Conserved miRNAs and Their Response to Salt Stress in Wild Eggplant Solanum linnaeanum Roots


Abstract
:1. Introduction
2. Results and Discussion
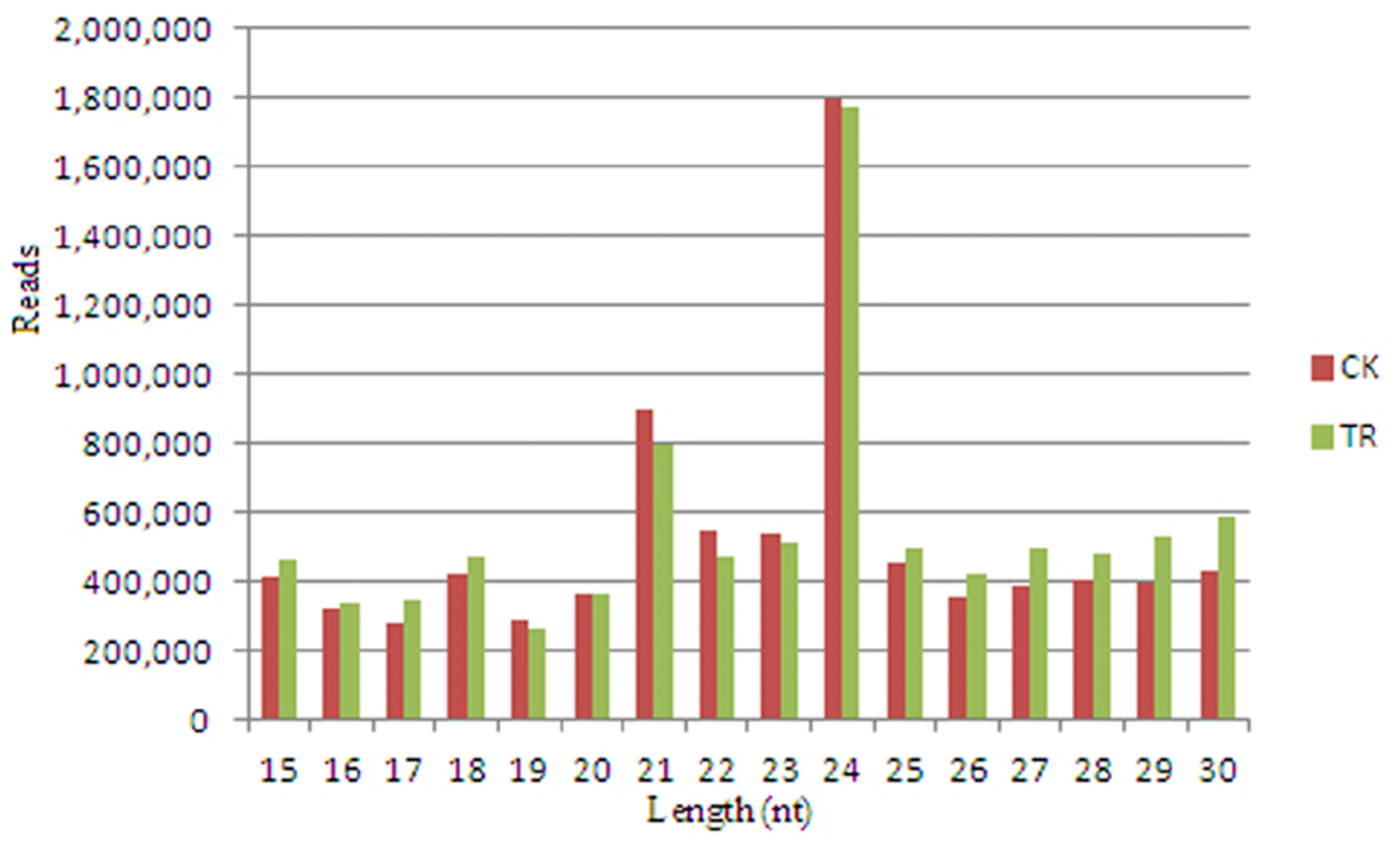
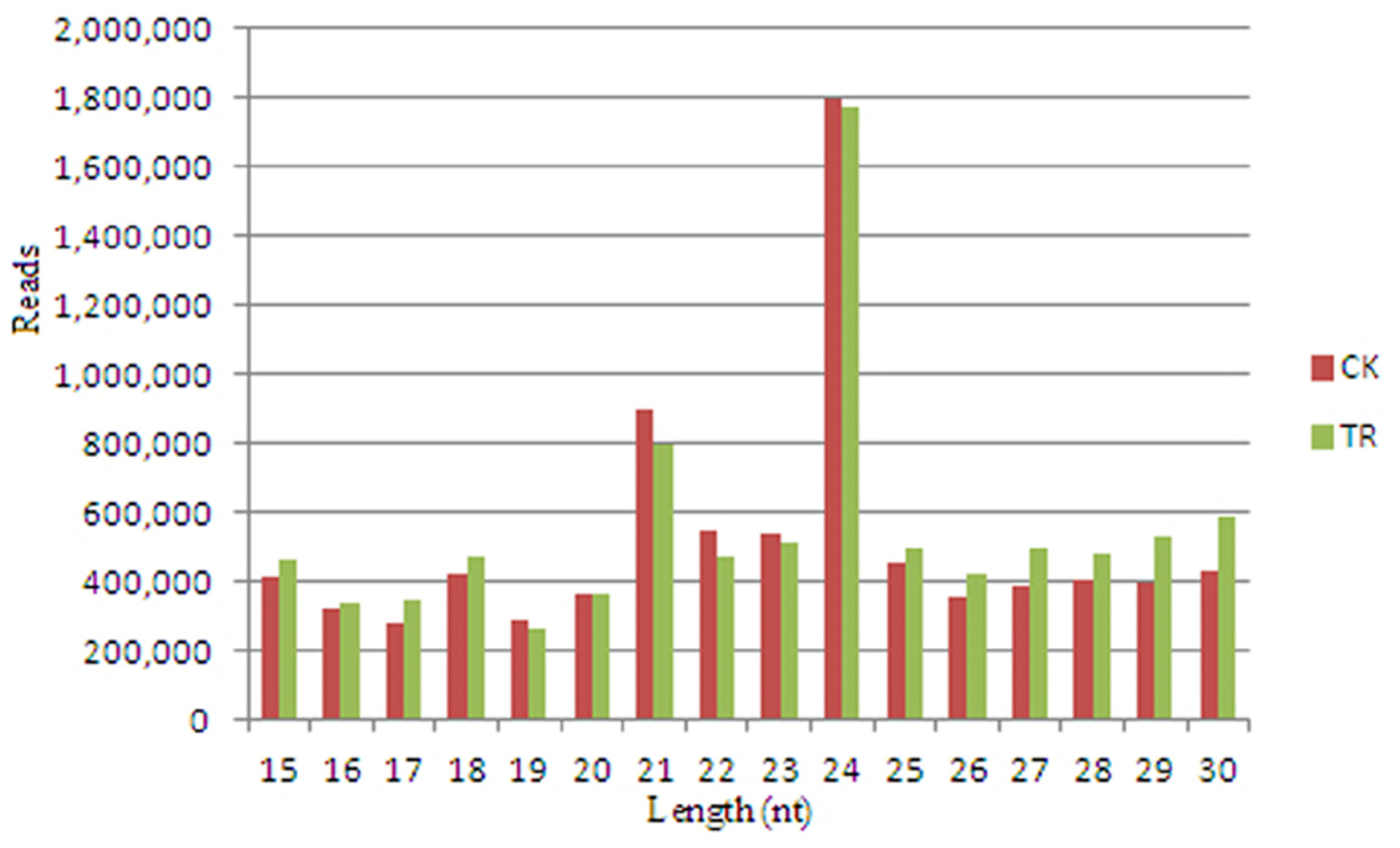
2.1. Deep Sequencing Results of Small RNAs from S. linnaeanum Roots
2.2. Conserved miRNAs in S. linnaeanum Roots
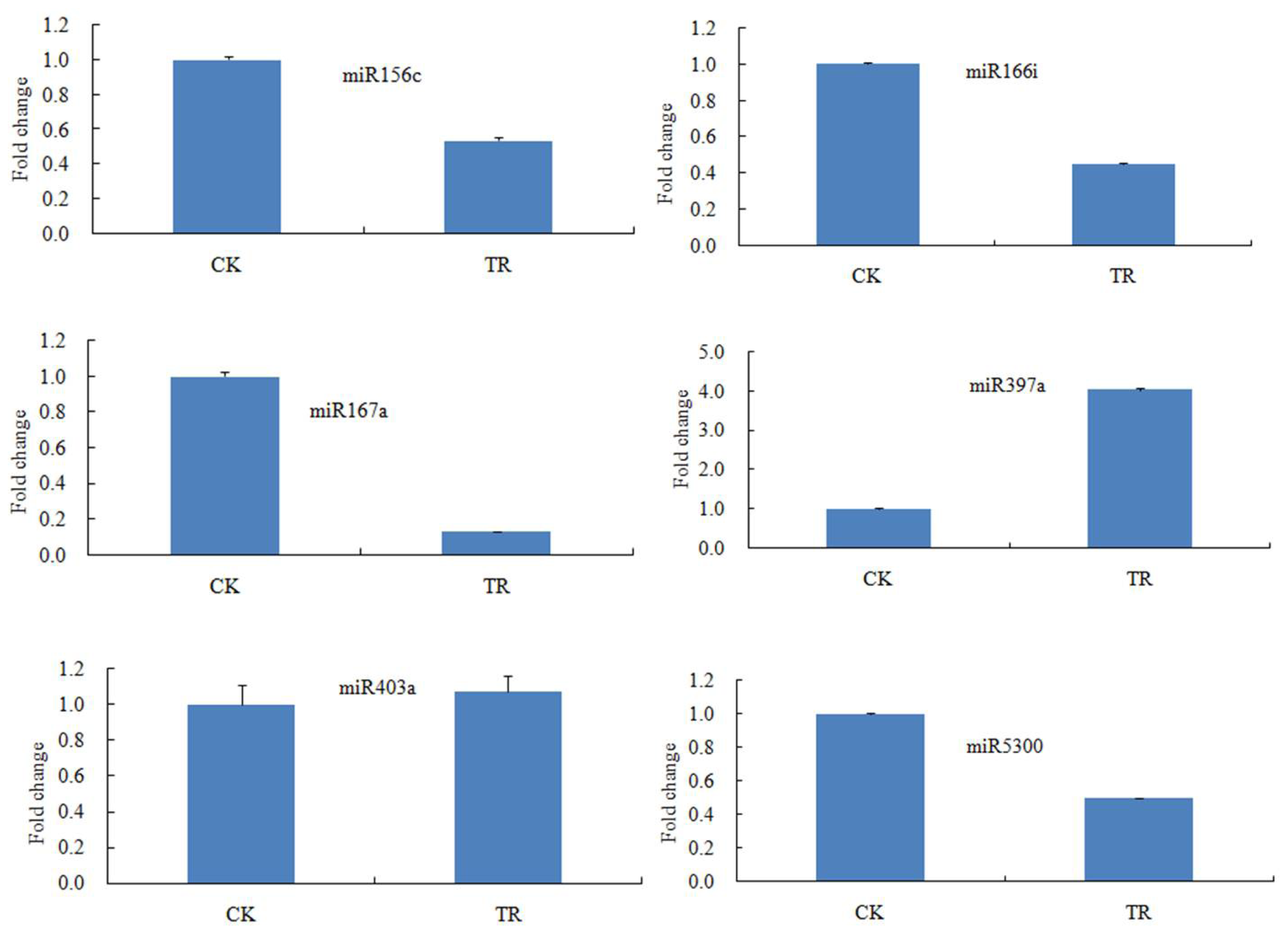
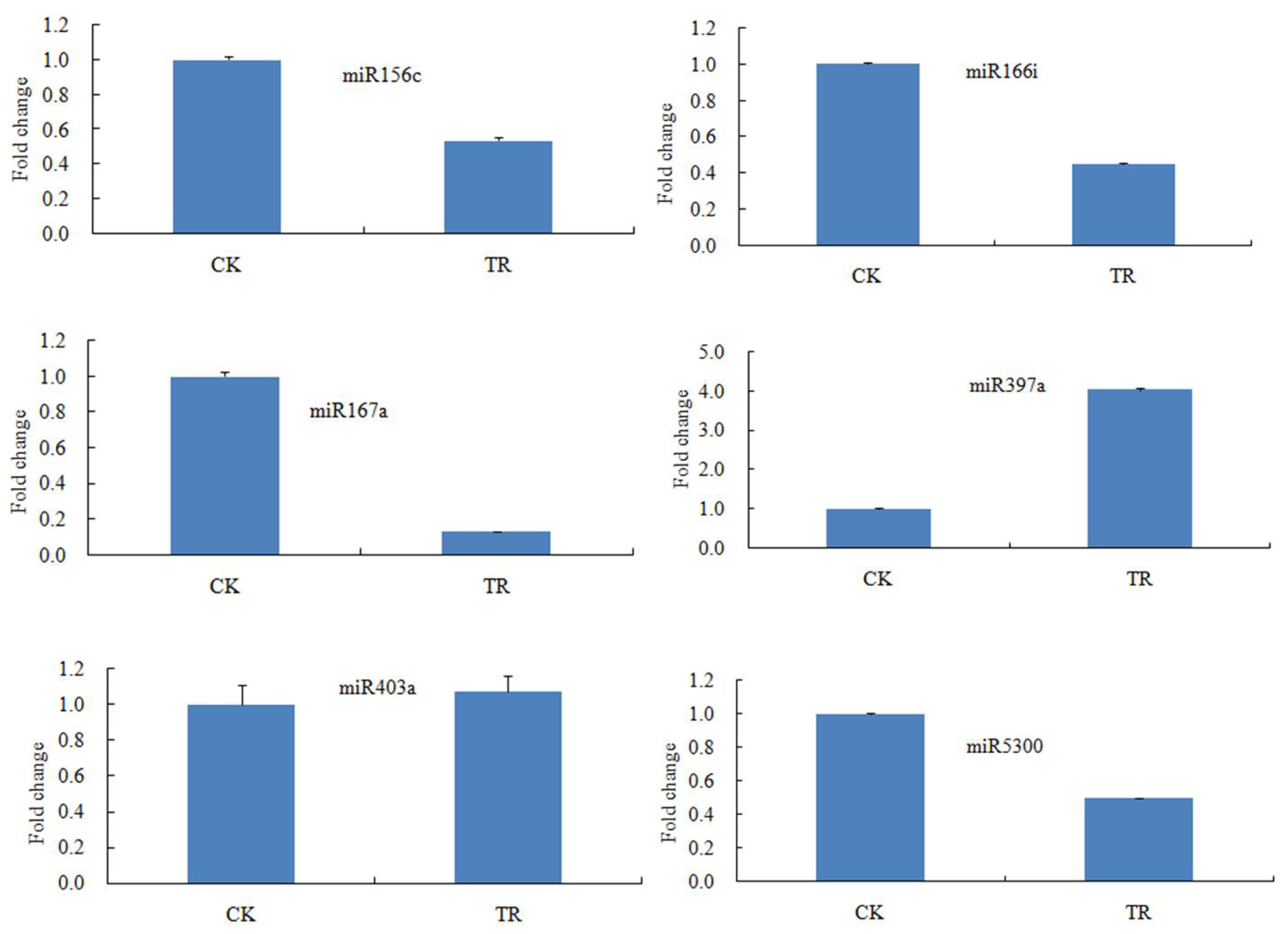
2.3. Validation of miRNAs in S. linnaeanum Roots
2.4. NaCl-Responsive miRNAs in S. linnaeanum Roots
3. Experimental Section
3.1. Plant Materials and NaCl Treatment
3.2. Small RNA Library Construction and Sequencing
3.3. Analysis of Small RNA Sequencing Data
3.4. miRNA Validation by Quantitative Real-Time PCR
3.5. Prediction of miRNA Target Genes
4. Conclusions
Supplementary Information
ijms-15-00839-s001.pdfAcknowledgments
Conflicts of Interest
References
- Wang, W.; Vinocur, B.; Altman, A. Plant responses to drought, salinity and extreme temperatures: Towards genetic engineering for stress tolerance. Planta 2003, 218, 1–14. [Google Scholar]
- Borsani, O.; Zhu, J.; Verslues, P.E.; Sunkar, R.; Zhu, J.K. Endogenous siRNAs derived from a pair of natural cis-antisense transcripts regulate salt tolerance in Arabidopsis. Cell 2005, 123, 1279–1291. [Google Scholar]
- Vinocur, B.; Altman, A. Recent advances in engineering plant tolerance to abiotic stress: Achievements and limitations. Curr. Opin. Biotechnol 2005, 16, 123–132. [Google Scholar]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar]
