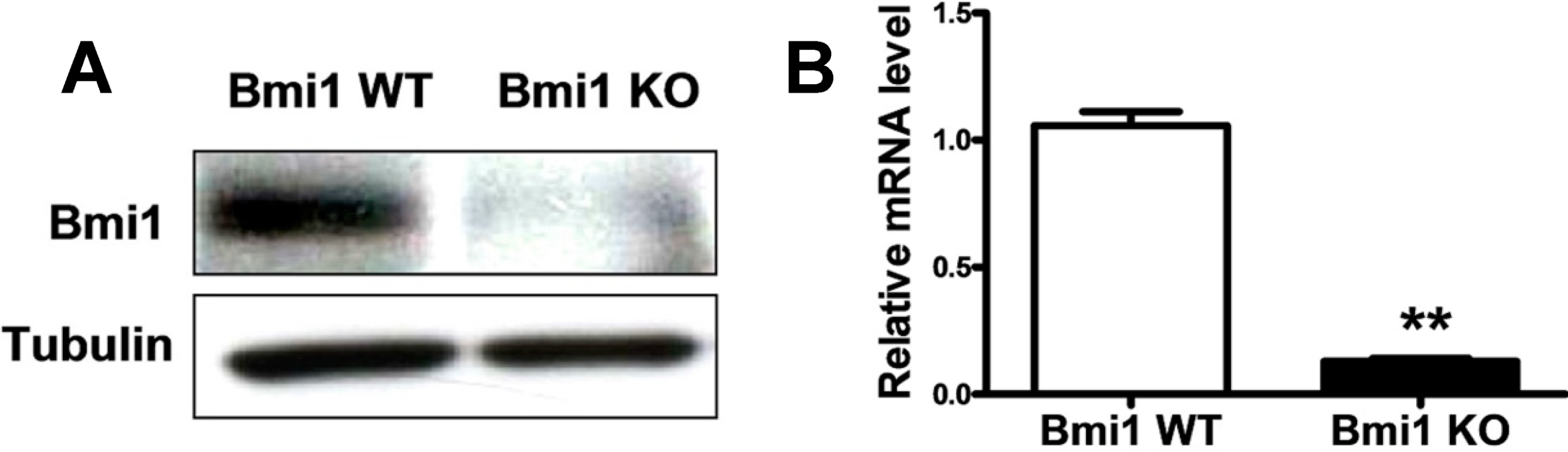
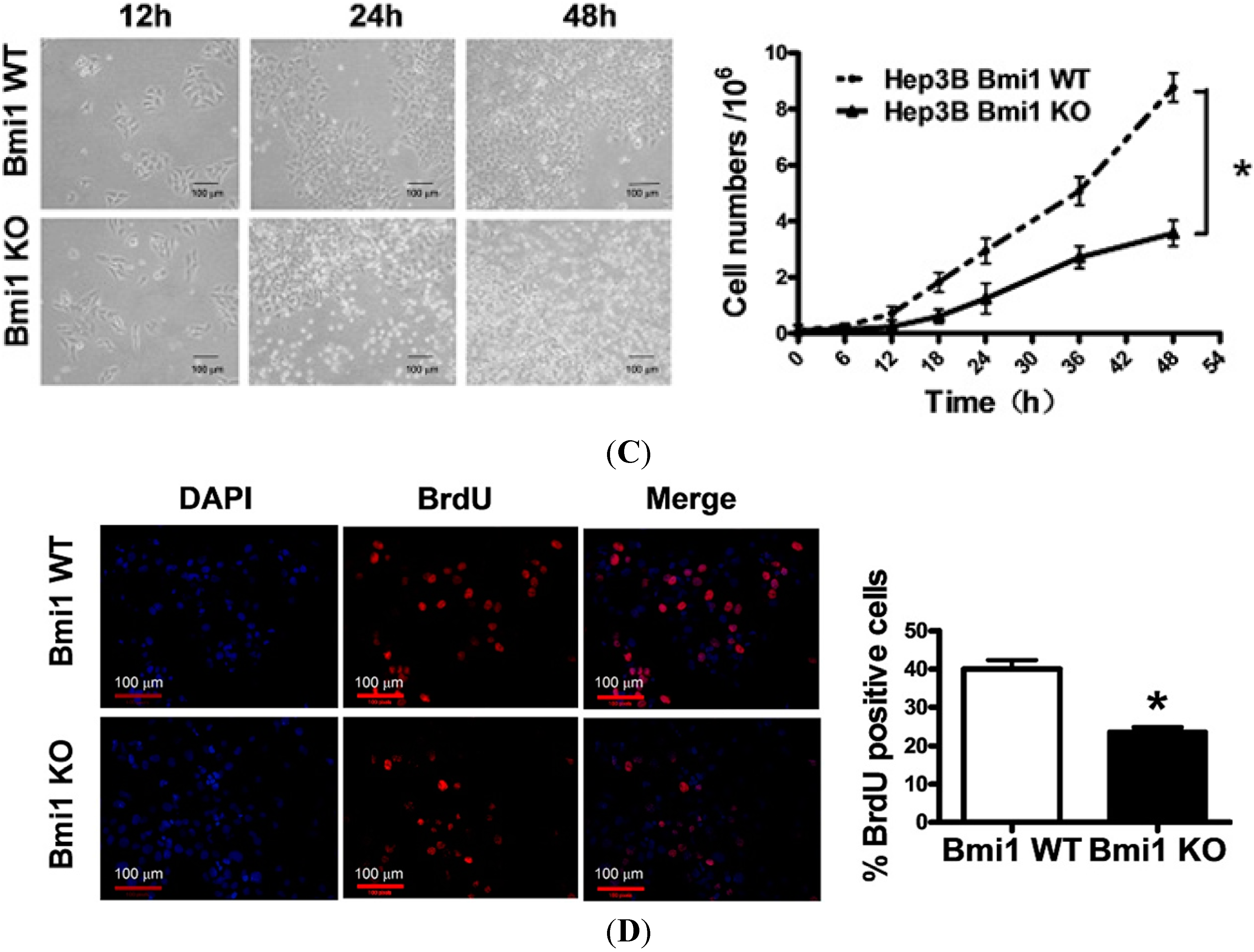
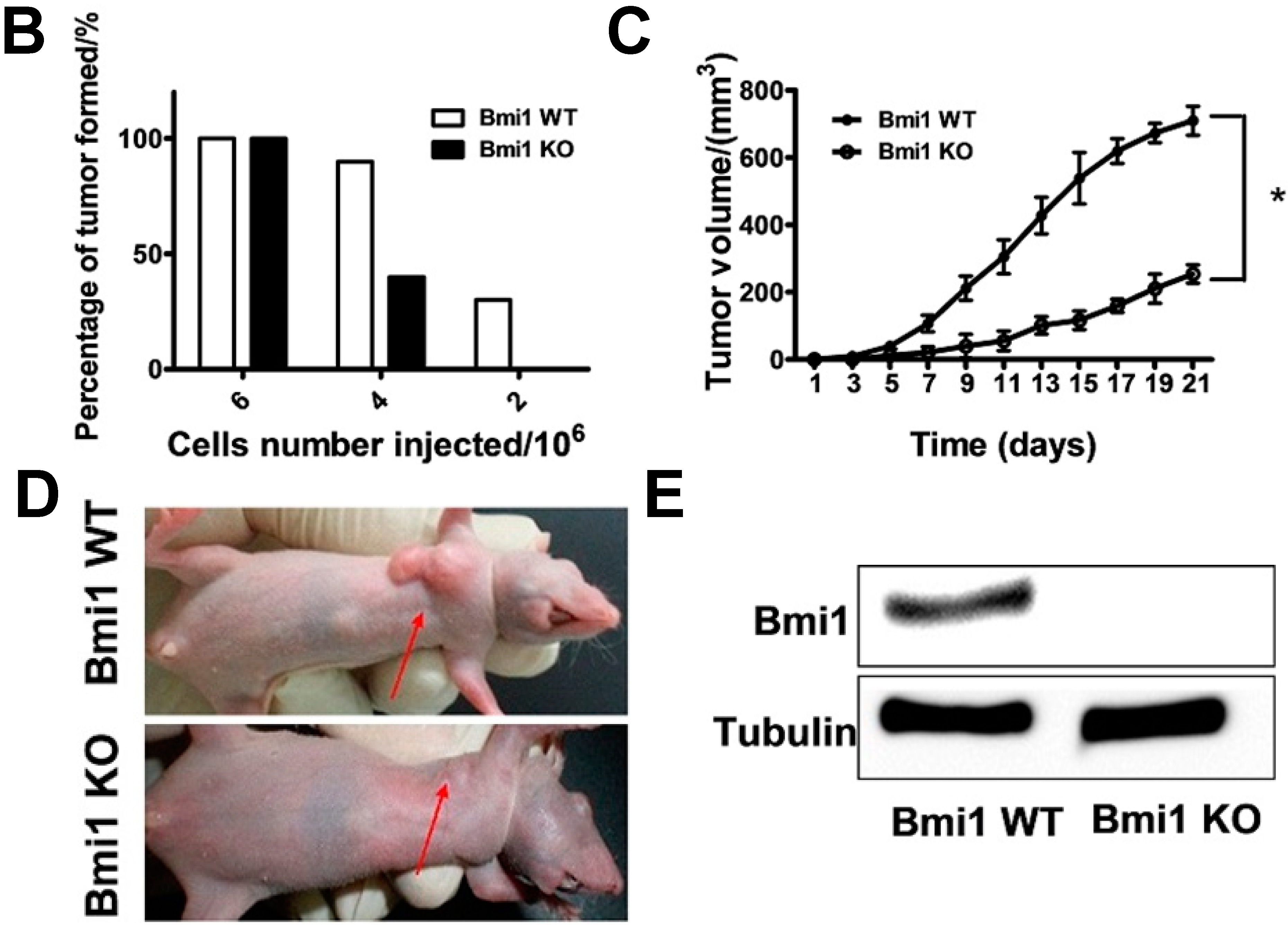
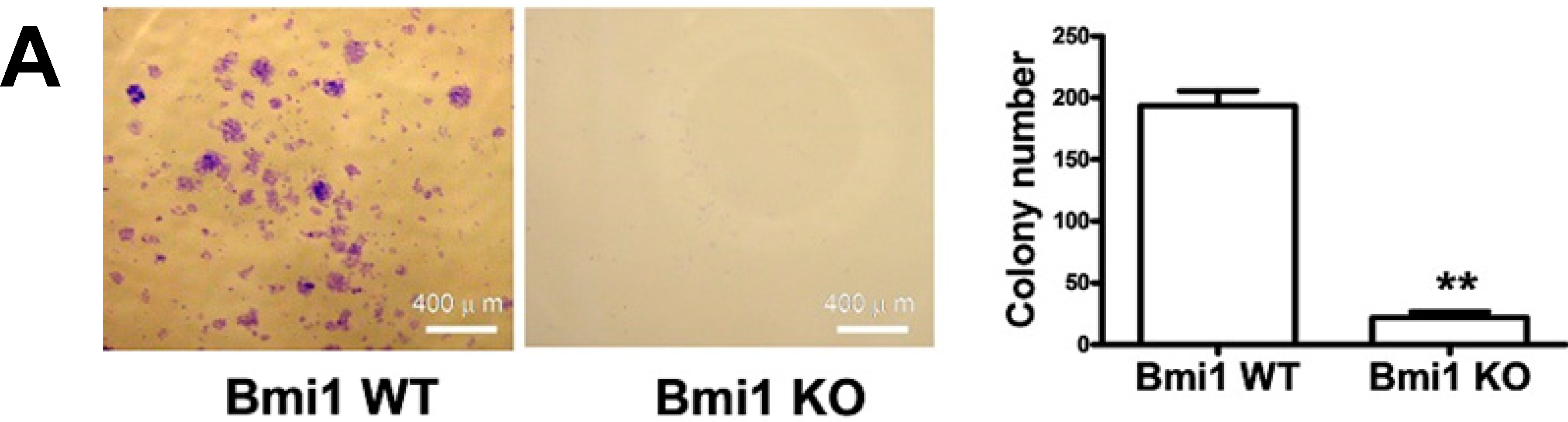
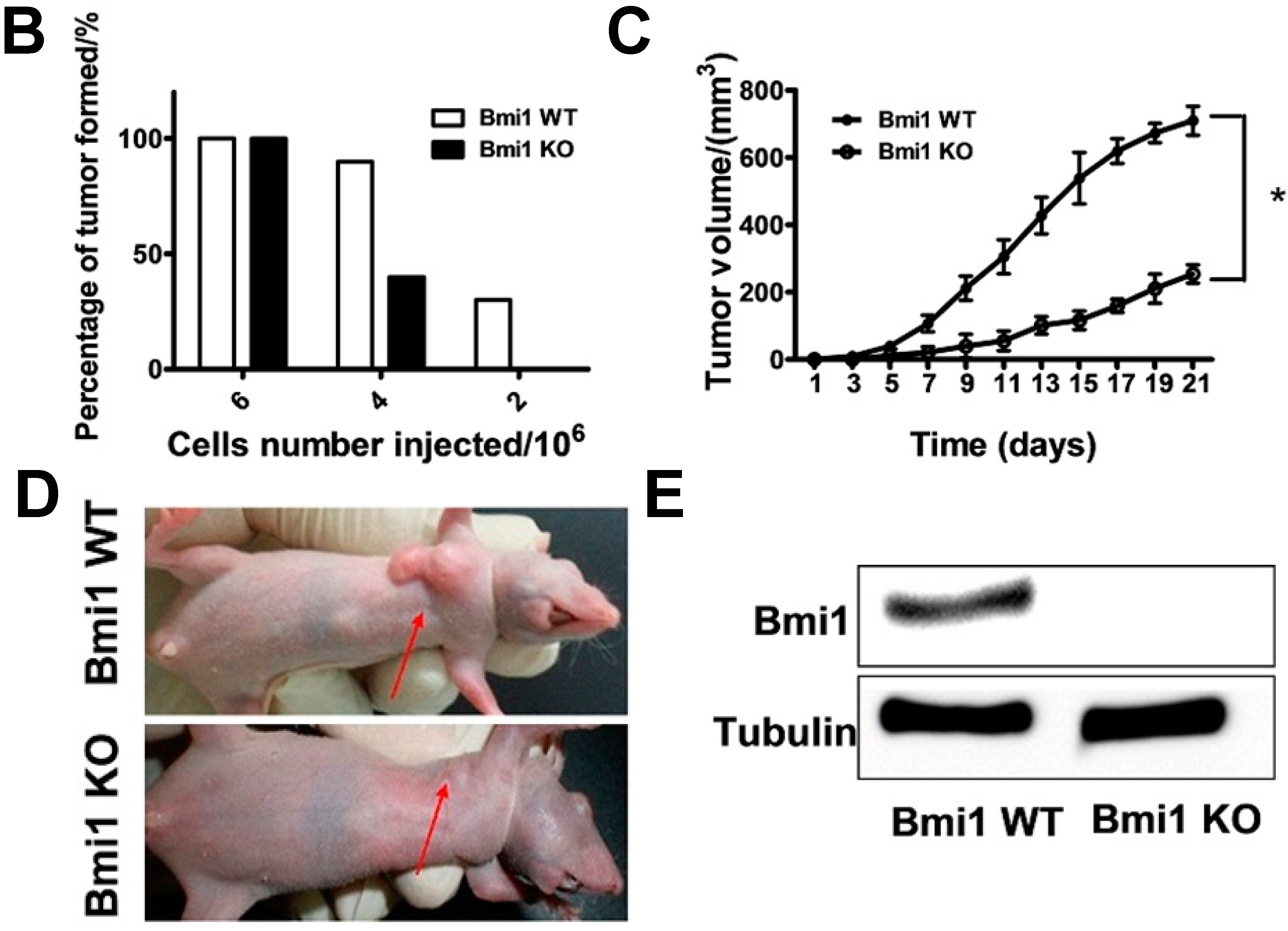
2.2. Bmi1 Knockdown Impairs the Tumor Formation Ability of HCC Cells in Nude Mice
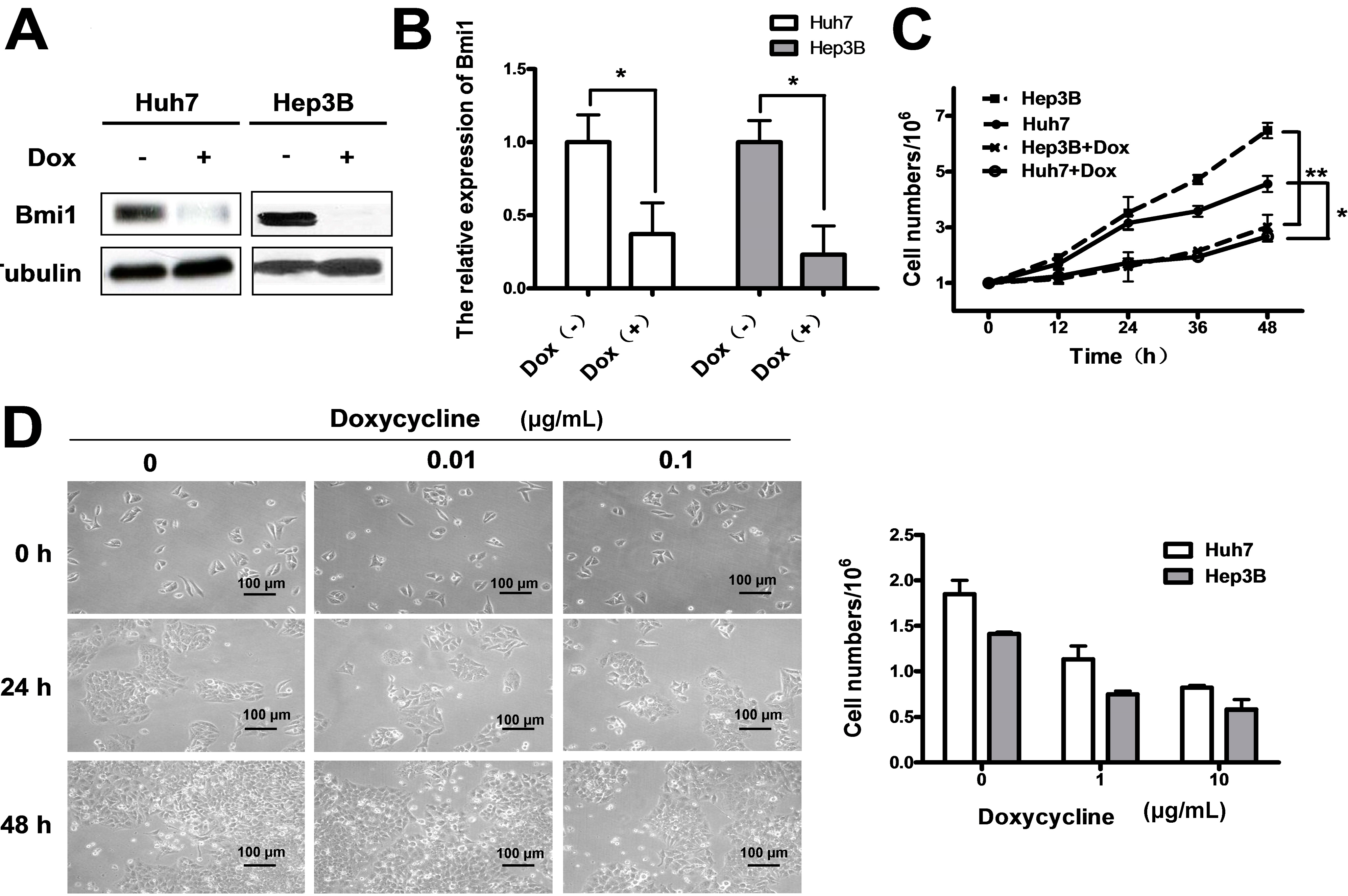
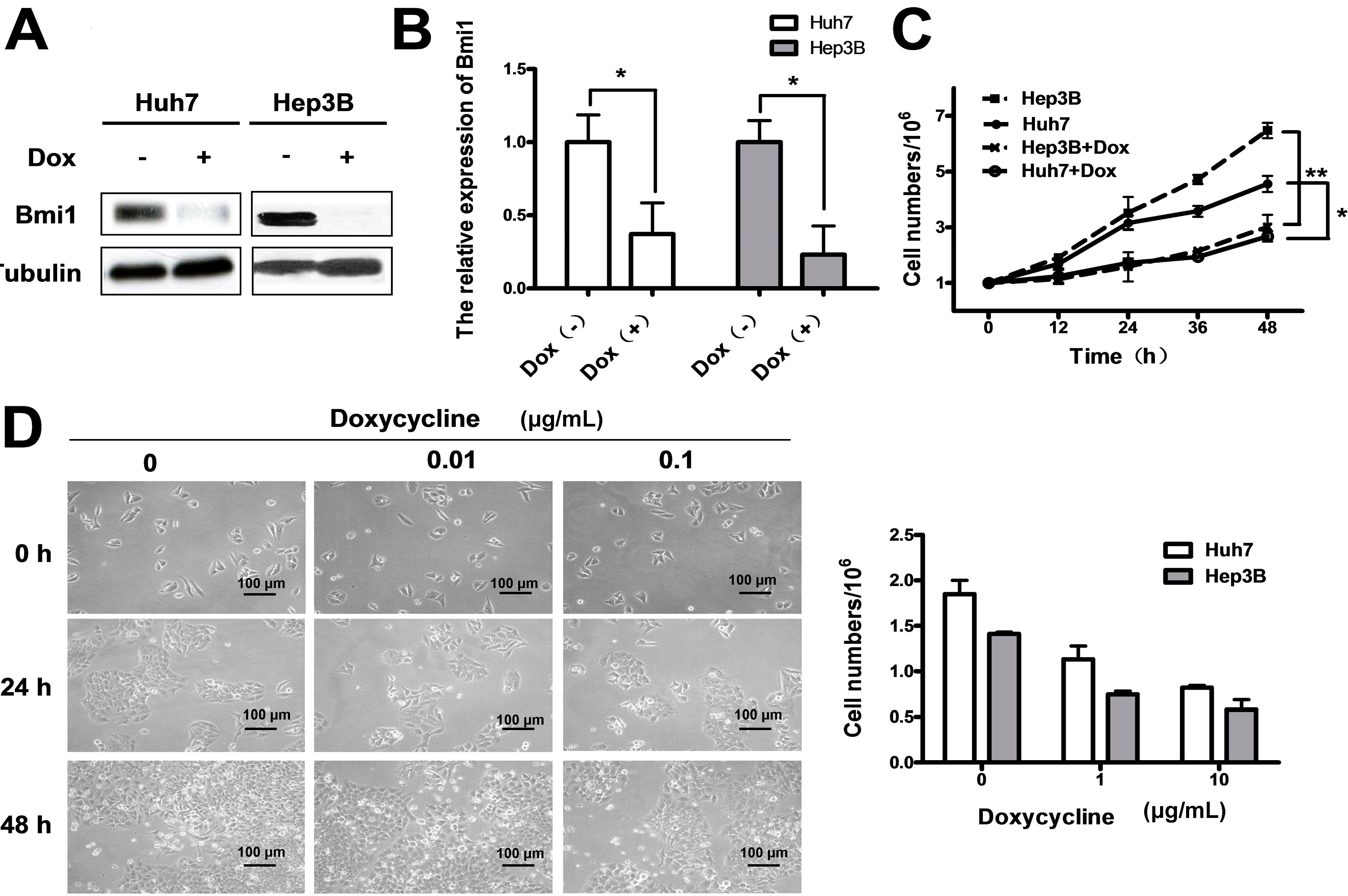
2.3. Conditional Knockdown of Bmi1 by Doxycycline Inhibits the HCC Cell Growth in Vitro
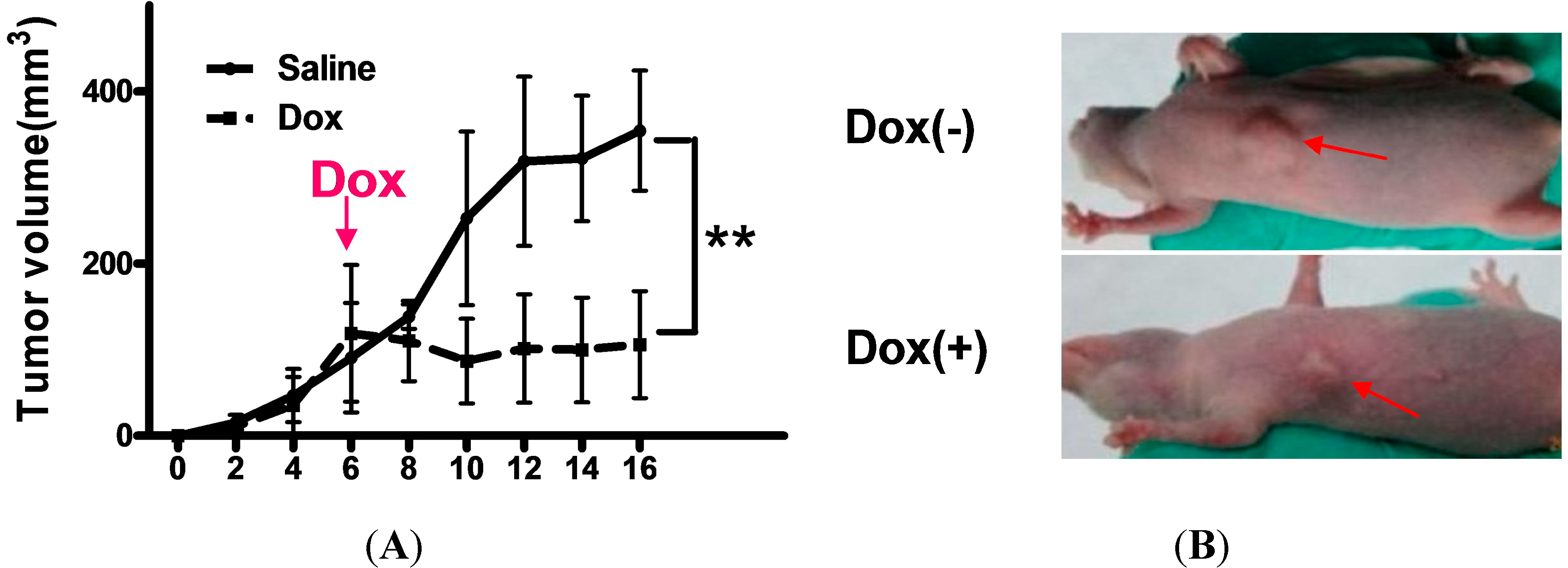
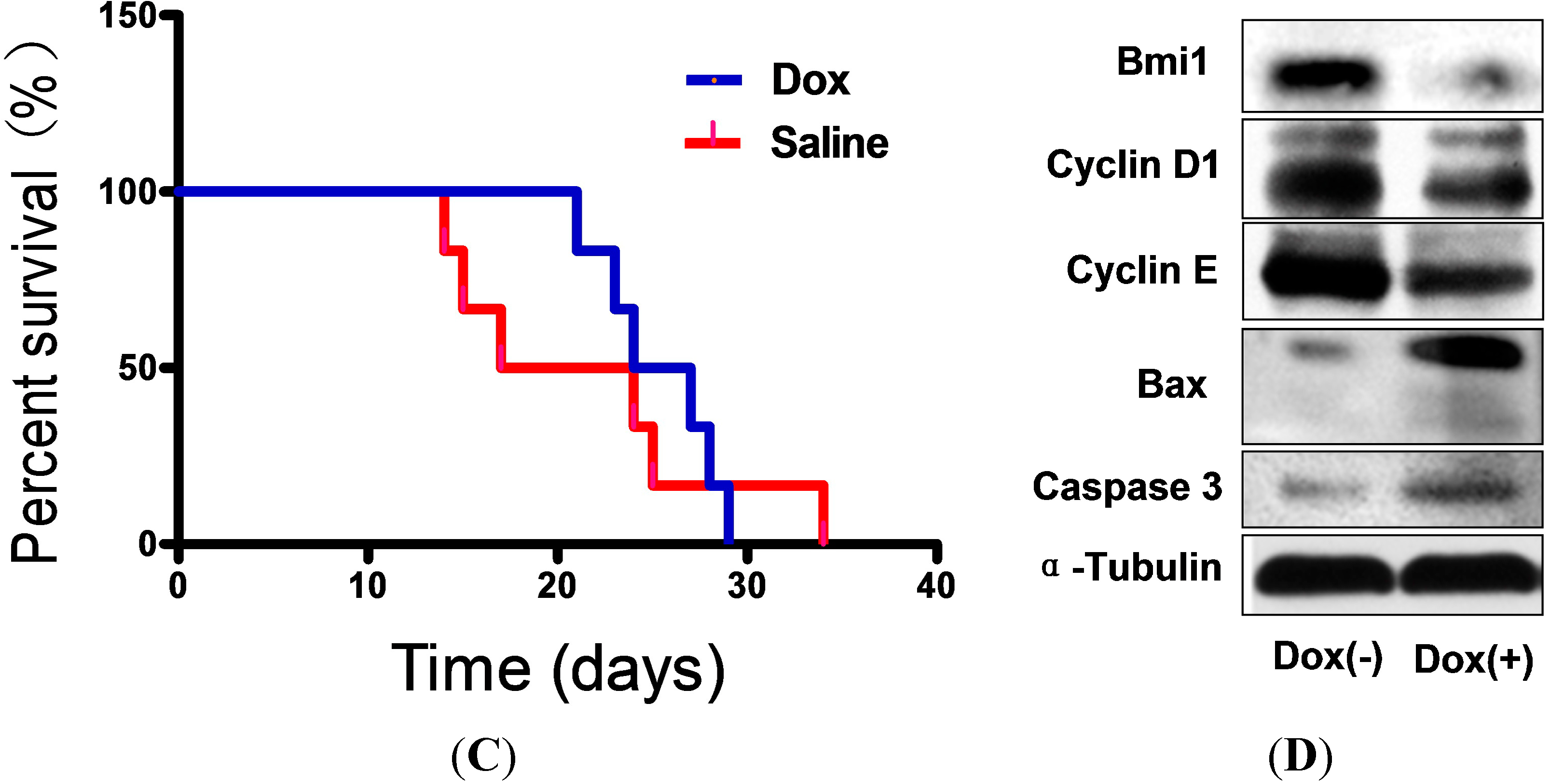
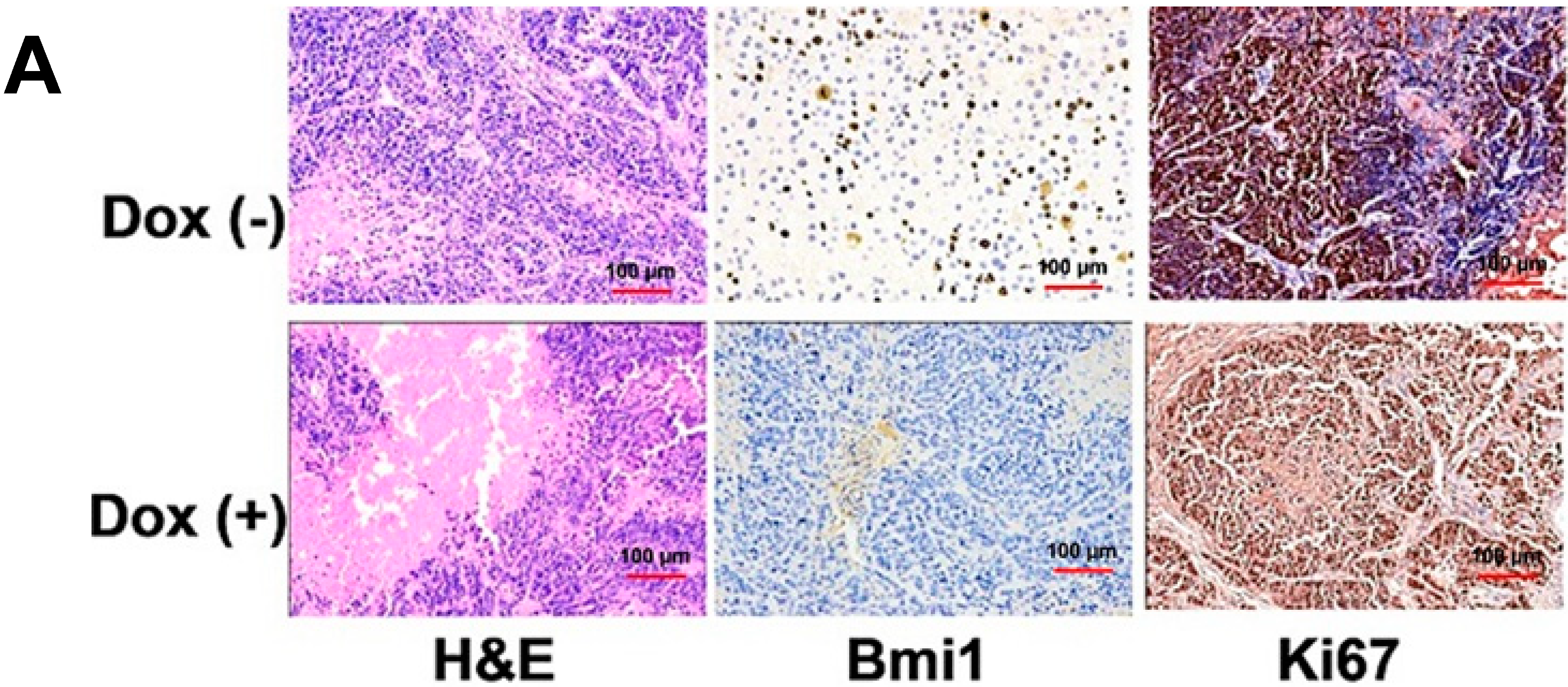
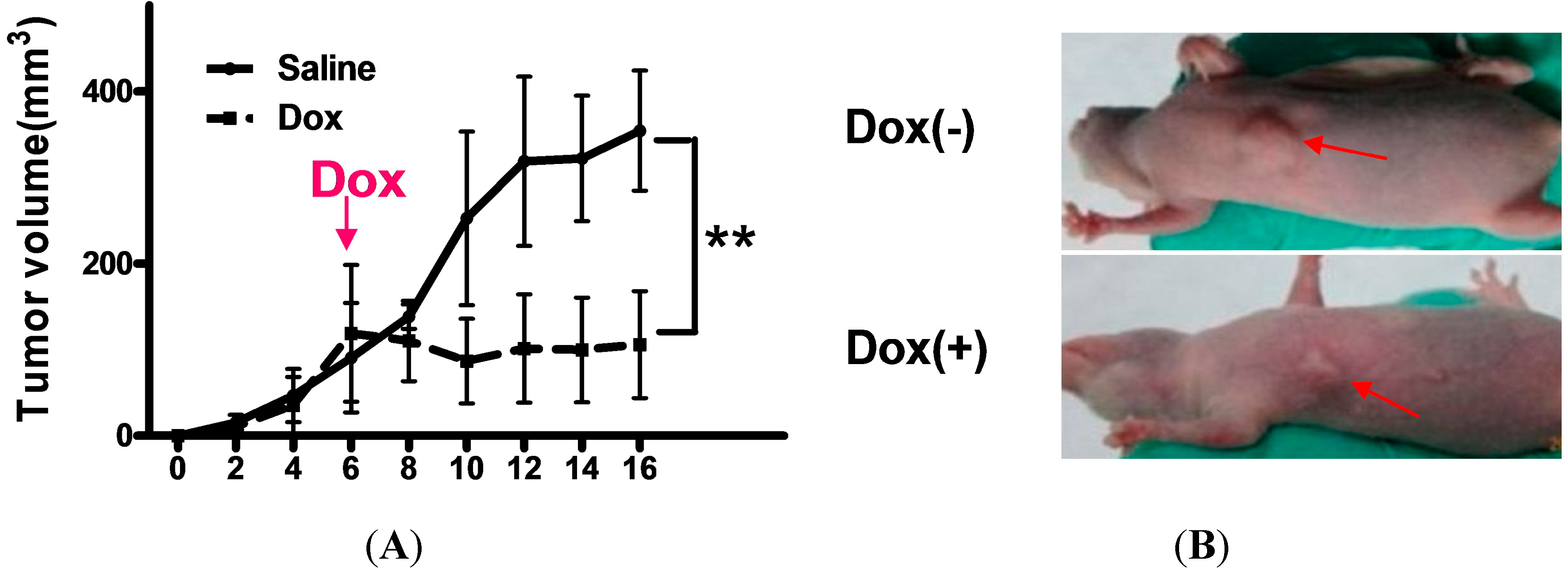
2.4. Shutdown of Bmi1 Represses the Tumor Growth in the Fast Growing Period in Nude Mice



2.5. Discussion
3. Experimental Section
3.1. Cell Culture, Short Hairpin RNA Constructs, Lentivirus and Infection
3.2. RNA Extraction and Real-Time qPCR
3.3. Protein Lysates and Western Blotting
3.4. Cell Proliferation and Cell Cycle Assays
3.5. Analysis of Clonogenicity in Vitro
3.6. Mice and Xenograft Mouse Model
3.7. Histology, Immunohistochemistry and Immunofluorescence
3.8. Microscopy
3.9. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Roberts, L.R. Sorafenib in liver cancer—Just the beginning. N. Engl. J. Med. 2008, 359, 420–422. [Google Scholar]
