Obligatory Role of Intraluminal O2− in Acute Endothelin-1 and Angiotensin II Signaling to Mediate Endothelial Dysfunction and MAPK Activation in Guinea-Pig Hearts

Abstract
:1. Introduction
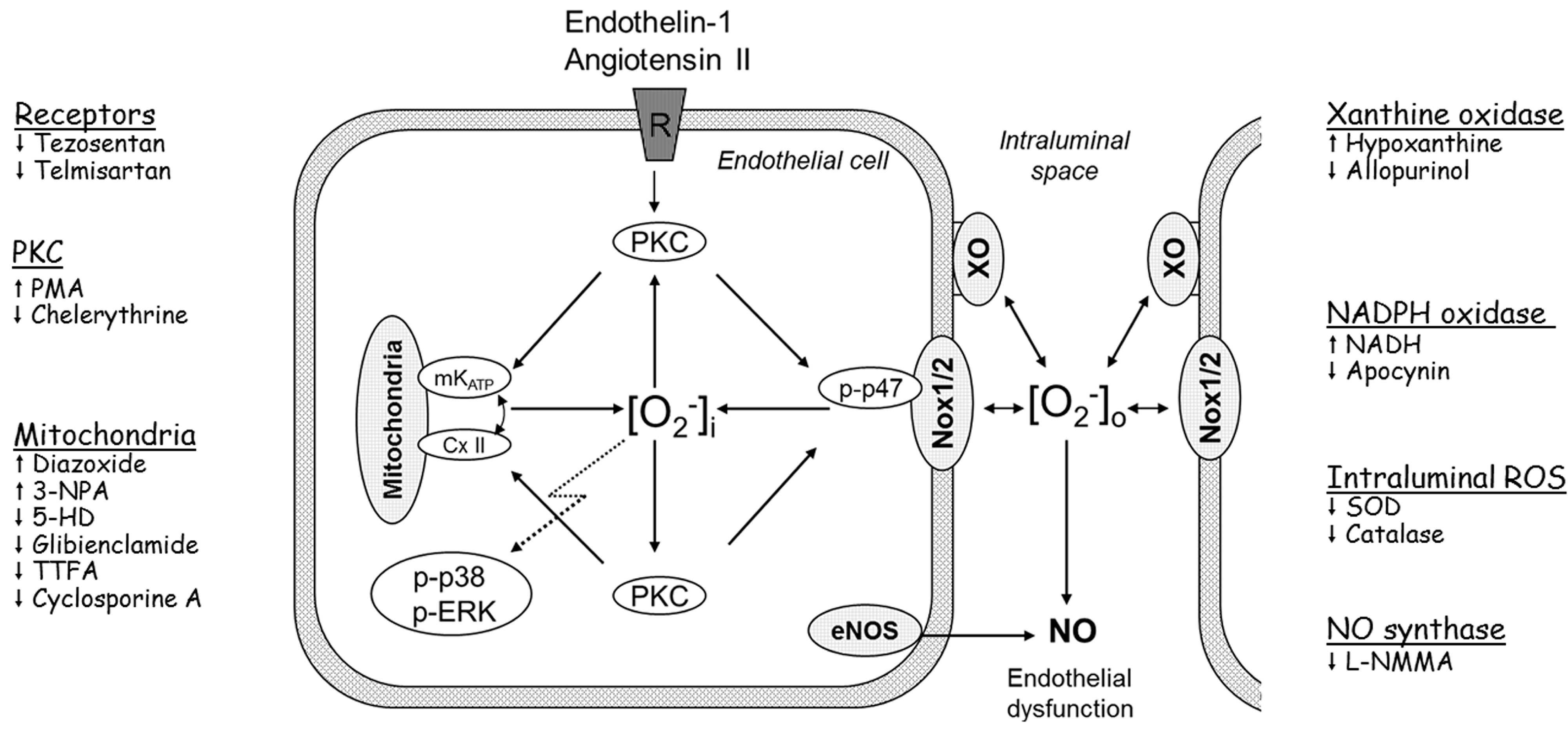
2. Results and Discussion
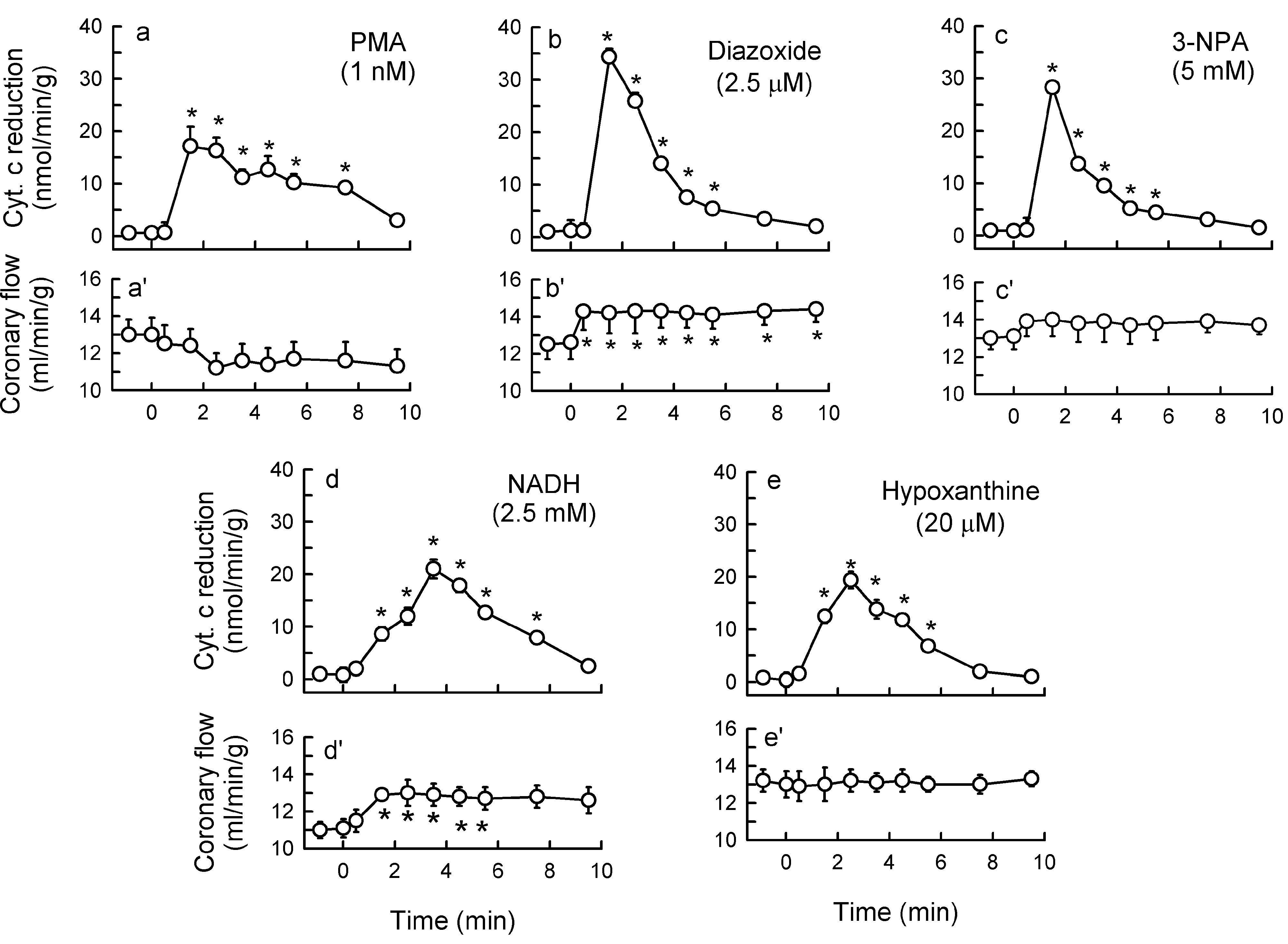
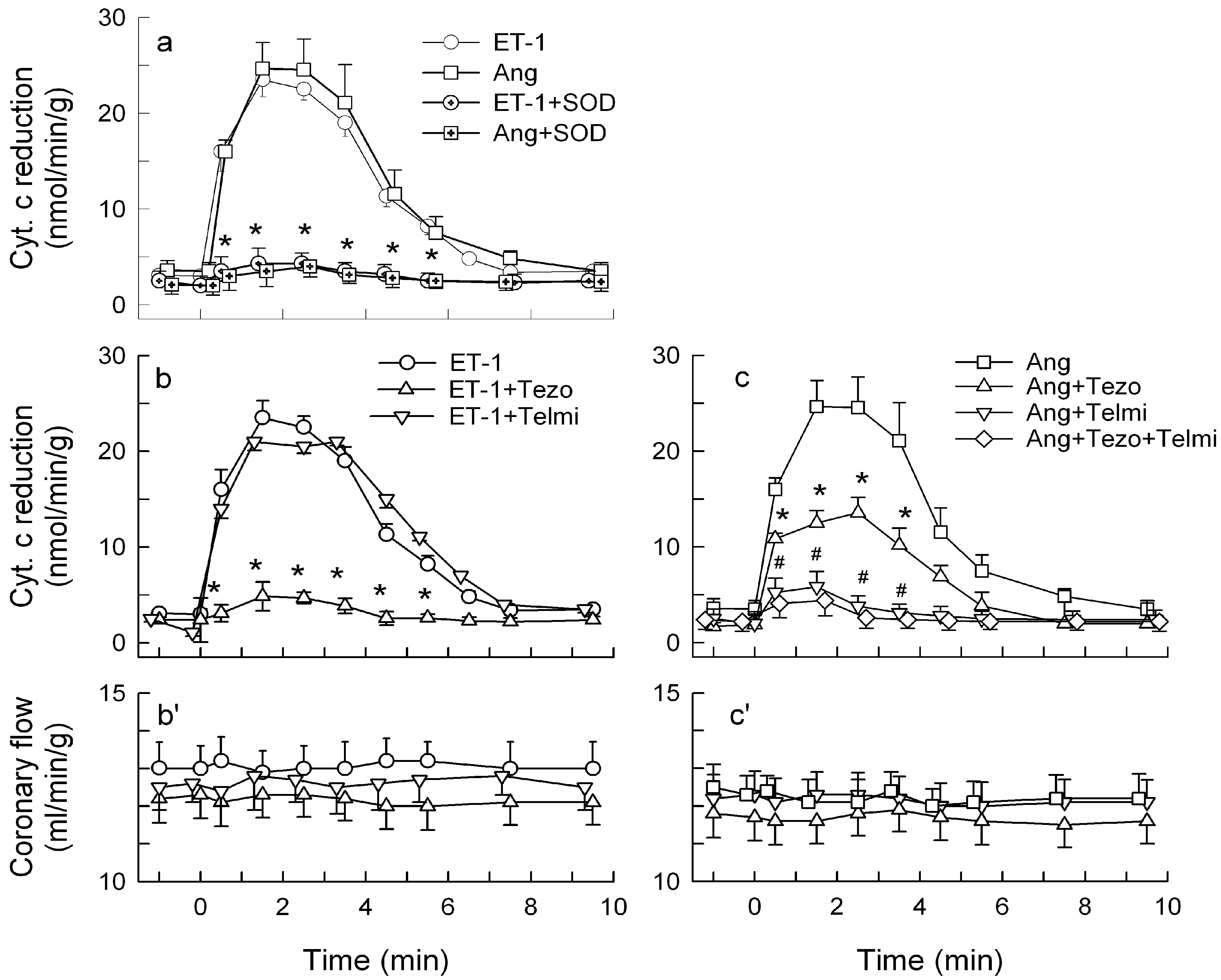
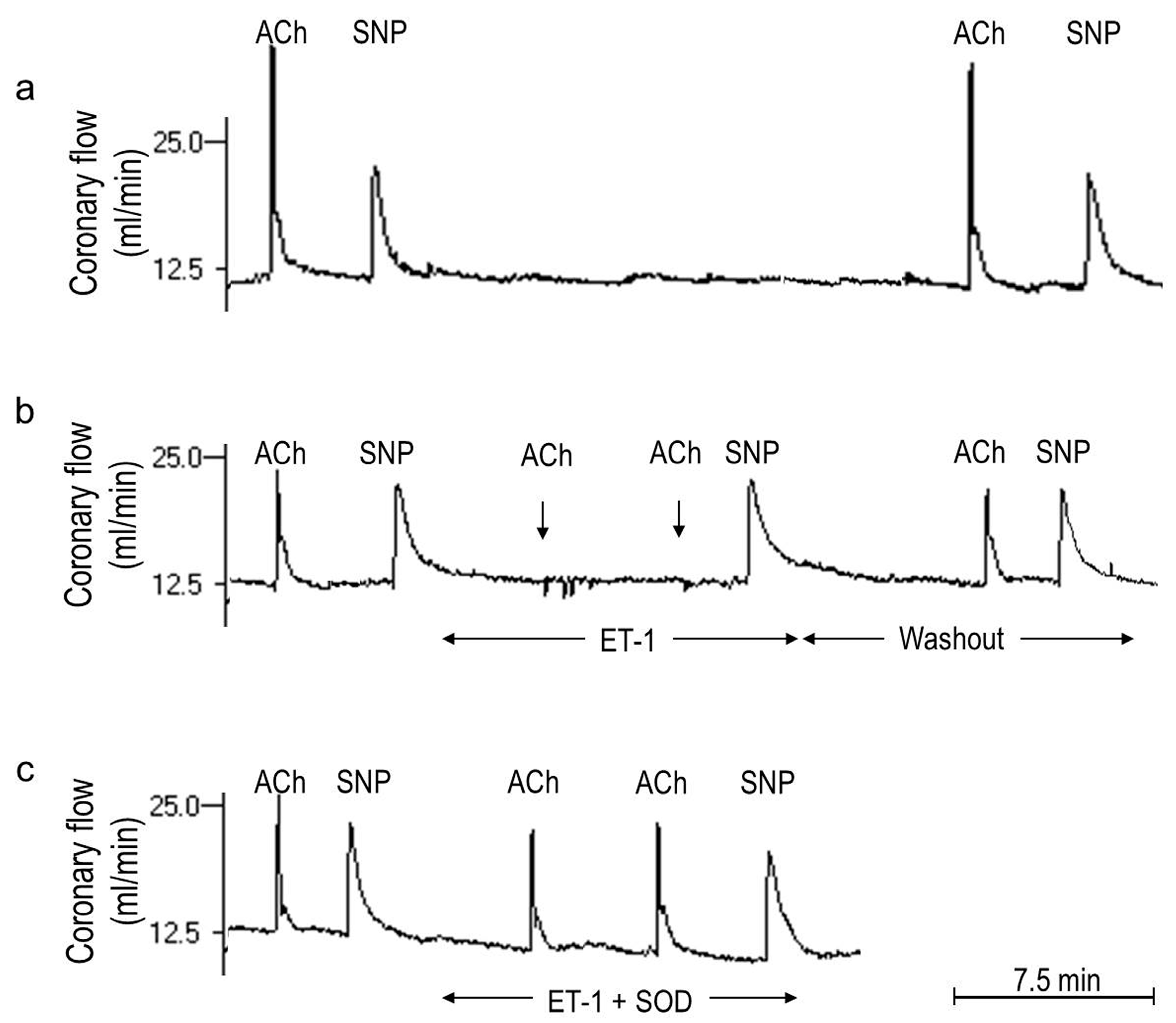
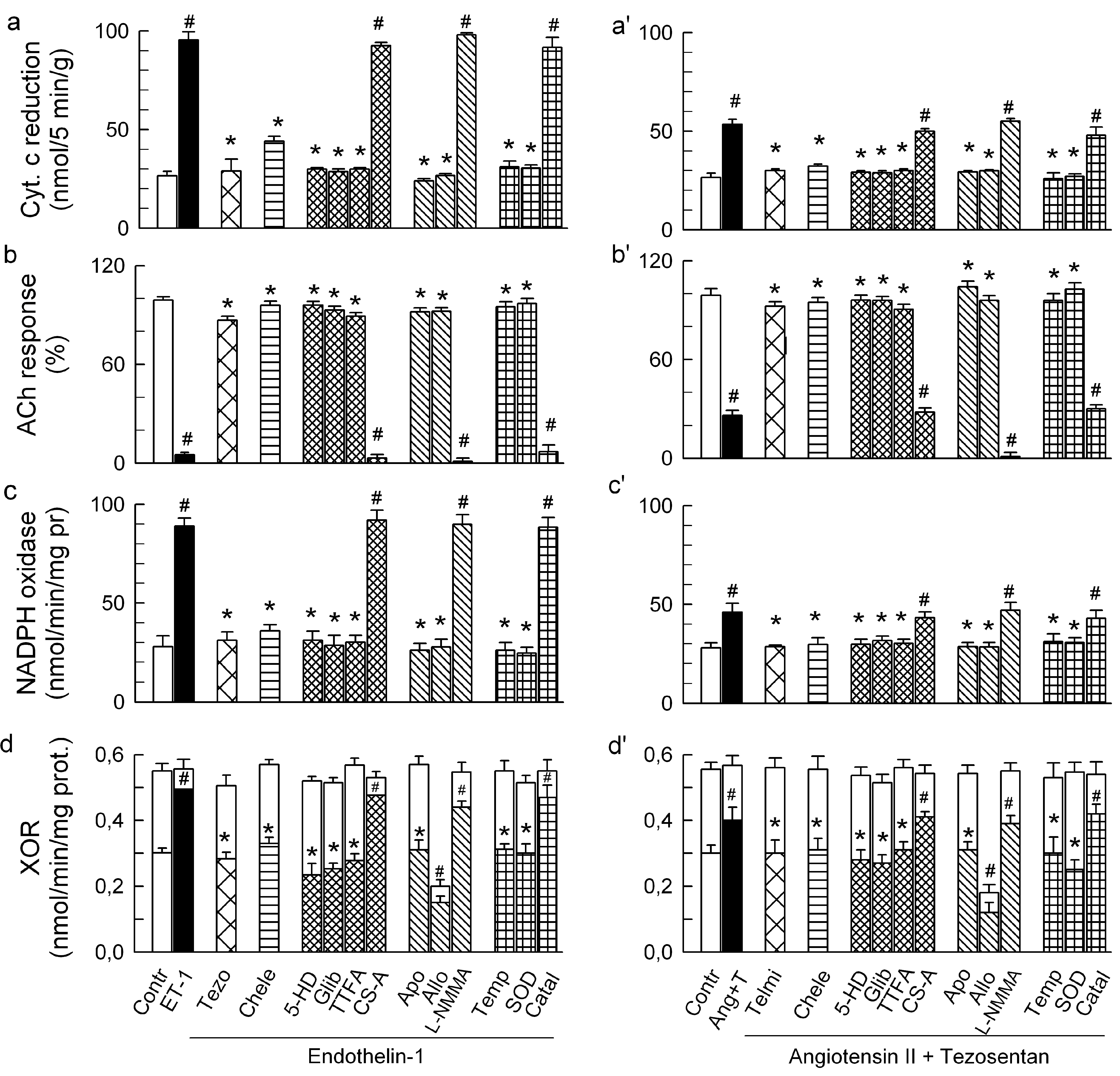
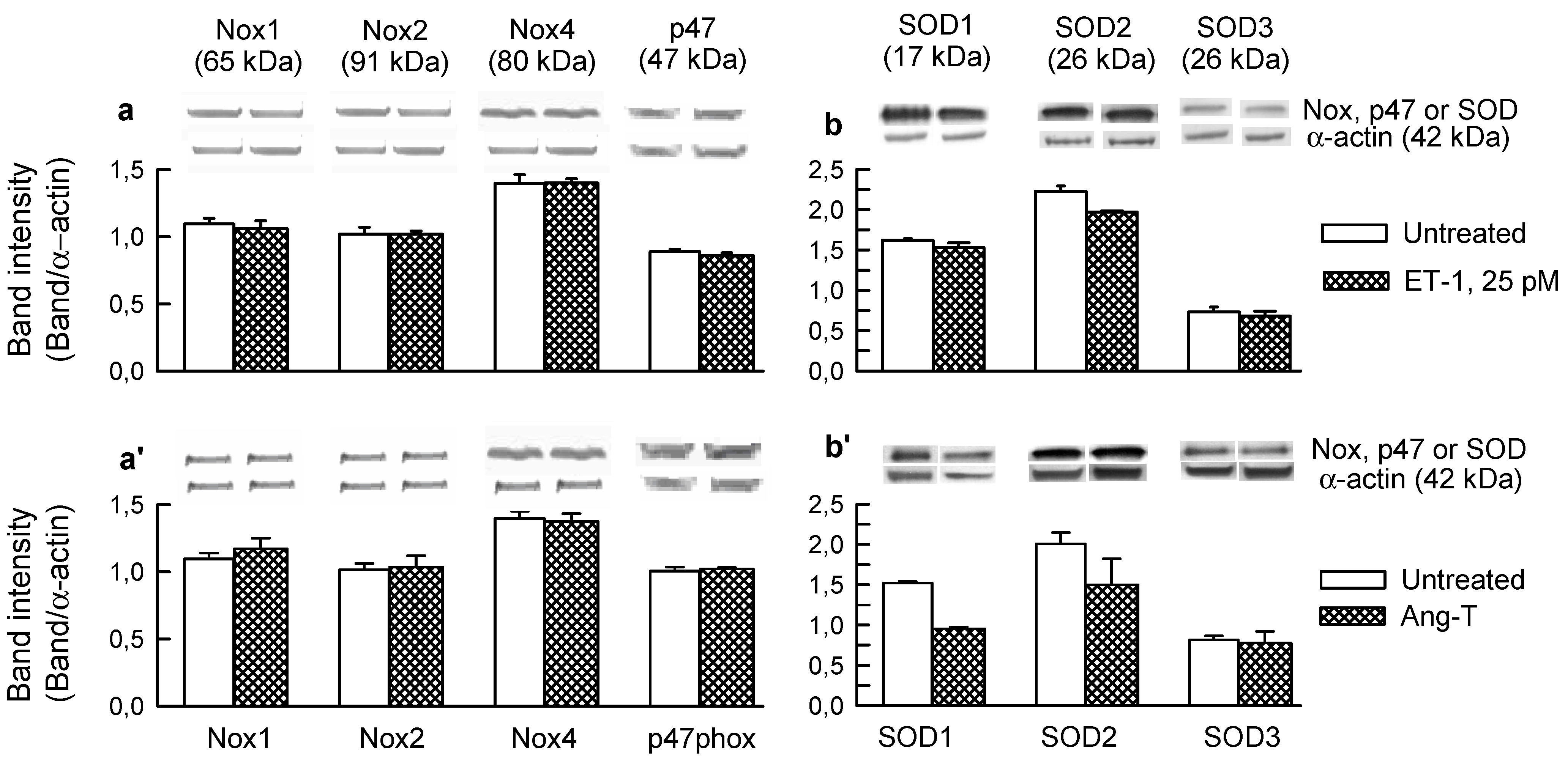
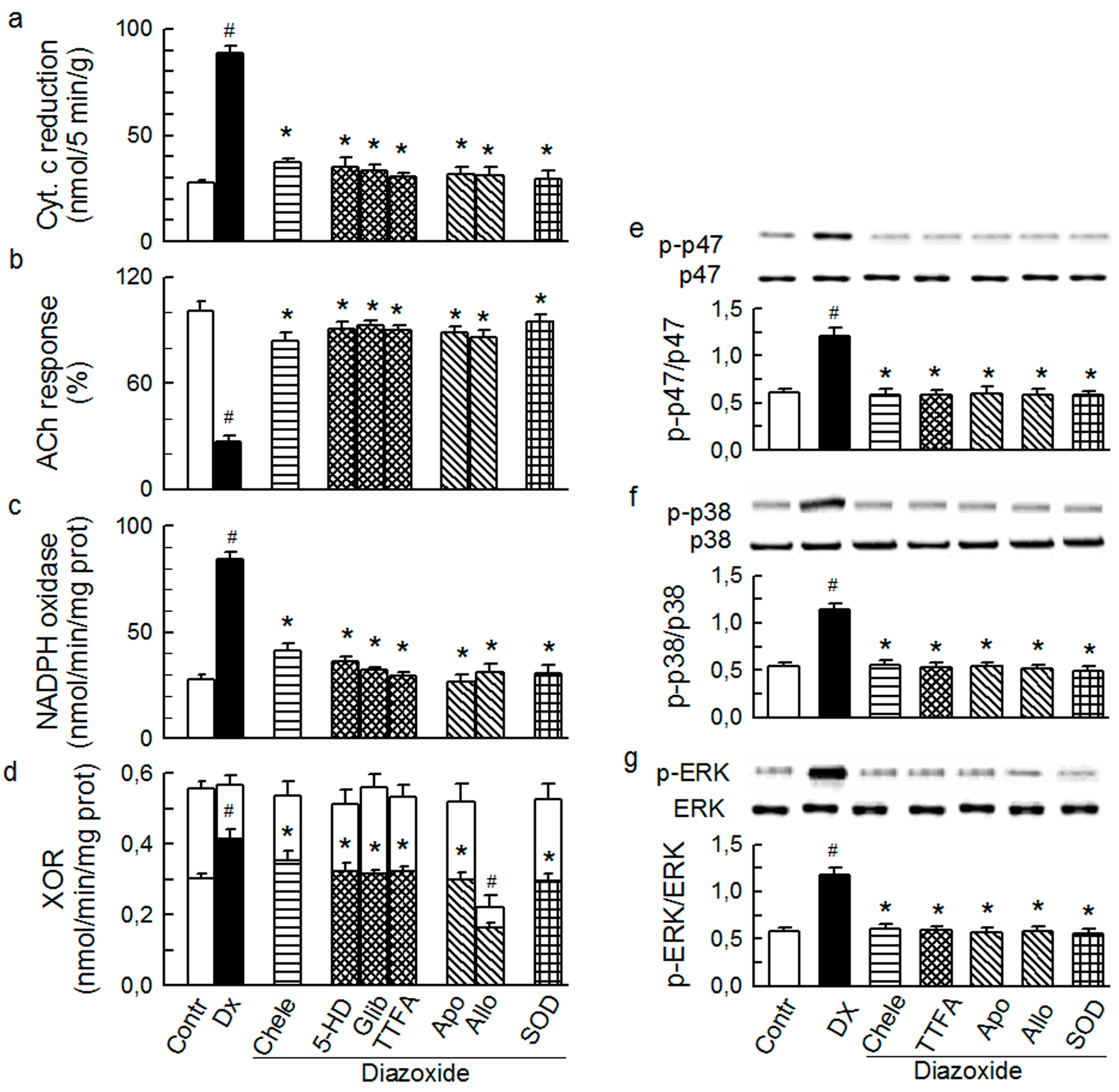
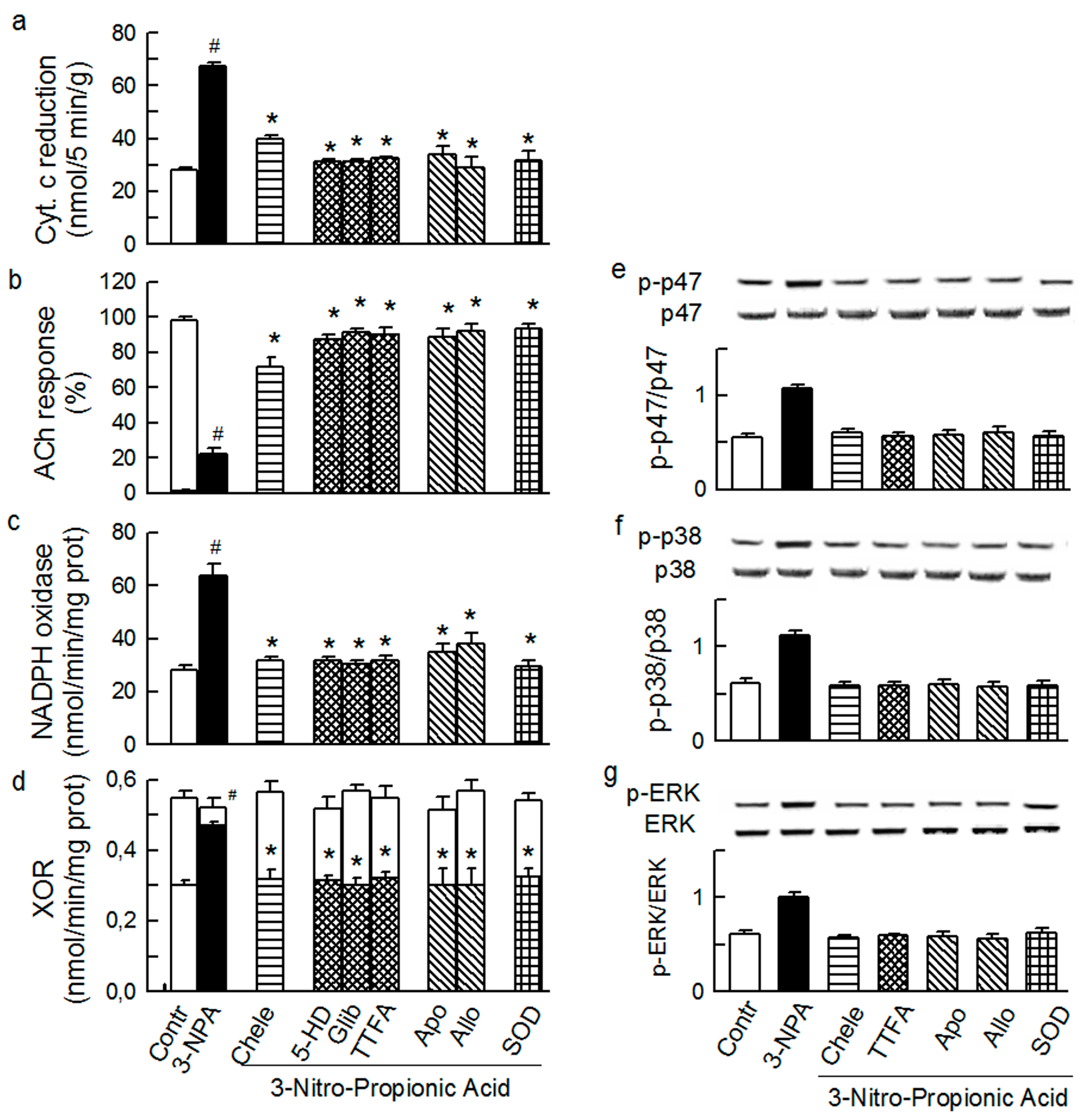
2.1. O2− Generation by ET-1 and AT-II and ET-1-Dependent Effects of AT-II

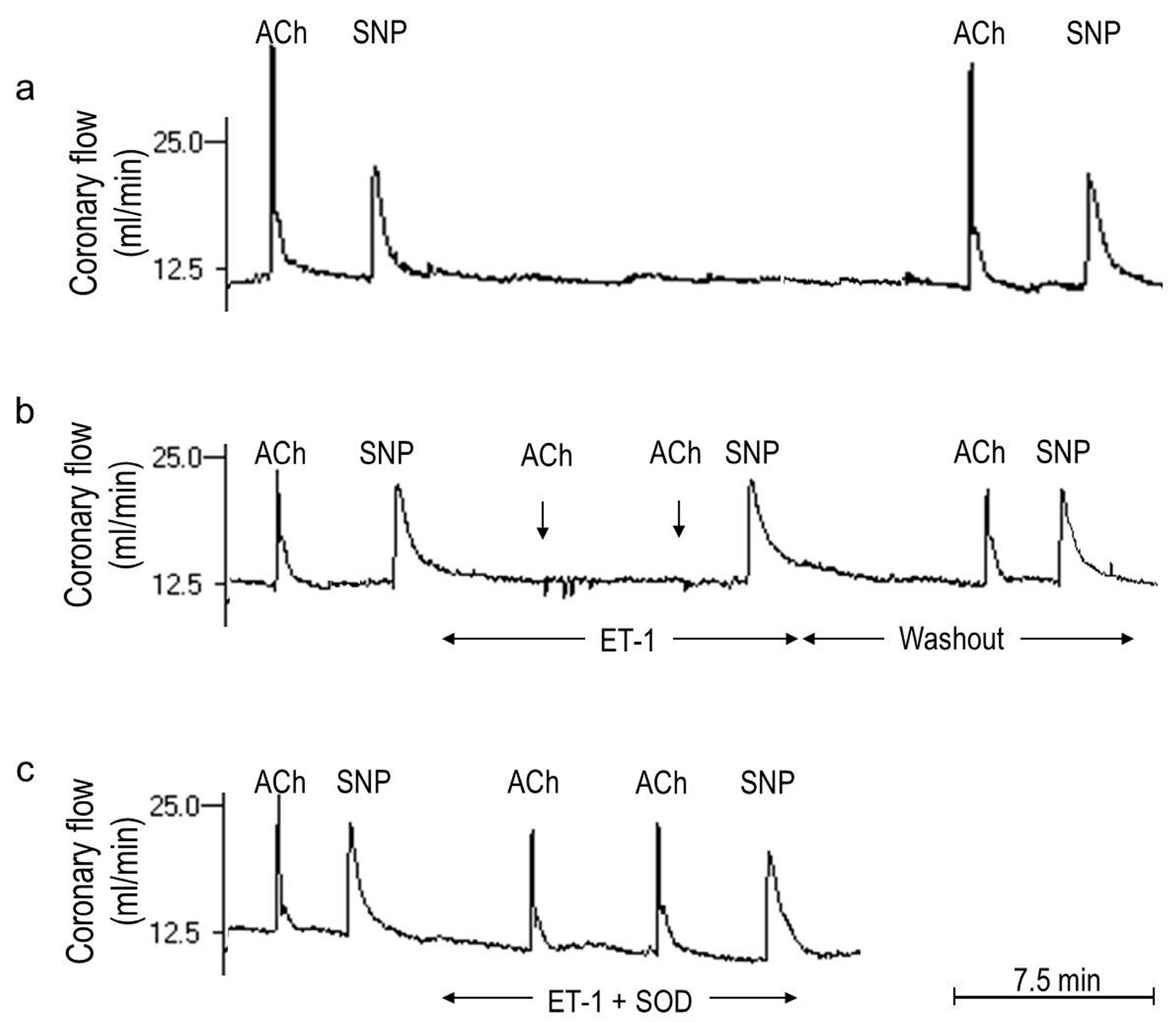
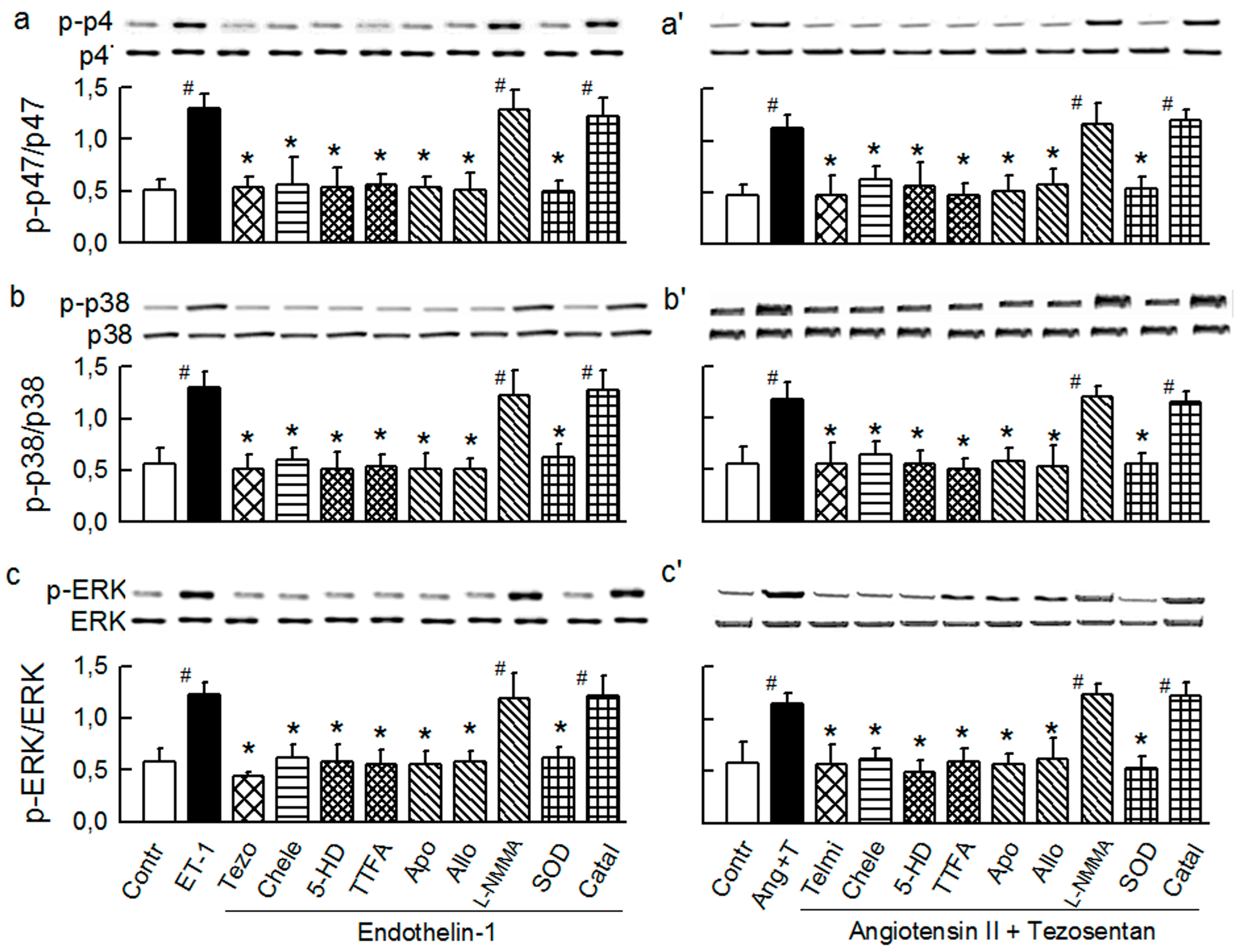
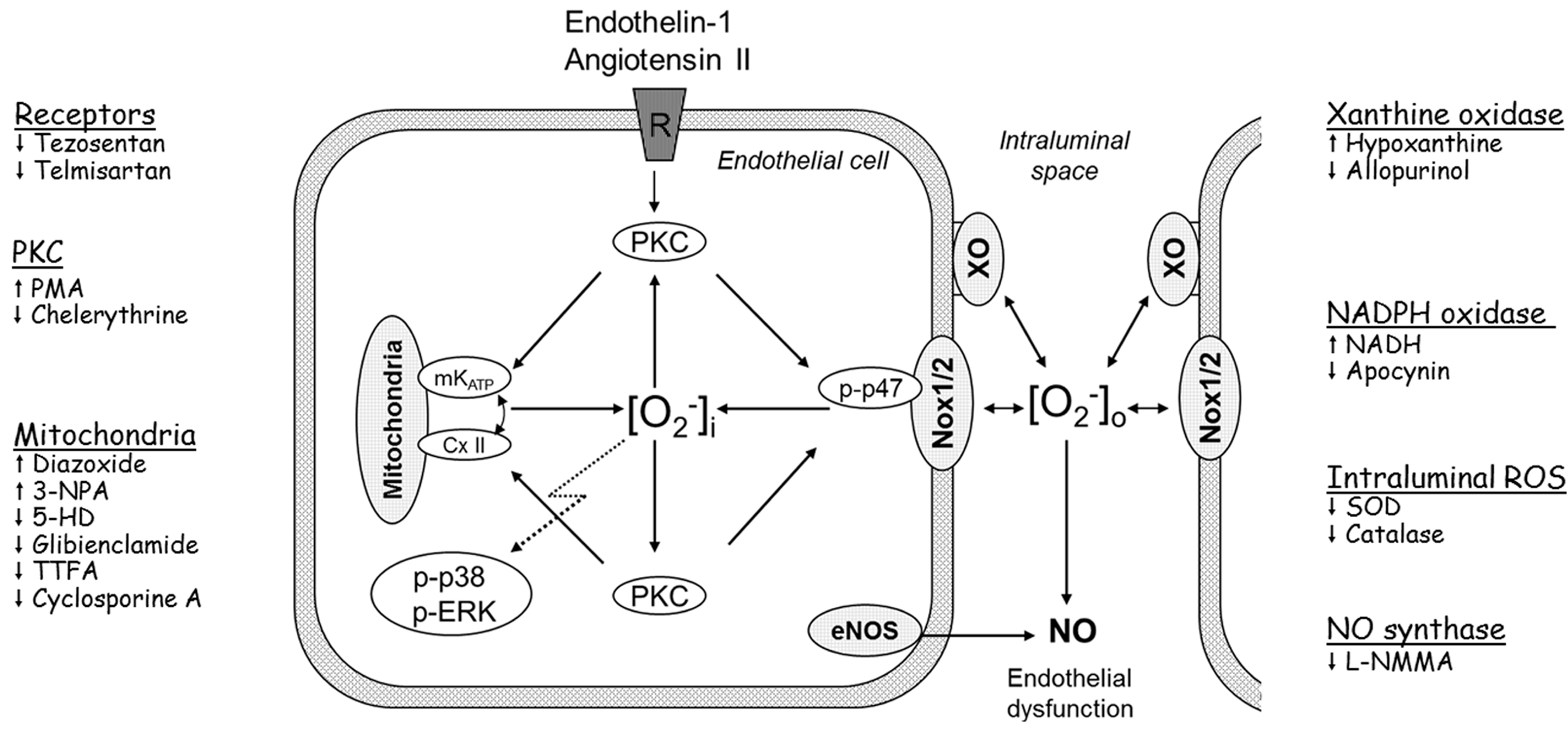
2.2. Players in the Signaling by ET-1 and Ang-T to Trigger O2−, Endothelial Dysfunction, and MAPKs Phosphorylation
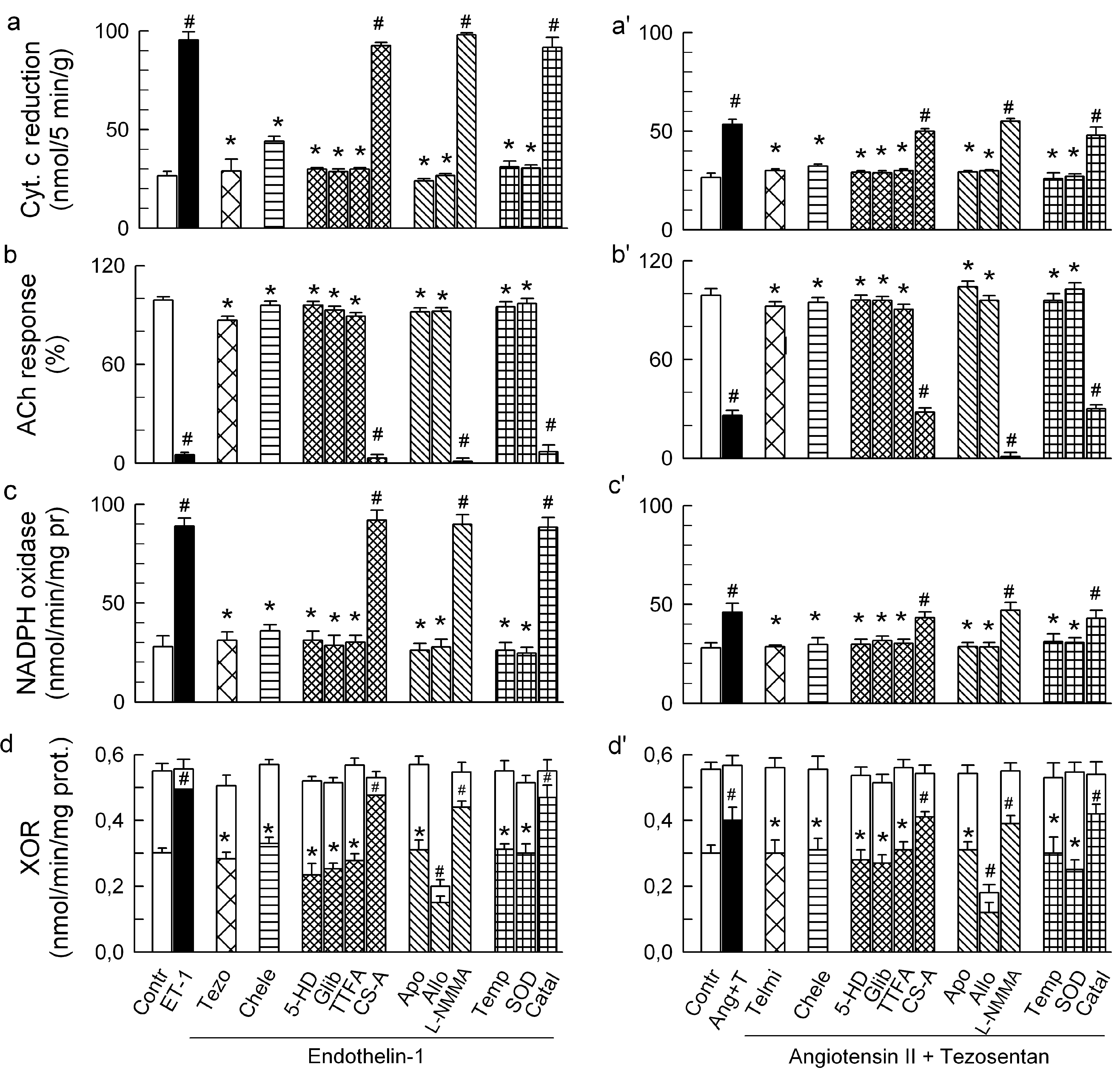
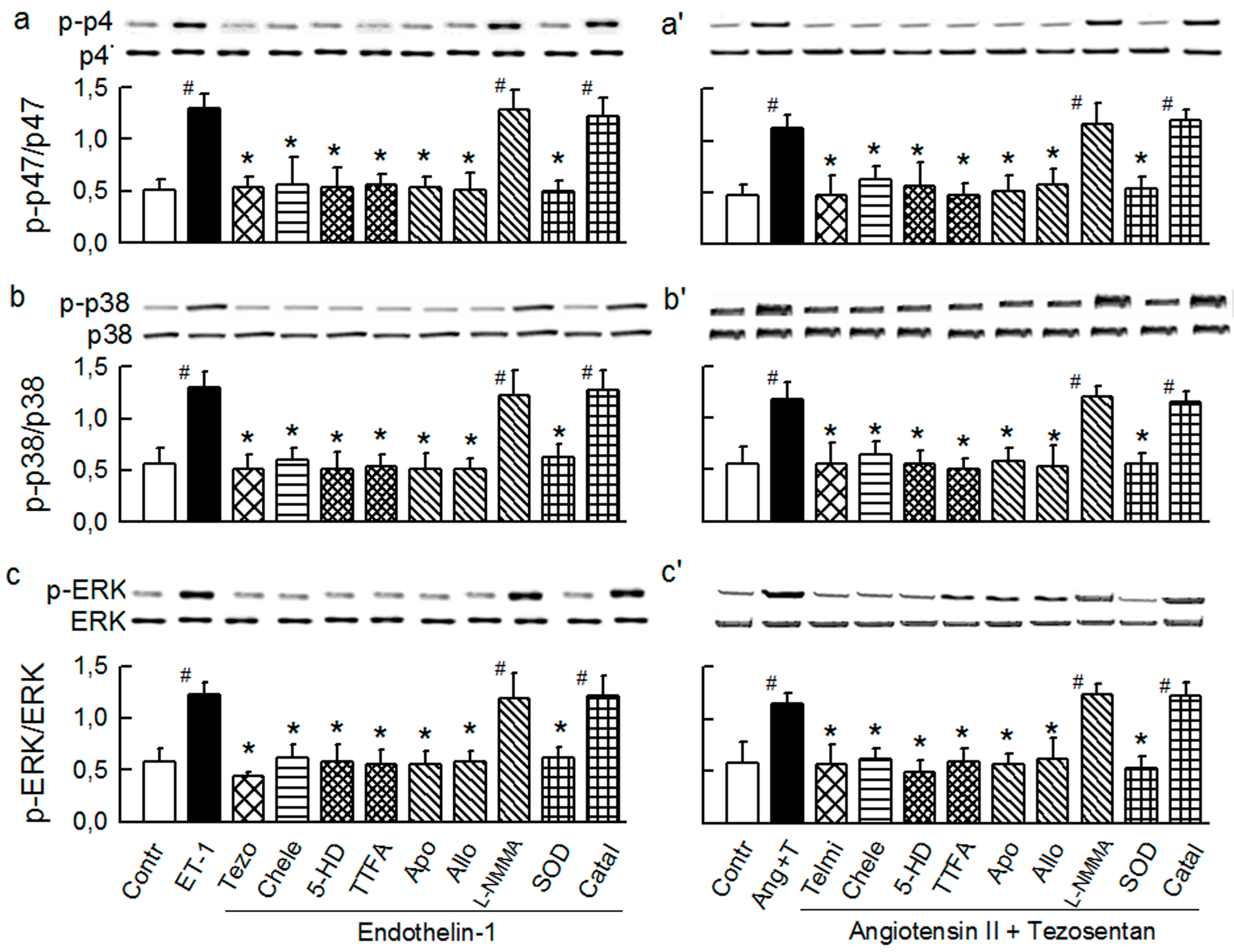
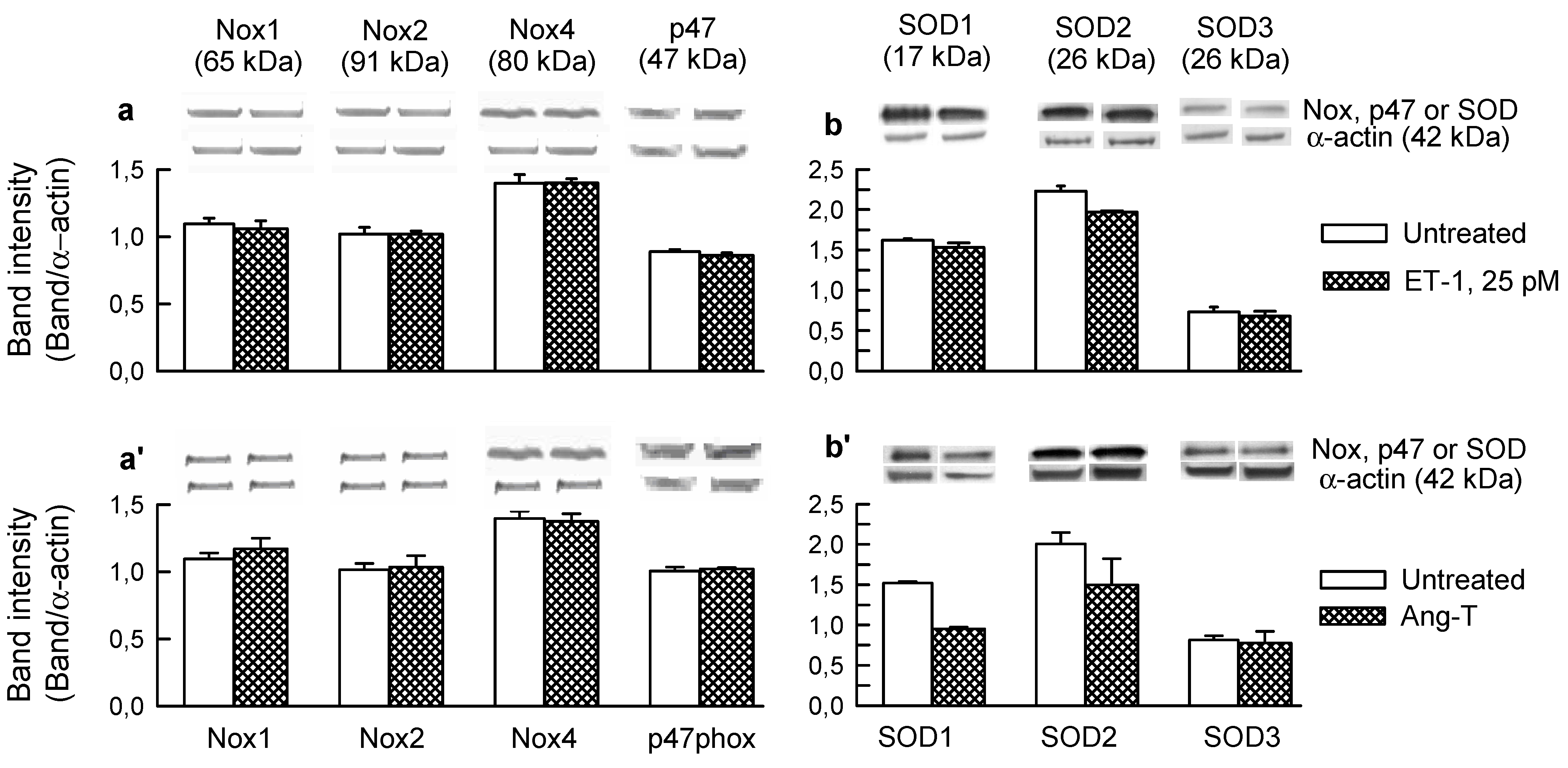

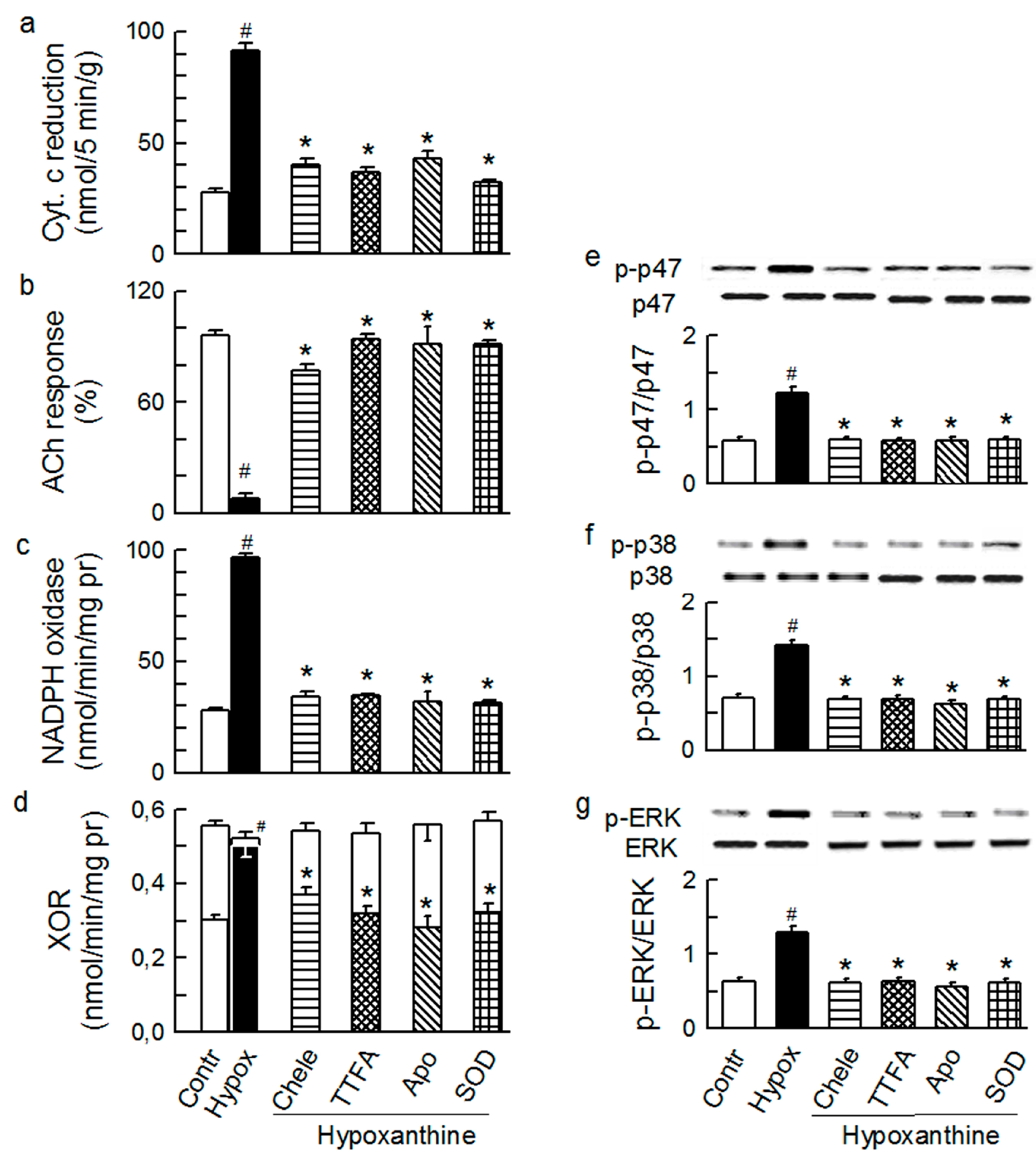
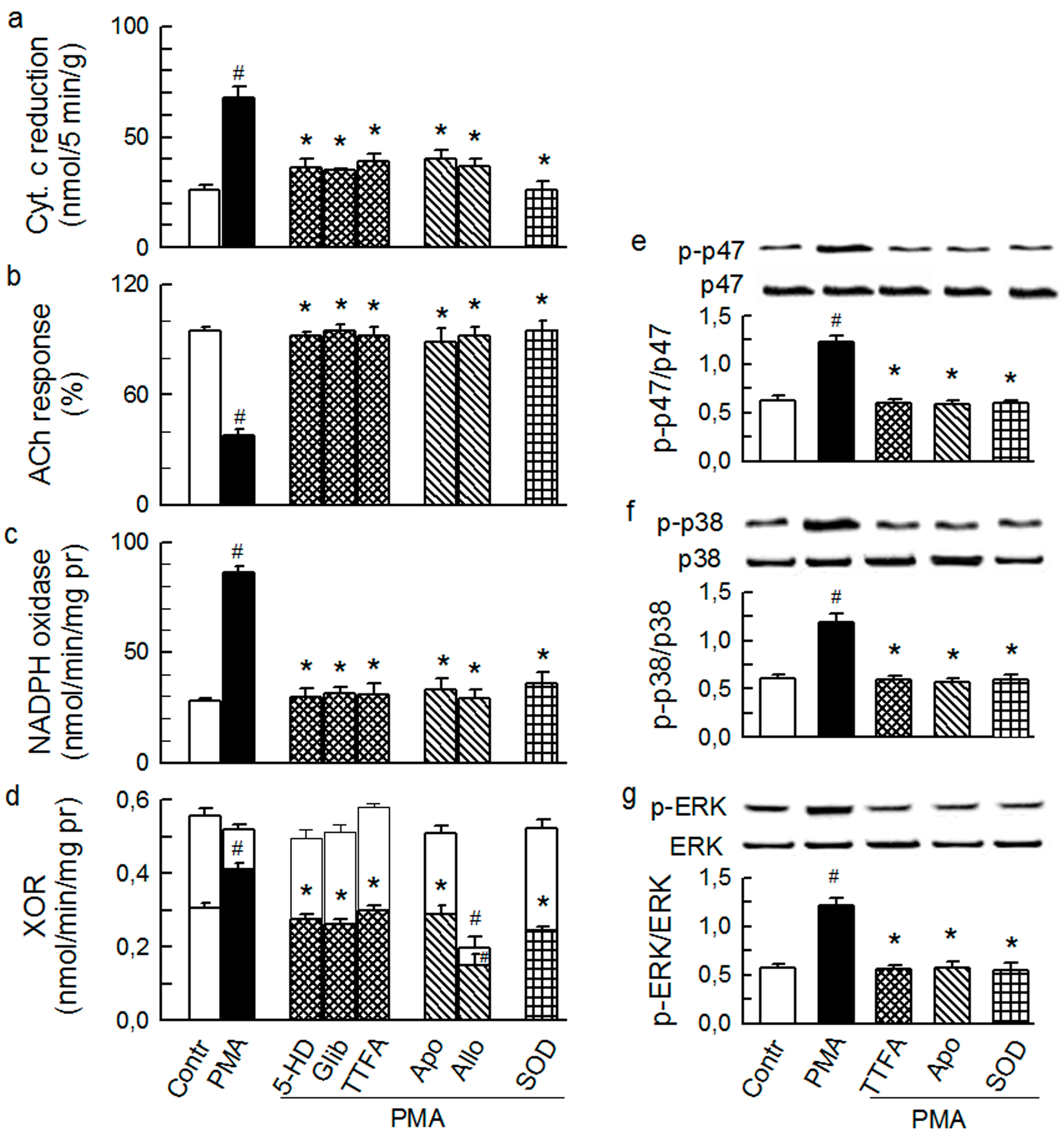
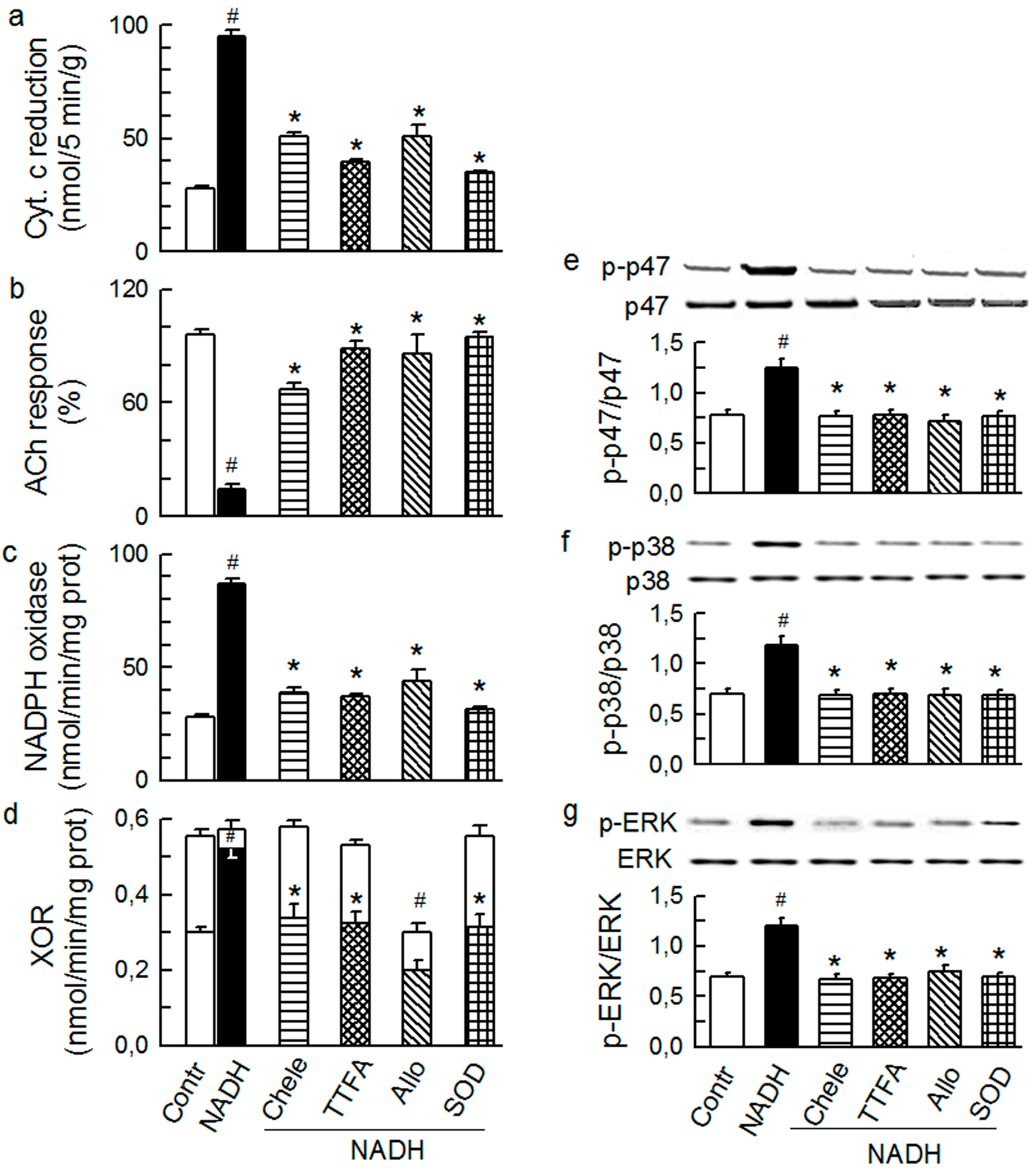
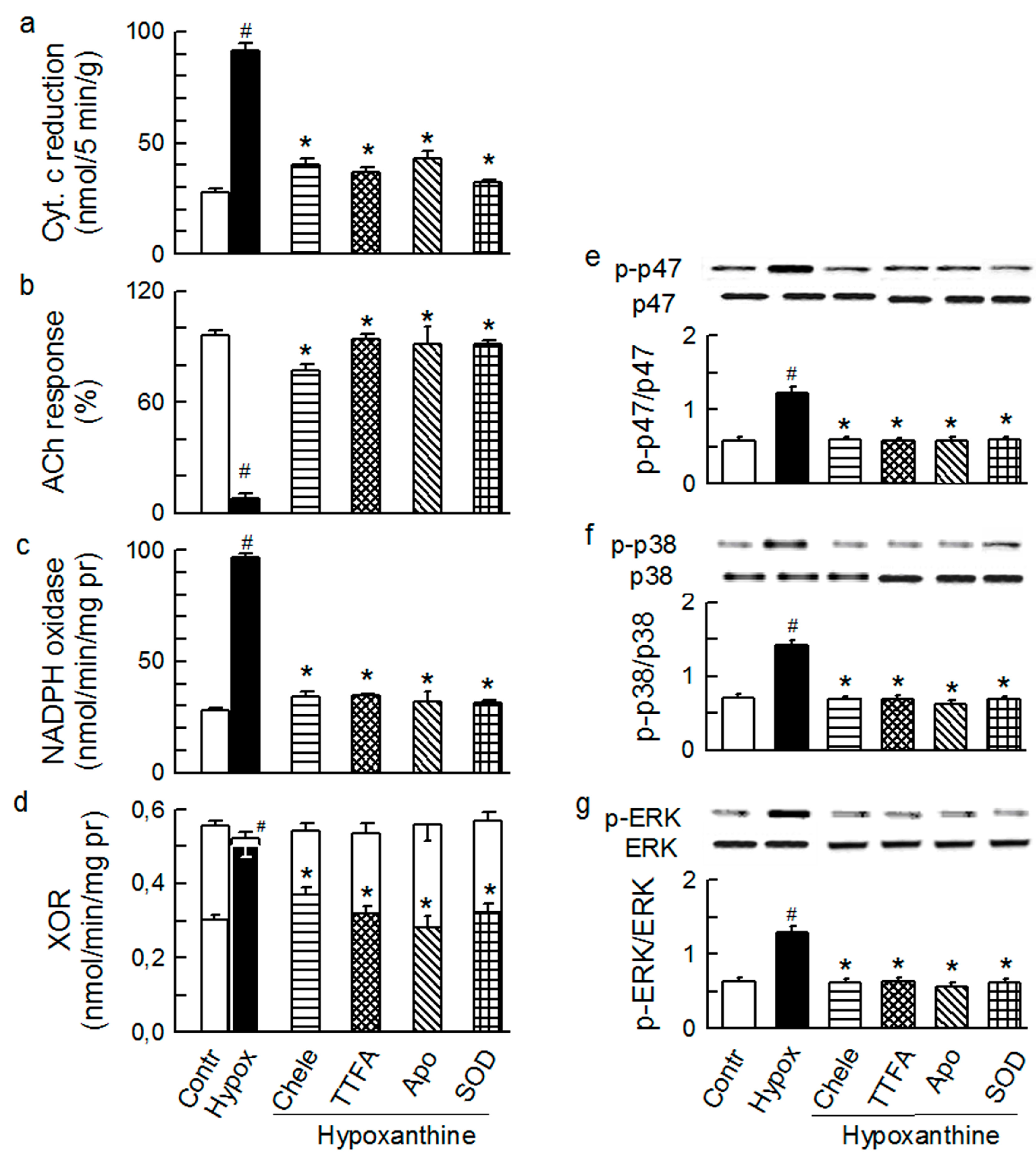
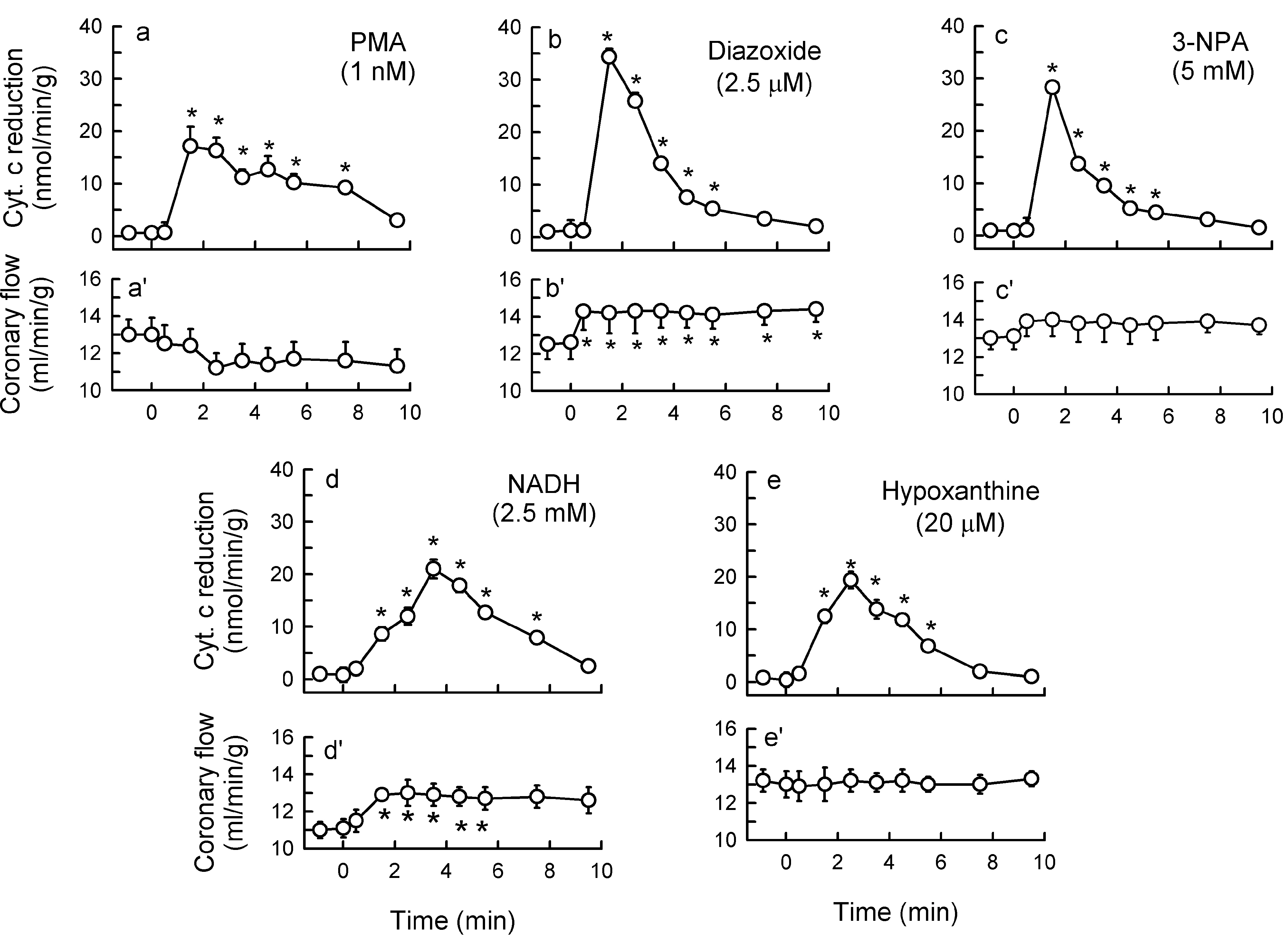
2.3. Mitochondria-Nox Cross-Talk and Its Reliance on PKC and Intraluminal O2−
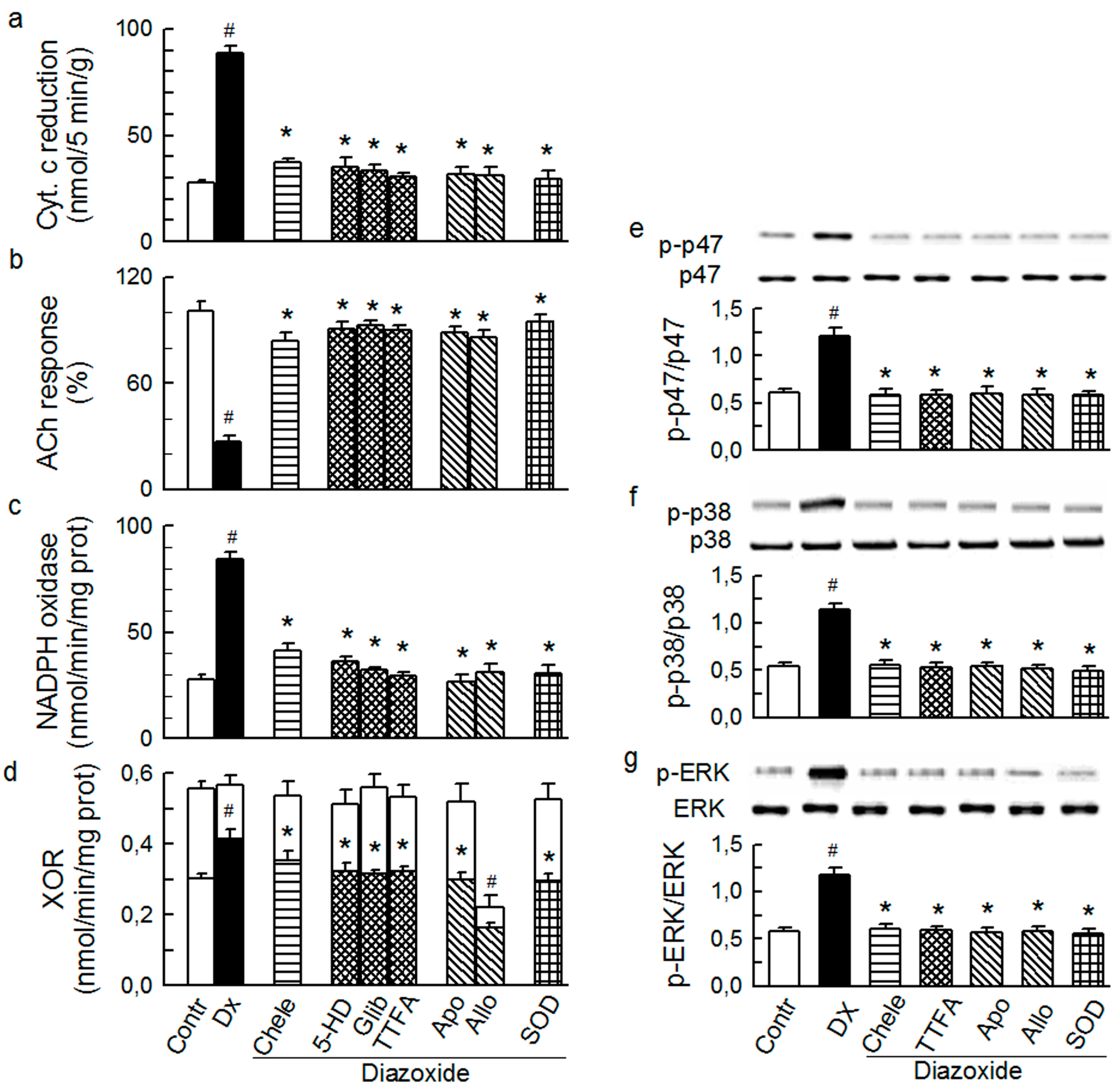
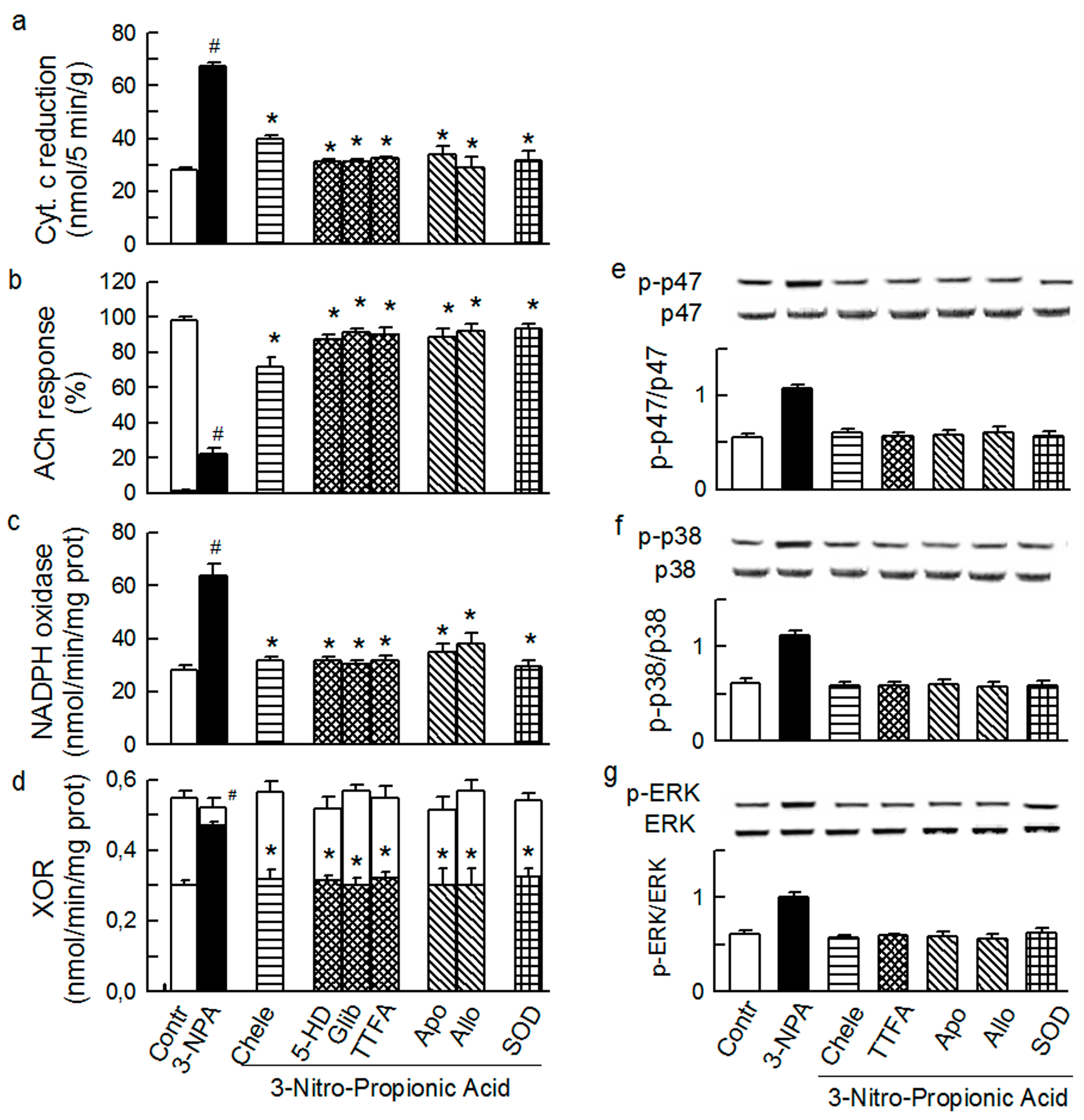
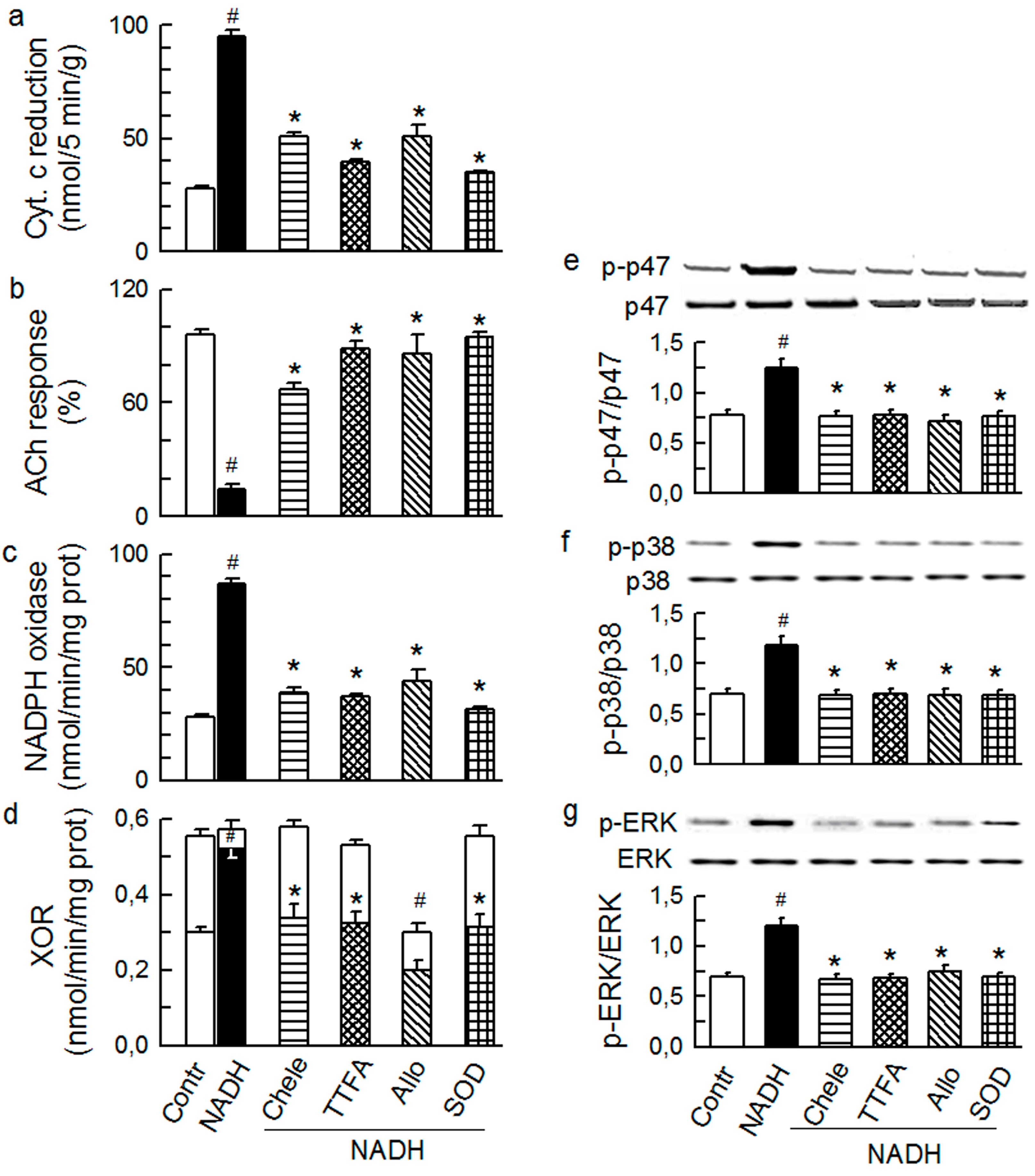
2.4. Cross-Talk between Cytoplasmic and Intraluminal Pool of ROS
3. Experimental Section
3.1. Chemicals
3.2. Heart Preparation and Protocols
3.3. Endothelium-Dependent Vasodilatation
3.4. Superoxide
3.5. Western Immunoblot Analyses
3.6. NADPH and Xanthine Oxidases Activity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary | Host | Dilution | Cat. No. | Company | Secondary | Dilution | Cat. No. | Company |
|---|---|---|---|---|---|---|---|---|
| ERK ½ | Mouse | 1:1000 | 135900 | SCB 1 | Rabbit anti-mouse AP 7 | 1:500 | 358915 | SCB |
| p-ERK ½ | Rabbit | 1:1000 | 16982 | SCB | Mouse anti-rabbit AP | 1:500 | 2358 | SCB |
| p38 | Rabbit | 1:1000 | 7149 | SCB | Mouse anti-rabbit AP | 1:500 | 2358 | SCB |
| p-p38 | Rabbit | 1:1000 | 17852 | SCB | Mouse anti-rabbit AP | 1:500 | 2358 | SCB |
| p47phox (total) | Rabbit | 1:1000 | 7660 | SCB | Mouse anti-rabbit AP | 1:500 | 2358 | SCB |