- Jones-Rhoades, M.W.; Bartel, D.P.; Bartel, B. MicroRNAs and their regulatory roles in plants. Annu. Rev. Plant Biol 2006, 57, 19–53. [Google Scholar]
- Voinnet, O. Origin, biogenesis, and activity of plant microRNAs. Cell 2009, 136, 669–687. [Google Scholar]
- Liu, P.P.; Montgomery, T.A.; Fahlgren, N.; Kasschau, K.D.; Nonogaki, H.; Carrington, J.C. Repression of AUXIN RESPONSE FACTOR10 by microRNA160 is critical for seed germination and post-germination stages. Plant J 2007, 52, 133–146. [Google Scholar]
- Reyes, J.L.; Chua, N.H. ABA induction of miR159 controls transcript levels of two MYB factors during Arabidopsis seed germination. Plant J 2007, 49, 592–606. [Google Scholar]
- Gutierrez, L.; Bussell, J.D.; Pacurar, D.I.; Schwambach, J.; Pacurar, M.; Bellini, C. Phenotypic plasticity of adventitious rooting in Arabidopsis is controlled by complex regulation of AUXIN RESPONSE FACTOR transcripts and microRNA abundance. Plant Cell 2009, 21, 3119–3132. [Google Scholar]
- Sunkar, R.; Chinnusamy, V.; Zhu, J.; Zhu, J.K. Small RNAs as big players in plant abiotic stress responses and nutrient deprivation. Trends Plant Sci 2007, 12, 301–309. [Google Scholar]
- Bazzini, A.A.; Hopp, H.E.; Beachy, R.N.; Asurmendi, S. Infection and coaccumulation of tobacco mosaic virus proteins alter microRNA levels, correlating with symptom and plant development. Proc. Natl. Acad. Sci. USA 2007, 104, 12157–12162. [Google Scholar]
- Lu, S.; Sun, Y.H.; Amerson, H.; Chiang, V.L. MicroRNAs in loblolly pine (Pinus taeda L.) and their association with fusiform rust gall development. Plant J 2007, 51, 1077–1098. [Google Scholar]