- Shariff, M.I.; Cox, I.J.; Gomaa, A.I.; Khan, S.A.; Gedroyc, W.; Taylor-Robinson, S.D. Hepatocellular carcinoma: Current trends in worldwide epidemiology, risk factors, diagnosis and therapeutics. Expert Rev. Gastroenterol. Hepatol. 2009, 3, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, E.; Unoura, M.; Kaneko, S.; Kobayashi, K. Risk factors for development of hepatocellular carcinoma in patients with liver cirrhosis associated with hepatitis C virus. Nihon Rinsho 1995, 53, 727–730. [Google Scholar] [PubMed]
- Zaman, S.N.; Melia, W.M.; Johnson, R.D.; Portmann, B.C.; Johnson, P.J.; Williams, R. Risk factors in development of hepatocellular carcinoma in cirrhosis: Prospective study of 613 patients. Lancet 1985, 1, 1357–1360. [Google Scholar] [CrossRef] [PubMed]
- Feo, F.; Pascale, R.M.; Simile, M.M.; de Miglio, M.R.; Muroni, M.R.; Calvisi, D. Genetic alterations in liver carcinogenesis: Implications for new preventive and therapeutic strategies. Crit. Rev. Oncog. 2000, 11, 19–62. [Google Scholar] [CrossRef] [PubMed]
- Thorgeirsson, S.S.; Grisham, J.W. Molecular pathogenesis of human hepatocellular carcinoma. Nat. Genet. 2002, 31, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, S.; Marais, R.; Zhu, A.X. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene 2010, 29, 4989–5005. [Google Scholar] [CrossRef] [PubMed]
- Forner, A.; Llovet, J.M.; Bruix, J. Hepatocellular carcinoma. Lancet 2012, 379, 1245–1255. [Google Scholar] [CrossRef] [PubMed]
