Enhanced Antitumor Effects of Adenoviral-Mediated siRNA against GRP78 Gene on Adenosine-Induced Apoptosis in Human Hepatoma HepG2 Cells

Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
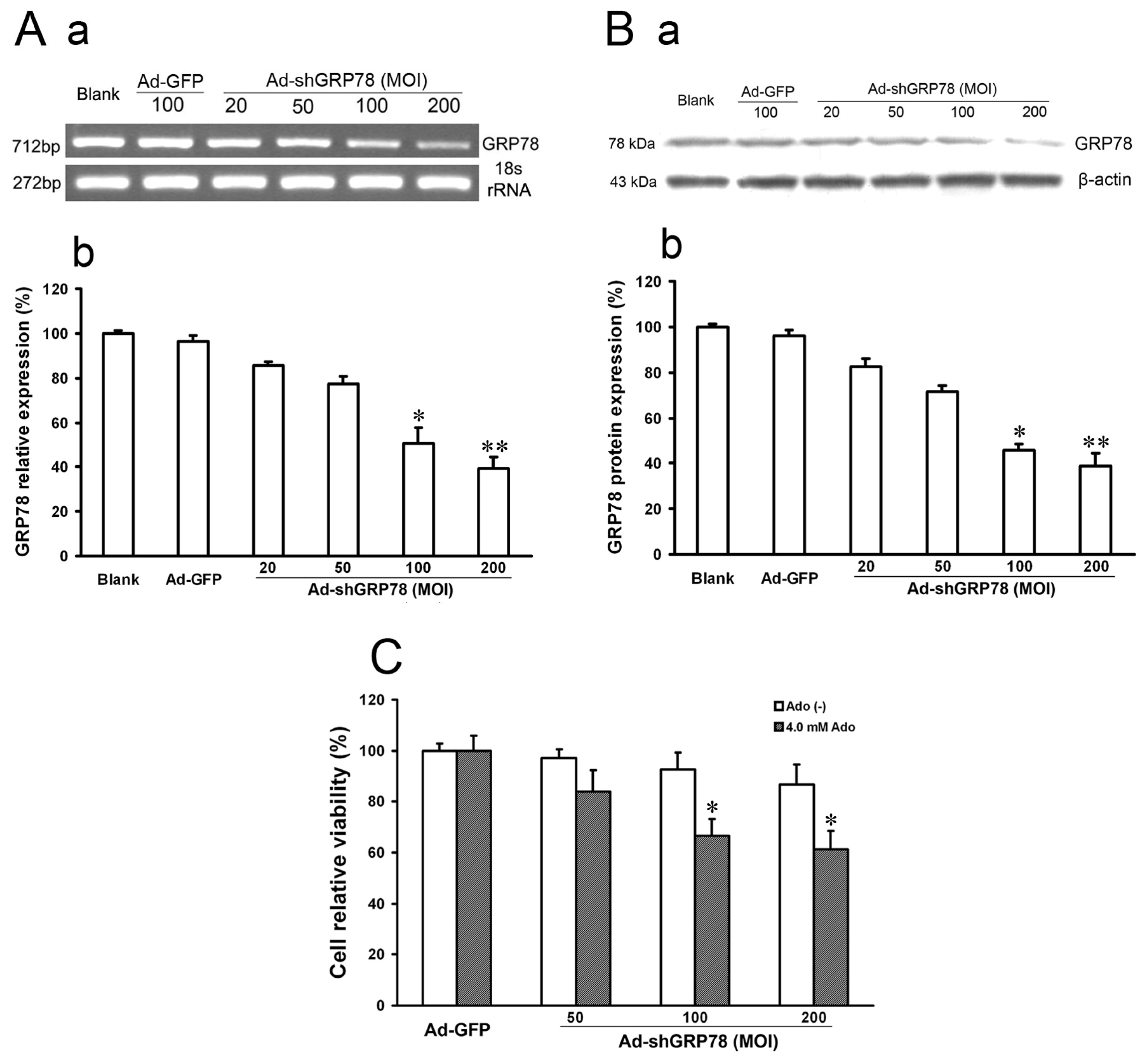
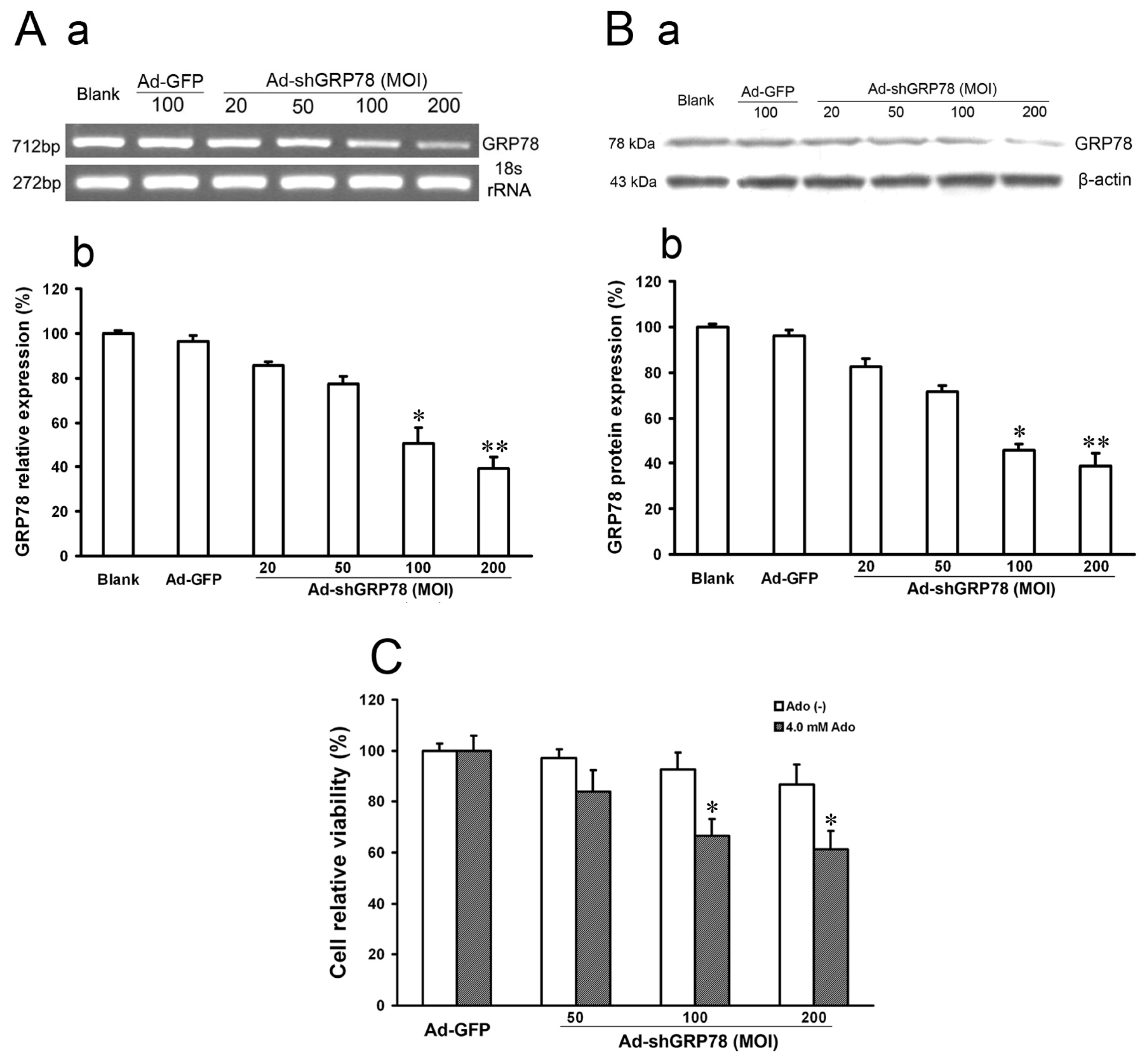
2.1.1. Verification of GRP78 Gene Knockdown and Evaluation of the Effects on Cell Growth
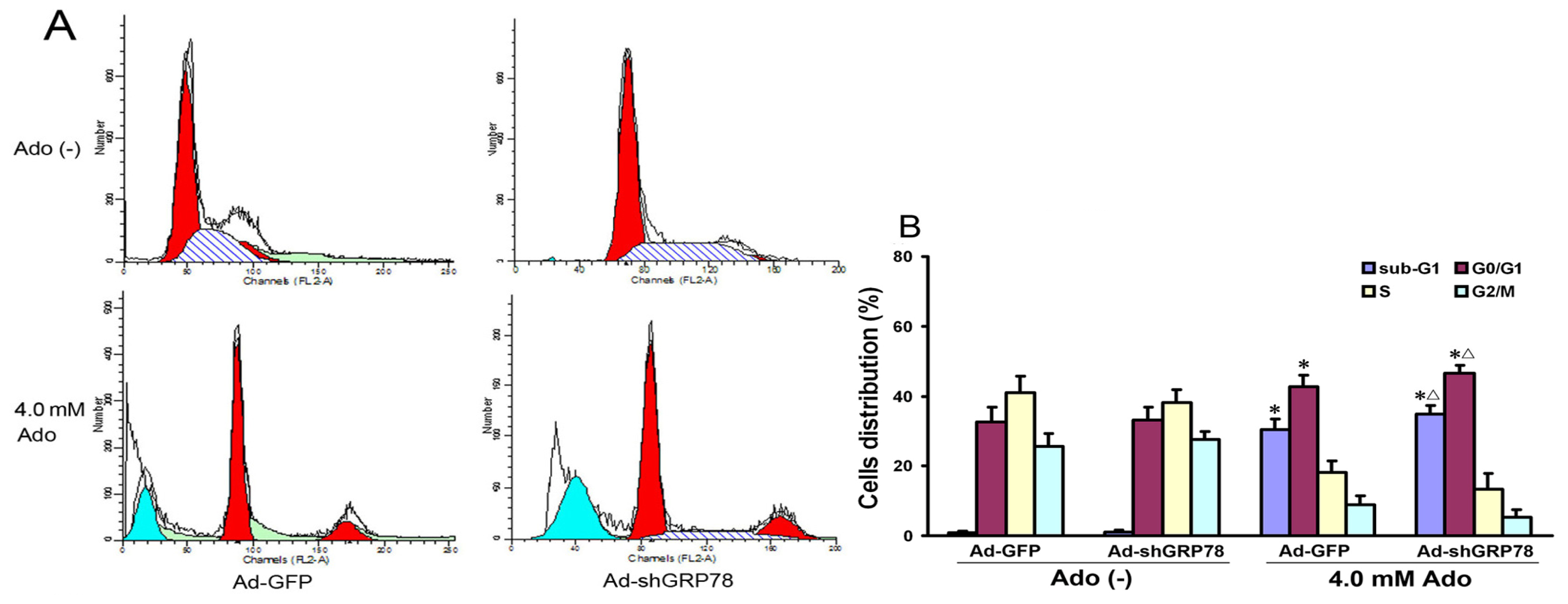
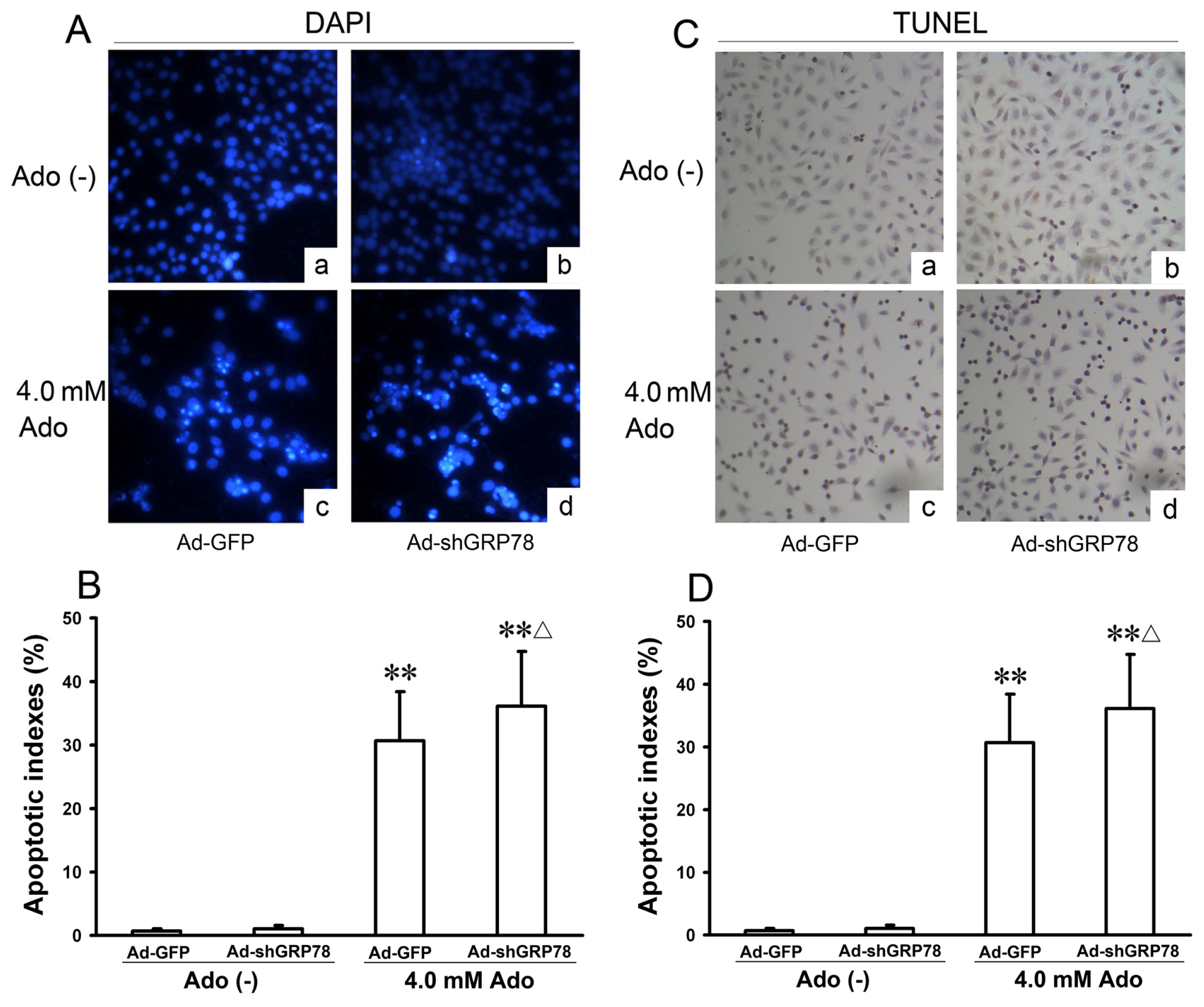
2.1.2. Effects of GRP78 Knockdown on Adenosine-Induced Alterations in Cell-Cycle Distribution and Apoptosis
2.1.3. Effects of Caspase-4 Knockdown on Adenosine-Induced Cell Growth Inhibition and Apoptosis
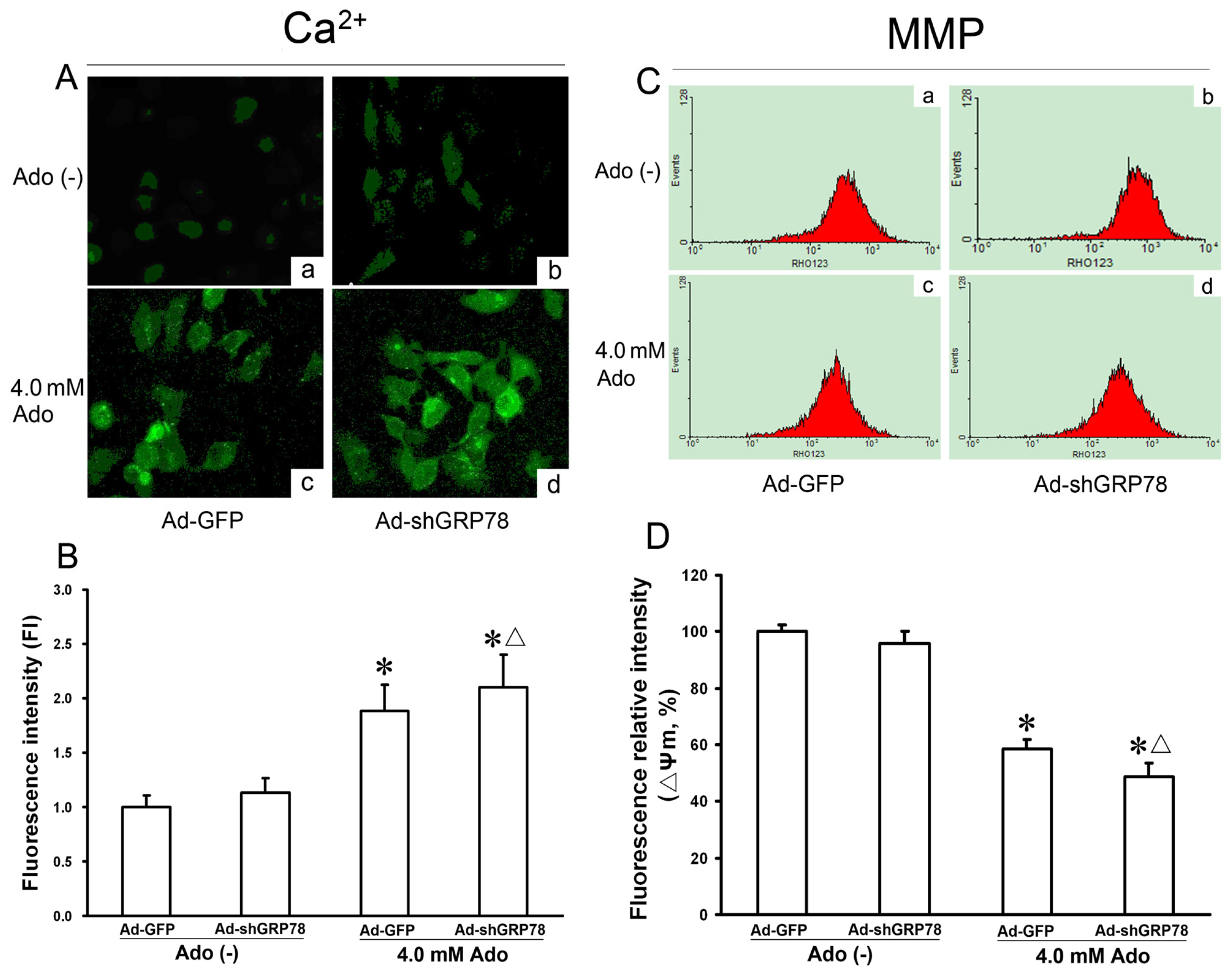
2.1.4. Effects of GRP78 Knockdown on Adenosine-Induced Cytosolic [Ca2+]i and Mitochondrial Membrane Potential (ΔΨm)
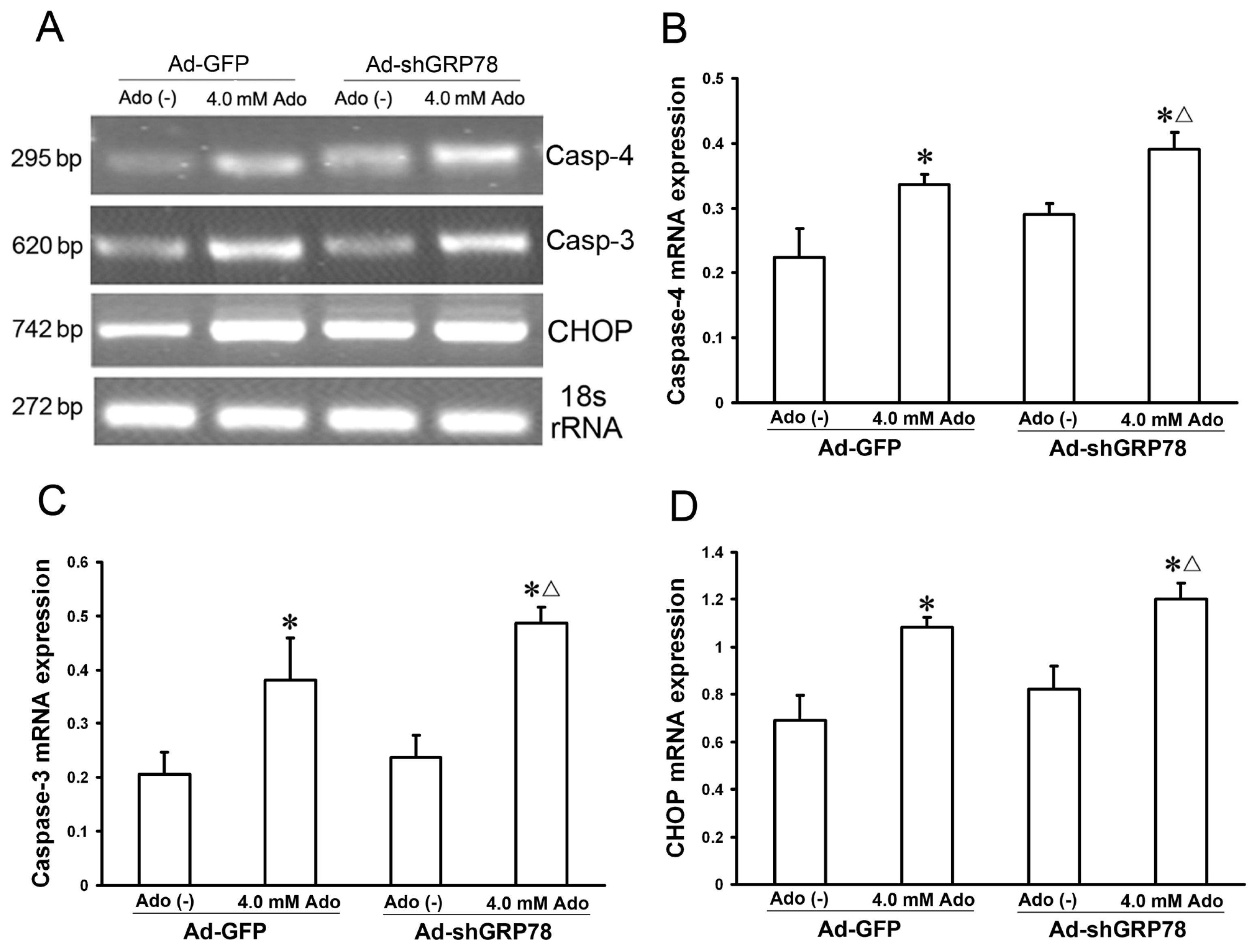
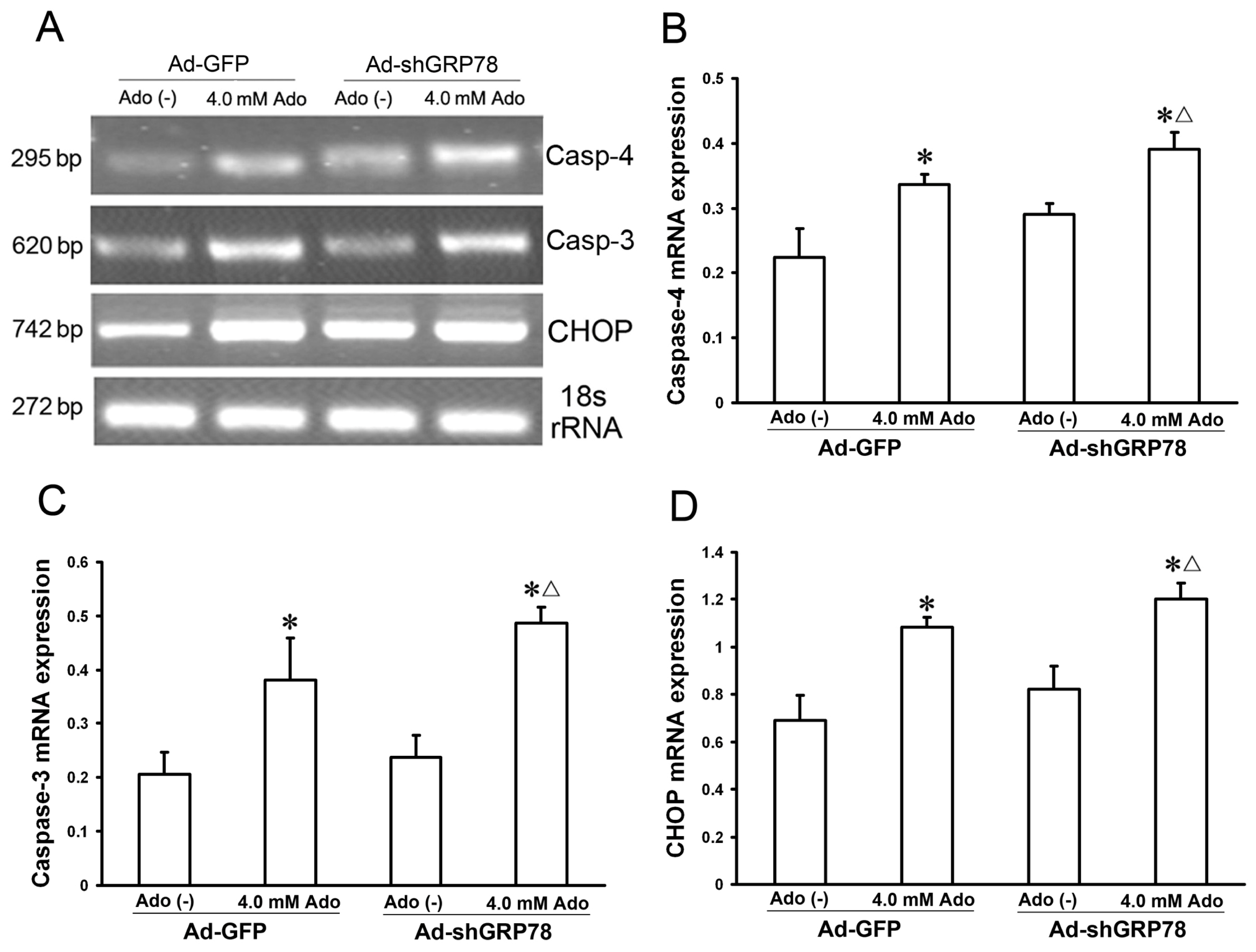
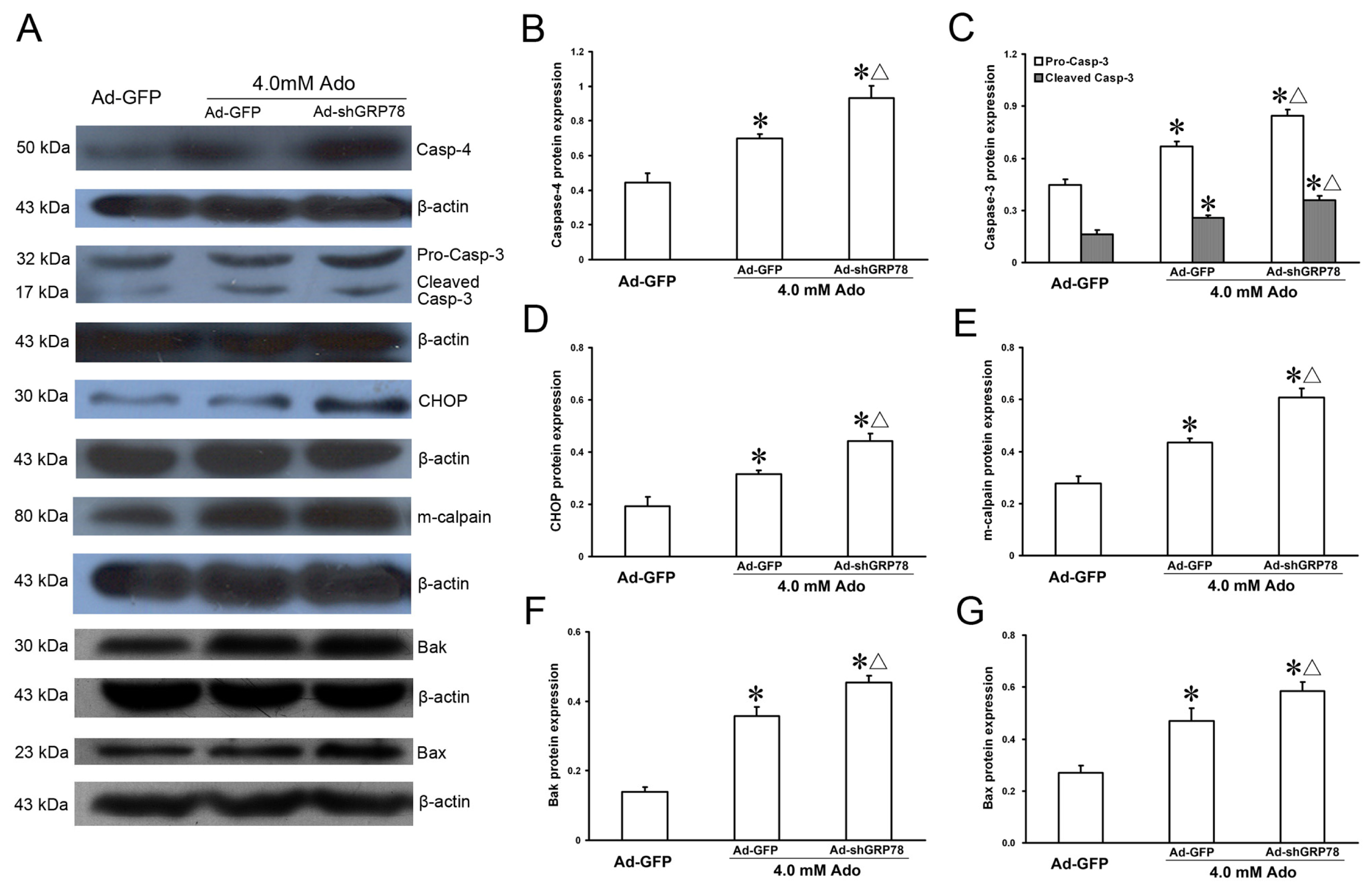
2.1.5. Effects of GRP78 Knockdown on the mRNA and Protein Expressions of ER Stress Relative Genes
2.2. Discussion
3. Materials and Methods
3.1. Reagents and Antibodies
3.2. Cell Culture and Experimental Groups
3.3. Construction of the Recombinant Adenovirus Vector and Adenovirus Transfection
3.4. Determination of Cell Viability by CCK-8 Assay