| p47phox (unphosphoryl.) | Rabbit | 1:1000 | 4502808 | Sigma 2 | Goat anti-rabbit AP | 1:1000 | 0418 | Sigma |
| p47phox (pSer345) | Rabbit | 1:1000 | 8391 | ABC 3 | Mouse anti-rabbit AP | 1:500 | 2358 | SCB |
| p47phox (pSer370) | Rabbit | 1:1000 | 4504288 | Sigma | Goat anti-rabbit AP | 1:1000 | 0418 | Sigma |
| Nox1 | Rabbit | 1:1000 | 55831 | Abcam 4 | Goat anti-rabbit AP | 1:1000 | 6722 | Abcam |
| Nox2 | Rabbit | 1:1000 | 31092 | Abcam | Goat anti-rabbit AP | 1:1000 | 6722 | Abcam |
| Nox4 | Rabbit | 1:1000 | 60940 | Abcam | Goat anti-rabbit AP | 1:1000 | 6722 | Abcam |
| SOD1 | Rabbit | 1:1000 | NB1001955 | NB 5 | Goat anti-rabbit HRP 8 | 1:5000 | P0448 | Dako 9 |
| SOD2 | Rabbit | 1:1000 | SOD111 | AD 6 | Goat anti-rabbit HRP | 1:5000 | P0448 | Dako |
| SOD3 | Rabbit | 1:1500 | 83108 | Abcam | Goat anti-rabbit HRP | 1:5000 | P0448 | Dako |
| α-Actin | Rabbit | 1:1000 | 15734 | Abcam | Mouse anti-rabbit AP | 1:500 | 2358 | SCB |
3.7. Statistics
4. Conclusions and Clinical Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Li, J.M.; Shah, A.M. Endothelial cell superoxide generation: Regulation and relevance for cardiovascular pathophysiology. Am. J. Physiol. 2004, 287, R1014–R1030. [Google Scholar]
- Mehta, P.K.; Griendling, K.K. Angiotensin II cell signaling: Physiological and pathological effects in the cardiovascular system. Am. J. Physiol. 2007, 292, C82–C97. [Google Scholar]
- Harrison, D.G.; Widder, J.; Grumbach, I.; Chen, W.; Weber, M.; Searles, C. Endothelial mechanotransduction, nitric oxide and vascular inflammation. J. Intern. Med. 2006, 259, 351–363. [Google Scholar]
- Kurzelewski, M.; Czarnowska, E.; Beresewicz, A. Endothelin in the mechanism of endothelial injury and neutrophil adhesion in the post-ischemic guinea pig heart. Eur. J. Pharmacol. 2002, 434, 95–107. [Google Scholar]
- Niccoli, G.; Lanza, G.A.; Shaw, S.; Romagnoli, E.; Gioia, D.; Burzotta, F.; Trani, C.; Mazzari, M.A.; Mongiardo, R.; de Vita, M.; et al. Endothelin-1 and acute myocardial infarction: A no-reflow mediator after successful percutaneous myocardial revascularization. Eur. Heart J. 2006, 27, 1793–1798. [Google Scholar]