- Levy, L.; Renard, C.A.; Wei, Y.; Buendia, M.A. Genetic alterations and oncogenic pathways in hepatocellular carcinoma. Ann. N. Y. Acad. Sci. 2002, 963, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.J.; Kieboom, K.; Marino, S.; de Pinho, R.A.; van Lohuizen, M. The oncogene and Polycomb-group gene Bmi1 regulates cell proliferation and senescence through the INK4a locus. Nature 1999, 397, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Park, I.K.; Qian, D.; Kiel, M.; Becker, M.W.; Pihalja, M.; Weissman, I.L.; Morrison, S.J.; Clarke, M.F. Bmi1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature 2003, 423, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.V.; Pardal, R.; Iwashita, T.; Park, I.K.; Clarke, M.F.; Morrison, S.J. Bmi1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature 2003, 425, 962–967. [Google Scholar] [CrossRef]
- Park, I.K.; Morrison, S.J.; Clarke, M.F. Bmi1, stem cells, and senescence regulation. J. Clin. Investig. 2004, 113, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.J.; Scheijen, B.; Voncken, J.W.; Kieboom, K.; Berns, A.; van Lohuizen, M. Bmi1 collaborates with c-Myc in tumorigenesis by inhibiting c-Myc-induced apoptosis via INK4a/ARF. Genes Dev. 1999, 13, 2678–2690. [Google Scholar] [CrossRef] [PubMed]
- Pietersen, A.M.; Horlings, H.M.; Hauptmann, M.; Langerod, A.; Ajouaou, A.; Cornelissen-Steijger, P.; Wessels, L.F.; Jonkers, J.; van de Vijver, M.J.; van Lohuizen, M. EZH2 and Bmi1 inversely correlate with prognosis and TP53 mutation in breast cancer. Breast Cancer Res. 2008, 10, R109. [Google Scholar]