3.5. Cell Cycle Phase Determination by Flow Cytometry
3.6. Identification of Cell Apoptosis by DAPI Staining and TUNEL Assay
3.7. Measurement of Intracellular Ca2+ Concentration
3.8. Detection of Mitochondrial Membrane Potential (ΔΨm) by Flow Cytometric Analysis
3.9. RNA Preparation and Real-Time PCR Assay
3.10. Protein Extraction and Western Blot Analysis
3.11. Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| GFP | green fluorescent protein |
| MOI | multiplicity of infection |
| Ad-GFP | an only expressing GFP empty adenovirus vector |
| Ad-shGRP78 | recombinant adenovirus carrying short hairpin RNA targeted against GRP78 |
| p-GFP | a plasmid vector carrying GFP |
| p-shCasp-4 | a plasmid vector carrying short hairpin RNA targeted against caspase-4 |
References
- Cao, H.; Phan, H.; Yang, L.X. Improved chemotherapy for hepatocellular carcinoma. Anticancer Res 2012, 32, 1379–1386. [Google Scholar]
- Bruix, J.; Sherman, M. Management of hepatocellular carcinoma: An update. Hepatology 2011, 53, 1020–1022. [Google Scholar]
- Lee, A.S. GRP78 induction in cancer: Therapeutic and prognostic implications. Cancer Res 2007, 67, 3496–3499. [Google Scholar]
- Gorman, A.M.; Healy, S.J.; Jager, R.; Samali, A. Stress management at the ER: Regulators of ER stress-induced apoptosis. Pharmacol. Ther 2012, 134, 306–316. [Google Scholar]