- Brunner, F.; Bras-Silva, C.; Cerdeira, A.S.; Leite-Moreira, A.F. Cardiovascular endothelins: Essential regulators of cardiovascular homeostasis. Pharmacol. Ther. 2006, 111, 508–531. [Google Scholar]
- Dammanahalli, K.J.; Sun, Z. Endothelins and NADPH oxidases in the cardiovascular system. Clin. Exp. Pharmacol. Physiol. 2008, 35, 2–6. [Google Scholar]
- Granger, D.N.; Rodrigues, S.F.; Yildirim, A.; Senchenkova, E.Y. Microvascular responses to cardiovascular risk factors. Microcirculation 2010, 17, 192–205. [Google Scholar]
- Lassegue, B.; Griendling, K.K. NADPH oxidases: Functions and pathologies in the vasculature. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 653–661. [Google Scholar]
- Duda, M.; Konior, A.; Klemenska, E.; Beresewicz, A. Preconditioning protects endothelium by preventing ET-1-induced activation of NADPH oxidase and xanthine oxidase in post-ischemic heart. J. Mol. Cell. Cardiol. 2007, 42, 400–410. [Google Scholar]
- Kimura, S.; Zhang, G.X.; Nishiyama, A.; Shokoji, T.; Yao, L.; Fan, Y.Y.; Rahman, M.; Abe, Y. Mitochondria-derived reactive oxygen species and vascular MAP kinases: Comparison of angiotensin II and diazoxide. Hypertension 2005, 45, 438–444. [Google Scholar]
- Vivekananthan, D.P.; Penn, M.S.; Sapp, S.K.; Hsu, A.; Topol, E.J. Use of antioxidant vitamins for the prevention of cardiovascular disease: Meta-analysis of randomised trials. Lancet 2003, 361, 2017–2023. [Google Scholar]
- Hausenloy, D.J.; Erik, B.H.; Condorelli, G.; Ferdinandy, P.; Garcia-Dorado, D.; Heusch, G.; Lecour, S.; van Laake, L.W.; Madonna, R.; Ruiz-Meana, M.; et al. Translating cardioprotection for patient benefit: Position paper from the Working Group of Cellular Biology of the Heart of the European Society of Cardiology. Cardiovasc. Res. 2013, 98, 7–27. [Google Scholar]
- Zhang, G.; Zhang, F.; Muh, R.; Yi, F.; Chalupsky, K.; Cai, H.; Li, P.L. Autocrine/paracrine pattern of superoxide production through NAD(P)H oxidase in coronary arterial myocytes. Am. J. Physiol. 2007, 292, H483–H495. [Google Scholar]
- Heitzer, T.; Schlinzig, T.; Krohn, K.; Meinertz, T.; Munzel, T. Endothelial dysfunction, oxidative stress, and risk of cardiovascular events in patients with coronary artery disease. Circulation 2001, 104, 2673–2678. [Google Scholar]