- Dhawan, S.; Tschen, S.I.; Bhushan, A. Bmi1 regulates the INK4a/ARF locus to control pancreatic beta-cell proliferation. Genes Dev. 2009, 23, 906–911. [Google Scholar] [CrossRef]
- Becker, M.; Korn, C.; Sienerth, A.R.; Voswinckel, R.; Luetkenhaus, K.; Ceteci, F.; Rapp, U.R. Polycomb group protein Bmi1 is required for growth of RAF driven non-small-cell lung cancer. PLoS One 2009, 4, e4230. [Google Scholar] [CrossRef]
- Dovey, J.S.; Zacharek, S.J.; Kim, C.F.; Lees, J.A. Bmi1 is critical for lung tumorigenesis and bronchioalveolar stem cell expansion. Proc. Natl. Acad. Sci. USA 2008, 105, 11857–11862. [Google Scholar] [CrossRef] [PubMed]
- Bruggeman, S.W.; Hulsman, D.; Tanger, E.; Buckle, T.; Blom, M.; Zevenhoven, J.; van Tellingen, O.; van Lohuizen, M. Bmi1 controls tumor development in an INK4a/ARF-independent manner in a mouse model for glioma. Cancer Cell 2007, 12, 328–341. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Bhattacharyya, S.; Szabolcs, A.; Rodriguez-Aguayo, C.; Jennings, N.B.; Lopez-Berestein, G.; Mukherjee, P.; Sood, A.K.; Bhattacharya, R. Enhancing chemotherapy response with Bmi1 silencing in ovarian cancer. PLoS One 2011, 6, e17918. [Google Scholar]
- Qin, Z.K.; Yang, J.A.; Ye, Y.L.; Zhang, X.; Xu, L.H.; Zhou, F.J.; Han, H.; Liu, Z.W.; Song, L.B.; Zeng, M.S. Expression of Bmi1 is a prognostic marker in bladder cancer. BMC Cancer 2009, 9, 61. [Google Scholar] [CrossRef] [PubMed]