- Saito, M.; Yaguchi, T.; Yasuda, Y.; Nakano, T.; Nishizaki, T. Adenosine suppresses CW2 human colonic cancer growth by inducing apoptosis via A(1) adenosine receptors. Cancer Lett 2010, 290, 211–215. [Google Scholar]
- Wu, L.F.; Li, G.P.; Feng, J.L.; Pu, Z.J. Molecular mechanisms of adenosine-induced apoptosis in human HepG2 cells. Acta Pharmacol. Sin 2006, 27, 477–484. [Google Scholar]
- Wu, L.F.; Wei, B.L.; Guo, Y.T.; Ye, Y.Q.; Li, G.P.; Pu, Z.J.; Feng, J.L. Apoptosis induced by adenosine involves endoplasmic reticulum stress in EC109 cells. Int. J. Mol. Med 2012, 30, 797–804. [Google Scholar]
- Shirali, S.; Aghaei, M.; Shabani, M.; Fathi, M.; Sohrabi, M.; Moeinifard, M. Adenosine induces cell cycle arrest and apoptosis via cyclinD1/Cdk4 and Bcl-2/Bax pathways in human ovarian cancer cell line OVCAR-3. Tumor Biol 2013, 34, 1085–1095. [Google Scholar]
- Wu, L.F.; Ye, Y.Q.; Huang, G.Y.; Li, H.B.; Li, G.P.; Pu, Z.J.; Wei, B.L.; Feng, J.L. Involvement of endoplasmic reticulum stress in adenosine-induced human hepatoma HepG2 cell apoptosis. Oncol. Rep 2011, 26, 73–79. [Google Scholar]
- Li, J.; Lee, A.S. Stress induction of GRP78/BiP and its role in cancer. Curr. Mol. Med 2006, 6, 45–54. [Google Scholar]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol 2012, 13, 89–102. [Google Scholar]