- Landmesser, U.; Spiekermann, S.; Preuss, C.; Sorrentino, S.; Fischer, D.; Manes, C.; Mueller, M.; Drexler, H. Angiotensin II induces endothelial xanthine oxidase activation: Role for endothelial dysfunction in patients with coronary disease. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 943–948. [Google Scholar]
- Matesanz, N.; Lafuente, N.; Azcutia, V.; Martin, D.; Cuadrado, A.; Nevado, J.; Rodriguez-Manas, L.; Sanchez-Ferrer, C.F.; Peiro, C. Xanthine oxidase-derived extracellular superoxide anions stimulate activator protein 1 activity and hypertrophy in human vascular smooth muscle via c-Jun N-terminal kinase and p38 mitogen-activated protein kinases. J. Hypertens. 2007, 25, 609–618. [Google Scholar]
- Tritto, I.; Dandrea, D.; Eramo, N.; Scognamiglio, A.; Desimone, C.; Violante, A.; Esposito, A.; Chiariello, M.; Ambrosio, G. Oxygen radicals can induce preconditioning in rabbit hearts. Circ. Res. 1997, 80, 743–748. [Google Scholar]
- Gomez-Cabrera, M.C.; Domenech, E.; Vina, J. Moderate exercise is an antioxidant: Upregulation of antioxidant genes by training. Free Radic. Biol. Med. 2008, 44, 126–131. [Google Scholar]
- Daiber, A. Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim. Biophys. Acta 1797, 897–906. [Google Scholar]
- Dikalov, S. Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 2011, 51, 1289–1301. [Google Scholar]
- Forstermann, U. Nitric oxide and oxidative stress in vascular disease. Pflugers Arch. 2010, 459, 923–939. [Google Scholar]
- McNally, J.S.; Davis, M.E.; Giddens, D.P.; Saha, A.; Hwang, J.; Dikalov, S.; Jo, H.; Harrison, D.G. Role of xanthine oxidoreductase and NAD(P)H oxidase in endothelial superoxide production in response to oscillatory shear stress. Am. J. Physiol. 2003, 285, H2290–H2297. [Google Scholar]
- Touyz, R.M.; Yao, G.; Viel, E.; Amiri, F.; Schiffrin, E.L. Angiotensin II and endothelin-1 regulate MAP kinases through different redox-dependent mechanisms in human vascular smooth muscle cells. J. Hypertens. 2004, 22, 1141–1149. [Google Scholar]
- Touyz, R.M.; Yao, G.; Schiffrin, E.L. c-Src induces phosphorylation and translocation of p47phox: Role in superoxide generation by angiotensin II in human vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 981–987. [Google Scholar]