- Yang, L.; Jue, D.; Li, W.; Zhang, R.; Chen, M.; Yang, Q. Identification of MiRNA from eggplant (Solanum melongena L.) by small RNA deep sequencing and their response to Verticillium dahliae infection. PLoS One 2013, 8, e72840. [Google Scholar]
- Tang, Z.; Zhang, L.; Xu, C.; Yuan, S.; Zhang, F.; Zheng, Y.; Zhao, C. Uncovering small RNA-mediated responses to cold stress in a wheat thermosensitive genic male-sterile line by deep sequencing. Plant Physiol 2012, 159, 721–738. [Google Scholar]
- Yu, X.; Wang, H.; Lu, Y.; de Ruiter, M.; Cariaso, M.; Prins, M.; van Tunen, A.; He, Y. Identification of conserved and novel microRNAs that are responsive to heat stress in Brassica rapa. J. Exp. Bot 2012, 63, 1025–1038. [Google Scholar]
- Barrera-Figueroa, B.E.; Gao, L.; Diop, N.N.; Wu, Z.; Ehlers, J.D.; Roberts, P.A.; Close, T.J.; Zhu, J.K.; Liu, R. Identification and comparative analysis of drought-associated microRNAs in two cowpea genotypes. BMC Plant Biol 2011, 11, 127. [Google Scholar]
- Li, B.; Qin, Y.; Duan, H.; Yin, W.; Xia, X. Genome-wide characterization of new and drought stress responsive microRNAs in Populus euphratica. J. Exp. Bot 2011, 62, 3765–3779. [Google Scholar]
- Ding, Y.; Chen, Z.; Zhu, C. Microarray-based analysis of cadmium-responsive microRNAs in rice (Oryza sativa). J. Exp. Bot 2011, 62, 3563–3573. [Google Scholar]
- Chen, L.; Wang, T.; Zhao, M.; Tian, Q.; Zhang, W.H. Identification of aluminum-responsive microRNAs in Medicago truncatula by genome-wide high-throughput sequencing. Planta 2012, 235, 375–386. [Google Scholar]