- Woehlbier, U.; Hetz, C. Modulating stress responses by the UPRosome: A matter of life and death. Trends Biochem. Sci 2011, 36, 329–337. [Google Scholar]
- Wu, J.; Kaufman, R.J. From acute ER stress to physiological roles of the Unfolded Protein Response. Cell Death Differ 2006, 13, 374–384. [Google Scholar]
- Roller, C.; Maddalo, D. The molecular chaperone GRP78/BiP in the development of chemoresistance: Mechanism and possible treatment. Front. Pharmacol 2013, 4, 10. [Google Scholar]
- Hitomi, J.; Katayama, T.; Eguchi, Y.; Kudo, T.; Taniguchi, M.; Koyama, Y.; Manabe, T.; Yamagishi, S.; Bando, Y.; Imaizumi, K.; et al. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Abeta-induced cell death. J. Cell Biol 2004, 165, 347–356. [Google Scholar]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep 2006, 7, 880–885. [Google Scholar]
- Pyrko, P.; Schonthal, A.H.; Hofman, F.M.; Chen, T.C.; Lee, A.S. The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas. Cancer Res 2007, 67, 9809–9816. [Google Scholar]
- Uckun, F.M.; Qazi, S.; Ozer, Z.; Garner, A.L.; Pitt, J.; Ma, H.; Janda, K.D. Inducing apoptosis in chemotherapy-resistant B-lineage acute lymphoblastic leukaemia cells by targeting HSPA5, a master regulator of the anti-apoptotic unfolded protein response signalling network. Br. J. Haematol 2011, 153, 741–752. [Google Scholar]