- Li, L.; Fink, G.D.; Watts, S.W.; Northcott, C.A.; Galligan, J.J.; Pagano, P.J.; Chen, A.F. Endothelin-1 increases vascular superoxide via endothelin(A)-NADPH oxidase pathway in low-renin hypertension. Circulation 2003, 107, 1053–1058. [Google Scholar]
- Dong, F.; Zhang, X.; Wold, L.E.; Ren, Q.; Zhang, Z.; Ren, J. Endothelin-1 enhances oxidative stress, cell proliferation and reduces apoptosis in human umbilical vein endothelial cells: Role of ETB receptor, NADPH oxidase and caveolin-1. Br. J. Pharmacol. 2005, 145, 323–333. [Google Scholar]
- Romero, M.; Jimenez, R.; Sanchez, M.; Lopez-Sepulveda, R.; Zarzuelo, M.J.; O’Valle, F.; Zarzuelo, A.; Perez-Vizcaino, F.; Duarte, J. Quercetin inhibits vascular superoxide production induced by endothelin-1: Role of NADPH oxidase, uncoupled eNOS and PKC. Atherosclerosis 2009, 202, 58–67. [Google Scholar]
- Lopez-Sepulveda, R.; Gomez-Guzman, M.; Zarzuelo, M.J.; Romero, M.; Sanchez, M.; Quintela, A.M.; Galindo, P.; O’Valle, F.; Tamargo, J.; Perez-Vizcaino, F.; et al. Red wine polyphenols prevent endothelial dysfunction induced by endothelin-1 in rat aorta: Role of NADPH oxidase. Clin. Sci. 2011, 120, 321–333. [Google Scholar]
- Downey, J.M.; Davis, A.M.; Cohen, M.V. Signaling pathways in ischemic preconditioning. Heart Fail. Rev. 2007, 12, 181–188. [Google Scholar]
- Garlid, K.D.; Costa, A.D.; Quinlan, C.L.; Pierre, S.V.; Dos, S.P. Cardioprotective signaling to mitochondria. J. Mol. Cell. Cardiol. 2009, 46, 858–866. [Google Scholar]
- Kimura, S.; Zhang, G.X.; Nishiyama, A.; Shokoji, T.; Yao, L.; Fan, Y.Y.; Rahman, M.; Suzuki, T.; Maeta, H.; Abe, Y. Role of NAD(P)H oxidase- and mitochondria-derived reactive oxygen species in cardioprotection of ischemic reperfusion injury by angiotensin II. Hypertension 2005, 45, 860–866. [Google Scholar]
- Doughan, A.K.; Harrison, D.G.; Dikalov, S.I. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: Linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ. Res. 2008, 102, 488–496. [Google Scholar]
- Wosniak, J., Jr.; Santos, C.X.; Kowaltowski, A.J.; Laurindo, F.R. Cross-talk between mitochondria and NADPH oxidase: Effects of mild mitochondrial dysfunction on angiotensin II-mediated increase in Nox isoform expression and activity in vascular smooth muscle cells. Antioxid. Redox. Signal. 2009, 11, 1265–1278. [Google Scholar]