- Liu, H.H.; Tian, X.; Li, Y.J.; Wu, C.A.; Zheng, C.C. Microarray-based analysis of stress-regulated microRNAs in Arabidopsis Thaliana. RNA 2008, 14, 836–843. [Google Scholar]
- Ding, D.; Zhang, L.; Wang, H.; Liu, Z.; Zhang, Z.; Zheng, Y. Differential expression of miRNAs in response to salt stress in maize roots. Ann. Bot 2009, 103, 29–38. [Google Scholar]
- Covarrubias, A.A.; Reyes, J.L. Post-transcriptional gene regulation of salinity and drought responses by plant microRNAs. Plant Cell Environ 2010, 33, 481–489. [Google Scholar]
- Collonnier, C.; Fock, I.; Kashyap, V.; Rotino, G.L.; Daunay, M.C.; Lian, Y.; Mariska, I.K.; Rajam, M.V.; Servaes, A.; Ducreux, G.; et al. Applications of biotechnology in eggplant. Plant Cell Tissue Organ 2001, 65, 91–107. [Google Scholar]
- Doganlar, S.; Frary, A.; Daunay, M.C.; Lester, R.N.; Tanksley, S.D. A comparative genetic linkage map of eggplant (Solanum melongena) and its implications for genome evolution in the solanaceae. Genetics 2002, 161, 1697–1711. [Google Scholar]
- Mueller, L.A.; Solow, T.H.; Taylor, N.; Skwarecki, B.; Buels, R.; Binns, J.; Lin, C.; Wright, M.H.; Ahrens, R.; Wang, Y.; et al. The SOL Genomics Network: A comparative resource for solanaceae biology and beyond. Plant Physiol 2005, 138, 1310–1317. [Google Scholar]
- Daniell, H.; Lee, S.B.; Grevich, J.; Saski, C.; Quesada-Vargas, T.; Guda, C.; Tomkins, J.; Jansen, R.K. Complete chloroplast genome sequences of Solanum bulbocastanum, Solanum lycopersicum and comparative analyses with other Solanaceae genomes. Theor. Appl. Genet 2006, 112, 1503–1518. [Google Scholar]