- Song, L.B.; Zeng, M.S.; Liao, W.T.; Zhang, L.; Mo, H.Y.; Liu, W.L.; Shao, J.Y.; Wu, Q.L.; Li, M.Z.; Xia, Y.F.; et al. Bmi1 is a novel molecular marker of nasopharyngeal carcinoma progression and immortalizes primary human nasopharyngeal epithelial cells. Cancer Res 2006, 66, 6225–6232. [Google Scholar] [CrossRef] [PubMed]
- Mohty, M.; Yong, A.S.; Szydlo, R.M.; Apperley, J.F.; Melo, J.V. The polycomb group Bmi1 gene is a molecular marker for predicting prognosis of chronic myeloid leukemia. Blood 2007, 110, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Miyagi, S.; Saraya, A.; Aoki, R.; Seki, A.; Morita, Y.; Yonemitsu, Y.; Yokosuka, O.; Taniguchi, H.; Nakauchi, H.; et al. The polycomb gene product Bmi1 contributes to the maintenance of tumor-initiating side population cells in hepatocellular carcinoma. Cancer Res. 2008, 68, 7742–7749. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.R.; Lee, S.; Ho, C.; Bommi, P.; Huang, S.A.; Cheung, S.T.; Dimri, G.P.; Chen, X. Bmi1 functions as an oncogene independent of INK4a/ARF repression in hepatic carcinogenesis. Mol. Cancer Res. 2009, 7, 1937–1945. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Seki, A.; Aoki, R.; Ichikawa, H.; Negishi, M.; Miyagi, S.; Oguro, H.; Saraya, A.; Kamiya, A.; Nakauchi, H.; et al. Bmi1 promotes hepatic stem cell expansion and tumorigenicity in both INK4a/ARF-dependent and -independent manners in mice. Hepatology 2010, 52, 1111–1123. [Google Scholar] [CrossRef] [PubMed]