- Dikalova, A.E.; Bikineyeva, A.T.; Budzyn, K.; Nazarewicz, R.R.; McCann, L.; Lewis, W.; Harrison, D.G.; Dikalov, S.I. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 2010, 107, 106–116. [Google Scholar]
- Wenzel, P.; Mollnau, H.; Oelze, M.; Schulz, E.; Wickramanayake, J.M.; Muller, J.; Schuhmacher, S.; Hortmann, M.; Baldus, S.; Gori, T.; et al. First evidence for a crosstalk between mitochondrial and NADPH oxidase-derived reactive oxygen species in nitroglycerin-triggered vascular dysfunction. Antioxid. Redox Signal. 2008, 10, 1435–1447. [Google Scholar]
- Rathore, R.; Zheng, Y.M.; Niu, C.F.; Liu, Q.H.; Korde, A.; Ho, Y.S.; Wang, Y.X. Hypoxia activates NADPH oxidase to increase [ROS]i and [Ca2+]i through the mitochondrial ROS-PKCepsilon signaling axis in pulmonary artery smooth muscle cells. Free Radic. Biol. Med. 2008, 45, 1223–1231. [Google Scholar]
- Nazarewicz, R.R.; Dikalova, A.E.; Bikineyeva, A.; Dikalov, S.I. Nox2 as a potential target of mitochondrial superoxide and its role in endothelial oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1131–H1140. [Google Scholar]
- Niwa, K.; Sakai, J.; Karino, T.; Aonuma, H.; Watanabe, T.; Ohyama, T.; Inanami, O.; Kuwabara, M. Reactive oxygen species mediate shear stress-induced fluid-phase endocytosis in vascular endothelial cells. Free Radic. Res. 2006, 40, 167–174. [Google Scholar]
- Li, W.G.; Miller, F.J., Jr.; Zhang, H.J.; Spitz, D.R.; Oberley, L.W.; Weintraub, N.L. H2O2-induced O2− production by a non-phagocytic NAD(P)H oxidase causes oxidant injury. J. Biol. Chem. 2001, 276, 29251–29256. [Google Scholar]
- Seshiah, P.N.; Weber, D.S.; Rocic, P.; Valppu, L.; Taniyama, Y.; Griendling, K.K. Angiotensin II stimulation of NAD(P)H oxidase activity: Upstream mediators. Circ. Res. 2002, 91, 406–413. [Google Scholar]
- De Giusti, V.C.; Correa, M.V.; Villa-Abrille, M.C.; Beltrano, C.; Yeves, A.M.; de Cingolani, G.E.; Cingolani, H.E.; Aiello, E.A. The positive inotropic effect of endothelin-1 is mediated by mitochondrial reactive oxygen species. Life Sci. 2008, 83, 264–271. [Google Scholar]