- Wu, F.; Eannetta, N.T.; Xu, Y.; Durrett, R.; Mazourek, M.; Jahn, M.M.; Tanksley, S.D. A COSII genetic map of the pepper genome provides a detailed picture of synteny with tomato and new insights into recent chromosome evolution in the genus Capsicum. Theor. Appl. Genet 2009, 118, 1279–1293. [Google Scholar]
- Wu, F.; Eannetta, N.T.; Xu, Y.; Tanksley, S.D. A detailed synteny map of the eggplant genome based on conserved ortholog set II (COSII) markers. Theor. Appl. Genet 2009, 118, 927–935. [Google Scholar]
- Rajagopalan, R.; Vaucheret, H.; Trejo, J.; Bartel, D.P. A diverse and evolutionarily fluid set of microRNAs in Arabidopsis thaliana. Genes Dev 2006, 20, 3407–3425. [Google Scholar]
- Fahlgren, N.; Howell, M.D.; Kasschau, K.D.; Chapman, E.J.; Sullivan, C.M.; Cumbie, J.S.; Givan, S.A.; Law, T.F.; Grant, S.R.; Dangl, J.L.; et al. High-throughput sequencing of Arabidopsis microRNAs: Evidence for frequent birth and death of MIRNA genes. PLoS One 2007, 2, e219. [Google Scholar]
- Szittya, G.; Moxon, S.; Santos, D.M.; Jing, R.; Fevereiro, M.P.; Moulton, V.; Dalmay, T. High-throughput sequencing of Medicago truncatula short RNAs identifies eight new miRNA families. BMC Genomics 2008, 9, 593. [Google Scholar]
- Morin, R.D.; Aksay, G.; Dolgosheina, E.; Ebhardt, H.A.; Magrini, V.; Mardis, E.R.; Sahinalp, S.C.; Unrau, P.J. Comparative analysis of the small RNA transcriptomes of Pinus contorta and Oryza sativa. Genome Res 2008, 18, 571–584. [Google Scholar]