- Sparmann, A.; van Lohuizen, M. Polycomb silencers control cell fate, development and cancer. Nat. Rev. Cancer 2006, 6, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Douglas, D.; Hsu, J.H.; Hung, L.; Cooper, A.; Abdueva, D.; van Doorninck, J.; Peng, G.; Shimada, H.; Triche, T.J.; Lawlor, E.R. Bmi1 promotes ewing sarcoma tumorigenicity independent of CDKN2A repression. Cancer Res. 2008, 68, 6507–6515. [Google Scholar] [CrossRef] [PubMed]
- Wiederschain, D.; Chen, L.; Johnson, B.; Bettano, K.; Jackson, D.; Taraszka, J.; Wang, Y.K.; Jones, M.D.; Morrissey, M.; Deeds, J.; et al. Contribution of polycomb homologues Bmi1 and Mel-18 to medulloblastoma pathogenesis. Mol. Cell Biol. 2007, 27, 4968–4979. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; He, L.; Kapoor, A.; Gillis, A.; Rybak, A.P.; Cutz, J.C.; Tang, D. Bmi1 promotes prostate tumorigenesis via inhibiting p16INK4a and p14ARF expression. Biochim. Biophys. Acta 2008, 1782, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Zheng, Y.W.; Kita, K.; Yokosuka, O.; Saisho, H.; Onodera, M.; Miyoshi, H.; Nakano, M.; Zen, Y.; Nakanuma, Y.; et al. Enhanced self-renewal capability in hepatic stem/progenitor cells drives cancer initiation. Gastroenterology 2007, 133, 937–950. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Z.P.; Xu, R.; Lv, Y.; Tian, T.; Wang, W.J.; Guo, H.; Nan, K.J. Bmi1 knockdown inhibits hepatocarcinogenesis. Int. J. Oncol. 2013, 42, 261–268. [Google Scholar] [PubMed]