- Scriven, P.; Coulson, S.; Haines, R.; Balasubramanian, S.; Cross, S.; Wyld, L. Activation and clinical significance of the unfolded protein response in breast cancer. Br. J. Cancer 2009, 101, 1692–1698. [Google Scholar]
- Pootrakul, L.; Datar, R.H.; Shi, S.R.; Cai, J.; Hawes, D.; Groshen, S.G.; Lee, A.S.; Cote, R.J. Expression of stress response protein Grp78 is associated with the development of castration-resistant prostate cancer. Clin. Cancer Res 2006, 12, 5987–5993. [Google Scholar]
- Dong, D.; Stapleton, C.; Luo, B.; Xiong, S.; Ye, W.; Zhang, Y.; Jhaveri, N.; Zhu, G.; Ye, R.; Liu, Z.; et al. A critical role for GRP78/BiP in the tumor microenvironment for neovascularization during tumor growth and metastasis. Cancer Res 2011, 71, 2848–2857. [Google Scholar]
- Roue, G.; Perez-Galan, P.; Mozos, A.; Lopez-Guerra, M.; Xargay-Torrent, S.; Rosich, L.; Saborit-Villarroya, I.; Normant, E.; Campo, E.; Colomer, D. The Hsp90 inhibitor IPI-504 overcomes bortezomib resistance in mantle cell lymphoma in vitro and in vivo by down-regulation of the prosurvival ER chaperone BiP/Grp78. Blood 2011, 117, 1270–1279. [Google Scholar]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet 2010, 11, 597–610. [Google Scholar]