- Chi, X.; Yang, Q.; Chen, X.; Wang, J.; Pan, L.; Chen, M.; Yang, Z.; He, Y.; Liang, X.; Yu, S. Identification and characterization of microRNAs from peanut (Arachis hypogaea L.) by high-throughput sequencing. PLoS One 2011, 6, e27530. [Google Scholar]
- Martínez, G.; Forment, J.; Llave, C.; Pallás, V.; Gómez, G. High-throughput sequencing, characterization and detection of new and conserved cucumber miRNAs. PLoS One 2011, 6, e19523. [Google Scholar]
- Guo, H.; Kan, Y.; Liu, W. Differential expression of miRNAs in response to topping in flue-cured tobacco (Nicotiana tabacum) roots. PLoS One 2011, 6, e28565. [Google Scholar]
- Song, C.; Wang, C.; Zhang, C.; Korir, N.K.; Yu, H.; Ma, Z.; Fang, J. Deep sequencing discovery of novel and conserved microRNAs in trifoliate orange (Citrus trifoliata). BMC Genomics 2010, 11, 431. [Google Scholar]
- Sunkar, R.; Zhu, J.K. Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell 2004, 16, 2001–2019. [Google Scholar]
- Cai, X.; Davis, E.J.; Ballif, J.; Liang, M.; Bushman, E.; Haroldsen, V.; Torabinejad, J.; Wu, Y. Mutant identification and characterization of the laccase gene family in Arabidopsis. J. Exp. Bot 2006, 57, 2563–2569. [Google Scholar]
- Li, Y.; Yan, J.; Kim, I.; Liu, C.; Huo, K.; Rao, H. Rad4 regulates protein turnover at a postubiquitylation step. Mol. Biol. Cell 2010, 21, 177–185. [Google Scholar]
- Pauluzzi, G.; Divol, F.; Puig, J.; Guiderdoni, E.; Dievart, A.; Périn, C. Surfing along the root ground tissue gene network. Dev. Biol 2012, 365, 14–22. [Google Scholar]
- Chung, M.Y.; Vrebalov, J.; Alba, R.; Lee, J.; McQuinn, R.; Chung, J.D.; Klein, P.; Giovannoni, J. A tomato (Solanum lycopersicum) APETALA2/ERF gene, SlAP2a, is a negative regulator of fruit ripening. Plant J 2010, 64, 936–947. [Google Scholar]