- Zhang, R.; Xu, L.B.; Yue, X.J.; Yu, X.H.; Wang, J.; Liu, C. Bmi1 gene silencing inhibits the proliferation and invasiveness of human hepatocellular carcinoma cells and increases their sensitivity to 5-fluorouracil. Oncol. Rep. 2013, 29, 967–974. [Google Scholar] [PubMed]
- Wu, Z.; Min, L.; Chen, D.; Hao, D.; Duan, Y.; Qiu, G.; Wang, Y. Overexpression of Bmi1 promotes cell growth and resistance to cisplatin treatment in osteosarcoma. PLoS One 2011, 6, e14648. [Google Scholar]
- Jagani, Z.; Wiederschain, D.; Loo, A.; He, D.; Mosher, R.; Fordjour, P.; Monahan, J.; Morrissey, M.; Yao, Y.M.; Lengauer, C.; et al. The Polycomb group protein Bmi1 is essential for the growth of multiple myeloma cells. Cancer Res. 2010, 70, 5528–5538. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Hoenerhoff, M.J.; Bommi, P.; Sainger, R.; Guo, W.J.; Dimri, M.; Band, H.; Band, V.; Green, J.E.; Dimri, G.P. Bmi1 cooperates with H-Ras to transform human mammary epithelial cells via dysregulation of multiple growth-regulatory pathways. Cancer Res. 2007, 67, 10286–10295. [Google Scholar] [CrossRef] [PubMed]
- Hoenerhoff, M.J.; Chu, I.; Barkan, D.; Liu, Z.Y.; Datta, S.; Dimri, G.P.; Green, J.E. Bmi1 cooperates with H-Ras to induce an aggressive breast cancer phenotype with brain metastases. Oncogene 2009, 28, 3022–3032. [Google Scholar] [CrossRef] [PubMed]