- Virrey, J.J.; Dong, D.; Stiles, C.; Patterson, J.B.; Pen, L.; Ni, M.; Schonthal, A.H.; Chen, T.C.; Hofman, F.M.; Lee, A.S. Stress chaperone GRP78/BiP confers chemoresistance to tumor-associated endothelial cells. Mol. Cancer Res 2008, 6, 1268–1275. [Google Scholar]
- Yung, H.W.; Charnock-Jones, D.S.; Burton, G.J. Regulation of AKT phosphorylation at Ser473 and Thr308 by endoplasmic reticulum stress modulates substrate specificity in a severity dependent manner. PLoS One 2011, 6, e17894. [Google Scholar]
- Wu, L.F.; Li, G.P.; Su, J.D.; Pu, Z.J.; Feng, J.L.; Ye, Y.Q.; Wei, B.L. Involvement of NF-κB activation in the apoptosis induced by extracellular adenosine in human hepatocellular carcinoma HepG2 cells. Biochem. Cell Biol 2010, 88, 705–714. [Google Scholar]
- Wang, N.S.; Unkila, M.T.; Reineks, E.Z.; Distelhorst, C.W. Transient expression of wild-type or mitochondrially targeted Bcl-2 induces apoptosis, whereas transient expression of endoplasmic reticulum-targeted Bcl-2 is protective against Bax-induced cell death. J. Biol. Chem 2001, 276, 44117–44128. [Google Scholar]
- Foyouzi-Youssefi, R.; Arnaudeau, S.; Borner, C.; Kelley, W.L.; Tschopp, J.; Lew, D.P.; Demaurex, N.; Krause, K.H. Bcl-2 decreases the free Ca2+ concentration within the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2000, 97, 5723–5728. [Google Scholar]