- Koyama, T.; Furutani, M.; Tasaka, M.; Ohme-Takagi, M. TCP transcription factors control the morphology of shoot lateral organs via negative regulation of the expression of boundary-specific genes in Arabidopsis. Plant Cell 2007, 19, 473–484. [Google Scholar]
- Audic, S.; Claverie, J.M. The significance of digital gene expression profiles. Genome Res 1997, 7, 986–995. [Google Scholar]
- Dai, X.; Zhao, P.X. psRNATarget: A plant small RNA target analysis server. Nucleic Acids Res 2011, 39, W155–W159. [Google Scholar]

{kind=link}
{kind=link}
| Category | CK | Percent (%) | TR | Percent (%) |
|---|---|---|---|---|
| Raw reads | 13,989,100 | 21,284,496 | ||
| Mappable reads | 8,462,890 | 100.00 | 8,999,145 | 100.00 |
| Mapped to miRNA | 466,136 | 5.51 | 437,144 | 4.86 |
| Mapped to mRNA | 723,297 | 8.55 | 793,475 | 8.82 |
| Mapped to RFam | 1,315,886 | 15.55 | 1,387,942 | 15.42 |
| Mapped to Repbase | 5,674 | 0.07 | 4,745 | 0.05 |
| Mapped to genome | 1,763,934 | 20.84 | 2,063,801 | 22.93 |
| No hit | 4,187,963 | 49.49 | 4,312,038 | 47.92 |
| miRNA | Log2(TR/CK) | p value | Predicted target | Putative function of target |
|---|---|---|---|---|
| sli-miR156b | −1.01 | 2.46 × 10−127 | SGN-U325281 | Squamosa promoter-binding protein |
| sli-miR156c | −1.18 | 2.72 × 10−52 | SGN-U317176 | Squamosa promoter-binding protein |
| sli-miR162b | −1.25 | 8.35 × 10−99 | Solyc10g005130.2.1 | Ribonuclease 3-like protein 3 |
| sli-miR164c | 1.03 | 3.57 × 10−16 | SGN-U327571 | Lipase-related |