- Lessard, J.; Sauvageau, G. Bmi1 determines the proliferative capacity of normal and leukaemic stem cells. Nature 2003, 423, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Tward, A.D.; Jones, K.D.; Yant, S.; Cheung, S.T.; Fan, S.T.; Chen, X.; Kay, M.A.; Wang, R.; Bishop, J.M. Distinct pathways of genomic progression to benign and malignant tumors of the liver. Proc. Natl. Acad. Sci. USA 2007, 104, 14771–14776. [Google Scholar] [CrossRef] [PubMed]
- Wojtowicz, J.M.; Kee, N. BrdU assay for neurogenesis in rodents. Nat. Protoc. 2006, 1, 1399–1405. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qi, S.; Li, B.; Yang, T.; Liu, Y.; Cao, S.; He, X.; Zhang, P.; Li, L.; Xu, C. Validation of Bmi1 as a Therapeutic Target of Hepatocellular Carcinoma in Mice. Int. J. Mol. Sci. 2014, 15, 20004-20021. https://doi.org/10.3390/ijms151120004
Qi S, Li B, Yang T, Liu Y, Cao S, He X, Zhang P, Li L, Xu C. Validation of Bmi1 as a Therapeutic Target of Hepatocellular Carcinoma in Mice. International Journal of Molecular Sciences. 2014; 15(11):20004-20021. https://doi.org/10.3390/ijms151120004
Chicago/Turabian StyleQi, Shibo, Bin Li, Tan Yang, Yong Liu, Shanshan Cao, Xingxing He, Peng Zhang, Lei Li, and Chuanrui Xu. 2014. "Validation of Bmi1 as a Therapeutic Target of Hepatocellular Carcinoma in Mice" International Journal of Molecular Sciences 15, no. 11: 20004-20021. https://doi.org/10.3390/ijms151120004