- Cingolani, H.E.; Villa-Abrille, M.C.; Cornelli, M.; Nolly, A.; Ennis, I.L.; Garciarena, C.; Suburo, A.M.; Torbidoni, V.; Correa, M.V.; Camilionde Hurtado, M.C.; et al. The positive inotropic effect of angiotensin II: Role of endothelin-1 and reactive oxygen species. Hypertension. 2006, 47, 727–734. [Google Scholar]
- Laplante, M.A.; de Champlain, J. The interrelation of the angiotensin and endothelin systems on the modulation of NAD(P)H oxidase. Can. J. Physiol. Pharmacol. 2006, 84, 21–28. [Google Scholar]
- Zhang, C.; Hein, T.W.; Wang, W.; Kuo, L. Divergent roles of angiotensin II AT1 and AT2 receptors in modulating coronary microvascular function. Circ. Res. 2003, 92, 322–329. [Google Scholar]
- Li, J.M.; Shah, A.M. Mechanism of endothelial cell NADPH oxidase activation by angiotensin II. Role of the p47phox subunit. J. Biol. Chem. 2003, 278, 12094–12100. [Google Scholar]
- Romero, M.; Jimenez, R.; Sanchez, M.; Lopez-Sepulveda, R.; Zarzuelo, A.; Tamargo, J.; Perez-Vizcaino, F.; Duarte, J. Vascular superoxide production by endothelin-1 requires Src non-receptor protein tyrosine kinase and MAPK activation. Atherosclerosis 2010, 212, 78–85. [Google Scholar]
- Konior, A.; Klemenska, E.; Brudek, M.; Podolecka, E.; Czarnowska, E.; Beresewicz, A. Seasonal O2− overproduction and endothelial activation in guinea-pig heart; seasonal oxidative stress in rats and humans. J. Mol. Cell. Cardiol. 2011, 50, 686–694. [Google Scholar]
- Ray, R.; Shah, A.M. NADPH oxidase and endothelial cell function. Clin. Sci. 2005, 109, 217–226. [Google Scholar]
- Knock, G.A.; Ward, J.P.T. Redox regulation of protein kinases as a modulator of vascular function. Antioxid. Redox Signal. 2011, 15, 1531–1547. [Google Scholar]
- Dikalov, S.I.; Nazarewicz, R.R.; Bikineyeva, A.; Hilenski, L.; Lassegue, B.; Griendling, K.; Harrison, D.G.; Dikalova, A. Nox2-induced production of mitochondrial superoxide in angiotensin II-mediated endothelial oxidative stress and hypertension. Antioxid. Redox Signal. 2013, 305, H1131–H1140. [Google Scholar]
- Bacsi, A.; Woodberry, M.; Widger, W.; Papaconstantinou, J.; Mitra, S.; Peterson, J.W.; Boldogh, I. Localization of superoxide anion production to mitochondrial electron transport chain in 3-NPA-treated cells. Mitochondrion 2006, 6, 235–244. [Google Scholar]