- Scorrano, L.; Oakes, S.A.; Opferman, J.T.; Cheng, E.H.; Sorcinelli, M.D.; Pozzan, T.; Korsmeyer, S.J. BAX and BAK regulation of endoplasmic reticulum Ca2+: A control point for apoptosis. Science 2003, 300, 135–139. [Google Scholar]
- He, H.; Lam, M.; McCormick, T.S.; Distelhorst, C.W. Maintenance of calcium homeostasis in the endoplasmic reticulum by Bcl-2. J. Cell Biol 1997, 138, 1219–1228. [Google Scholar]
- Pizzo, P.; Pozzan, T. Mitochondria-endoplasmic reticulum choreography: Structure and signaling dynamics. Trends Cell Biol 2007, 17, 511–517. [Google Scholar]
- Bromati, C.R.; Lellis-Santos, C.; Yamanaka, T.S.; Nogueira, T.C.; Leonelli, M.; Caperuto, L.C.; Gorjao, R.; Leite, A.R.; Anhe, G.F.; Bordin, S. UPR induces transient burst of apoptosis in islets of early lactating rats through reduced AKT phosphorylation via ATF4/CHOP stimulation of TRB3 expression. Am. J. Physiol. Regul. Integr. Comp. Physiol 2011, 300, R92–R100. [Google Scholar]
- Brunelle, J.K.; Letai, A. Control of mitochondrial apoptosis by the Bcl-2 family. J. Cell Sci 2009, 122, 437–441. [Google Scholar]
- Hetz, C.; Bernasconi, P.; Fisher, J.; Lee, A.H.; Bassik, M.C.; Antonsson, B.; Brandt, G.S.; Iwakoshi, N.N.; Schinzel, A.; Glimcher, L.H.; et al. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha. Science 2006, 312, 572–576. [Google Scholar]