| sli-miR166d | 1.97 | 1.02 × 10−45 | Solyc11g011150.1.1 | DNA repair protein Rad4 family |
| sli-miR167a | −1.45 | 4.26 × 10−166 | SGN-U313907 | Annexin 1 |
| sli-miR167b | −1.25 | 5.57 × 10−15 | Solyc03g095940.1.1 | LOB domain family protein |
| sli-miR171b | −1.18 | 1.89 × 10−47 | SGN-U333058 | Scarecrow transcription factor family protein |
| sli-miR171e | −1.08 | 1.24 × 10−12 | SGN-U333058 | Scarecrow transcription factor family protein |
| sli-miR172a | −1.66 | 1.94 × 10−44 | SGN-U563871 | Floral homeotic protein APETALA2 |
| sli-miR319a | −1.07 | 2.28 × 10−28 | SGN-U31990 | TCP family transcription factor |
| sli-miR397a | 1.91 | 1.04 × 10−43 | SGN-U327694 | Laccase |
| sli-miR399b | −1.14 | 9.82 × 10−14 | Solyc03g031410.1.1 | Unknown Protein |
| sli-miR5300 | −1.92 | 1.55 × 10−155 | SGN-U336733 | CC-NBS-LRR protein |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhuang, Y.; Zhou, X.-H.; Liu, J. Conserved miRNAs and Their Response to Salt Stress in Wild Eggplant Solanum linnaeanum Roots. Int. J. Mol. Sci. 2014, 15, 839-849. https://doi.org/10.3390/ijms15010839
Zhuang Y, Zhou X-H, Liu J. Conserved miRNAs and Their Response to Salt Stress in Wild Eggplant Solanum linnaeanum Roots. International Journal of Molecular Sciences. 2014; 15(1):839-849. https://doi.org/10.3390/ijms15010839
Chicago/Turabian StyleZhuang, Yong, Xiao-Hui Zhou, and Jun Liu. 2014. "Conserved miRNAs and Their Response to Salt Stress in Wild Eggplant Solanum linnaeanum Roots" International Journal of Molecular Sciences 15, no. 1: 839-849. https://doi.org/10.3390/ijms15010839