- Berry, C.E.; Hare, J.M. Xanthine oxidoreductase and cardiovascular disease: Molecular mechanisms and pathophysiological implications. J. Physiol. 2004, 555, 589–606. [Google Scholar]
- Beresewicz, A.; Maczewski, M.; Duda, M. Effect of classic preconditioning and diazoxide on endothelial function and O2− and NO generation in the post-ischemic guinea-pig heart. Cardiovasc. Res. 2004, 63, 118–129. [Google Scholar]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar]
- Baysal, B.E.; Ferrell, R.E.; Willett-Brozick, J.E.; Lawrence, E.C.; Myssiorek, D.; Bosch, A.; van der, M.A.; Taschner, P.E.; Rubinstein, W.S.; Myers, E.N.; et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 2000, 287, 848–851. [Google Scholar]
- Chambers, D.E.; Parks, D.A.; Patterson, G.; Roy, R.; McCord, J.M.; Yoshida, S.; Parmley, L.F.; Downey, J.M. Xanthine oxidase as a source of free radical damage in myocardial ischemia. J. Mol. Cell. Cardiol. 1985, 17, 145–152. [Google Scholar]
- Powers, S.K.; Duarte, J.; Kavazis, A.N.; Talbert, E.E. Reactive oxygen species are signalling molecules for skeletal muscle adaptation. Exp. Physiol. 2010, 95, 1–9. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wojtera, E.; Konior, A.; Fedoryszak-Kuśka, N.; Beręsewicz, A. Obligatory Role of Intraluminal O2− in Acute Endothelin-1 and Angiotensin II Signaling to Mediate Endothelial Dysfunction and MAPK Activation in Guinea-Pig Hearts. Int. J. Mol. Sci. 2014, 15, 19417-19443. https://doi.org/10.3390/ijms151119417
Wojtera E, Konior A, Fedoryszak-Kuśka N, Beręsewicz A. Obligatory Role of Intraluminal O2− in Acute Endothelin-1 and Angiotensin II Signaling to Mediate Endothelial Dysfunction and MAPK Activation in Guinea-Pig Hearts. International Journal of Molecular Sciences. 2014; 15(11):19417-19443. https://doi.org/10.3390/ijms151119417
Chicago/Turabian StyleWojtera, Emilia, Anna Konior, Natalia Fedoryszak-Kuśka, and Andrzej Beręsewicz. 2014. "Obligatory Role of Intraluminal O2− in Acute Endothelin-1 and Angiotensin II Signaling to Mediate Endothelial Dysfunction and MAPK Activation in Guinea-Pig Hearts" International Journal of Molecular Sciences 15, no. 11: 19417-19443. https://doi.org/10.3390/ijms151119417