- Morishima, N.; Nakanishi, K.; Takenouchi, H.; Shibata, T.; Yasuhiko, Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J. Biol. Chem 2002, 277, 34287–34294. [Google Scholar]
- Lakshmanan, U.; Porter, A.G. Caspase-4 interacts with TNF receptor-associated factor 6 and mediates lipopolysaccharide-induced NF-kappaB-dependent production of IL-8 and CC chemokine ligand 4 (macrophage-inflammatory protein-1). J. Immunol 2007, 179, 8480–8490. [Google Scholar]
- Tamura, K.; Kanno, T.; Fujita, Y.; Gotoh, A.; Nakano, T.; Nishizaki, T. A(2a) adenosine receptor mediates HepG2 cell apoptosis by downregulating Bcl-X(L) expression and upregulating Bid expression. J. Cell. Biochem 2012, 113, 1766–1775. [Google Scholar]
- Cook, K.L.; Shajahan, A.N.; Wärri, A.; Jin, L.; Hilakivi-Clarke, L.A.; Clarke, R. Glucose-regulated protein 78 controls cross-talk between apoptosis and autophagy to determine antiestrogen responsiveness. Cancer Res 2012, 72, 3337–3349. [Google Scholar]
- Su, S.F.; Chang, Y.W.; Andreu-Vieyra, C.; Fang, J.Y.; Yang, Z.; Han, B.; Lee, A.S.; Liang, G. miR-30d, miR-181a and miR-199a-5p cooperatively suppress the endoplasmic reticulum chaperone and signaling regulator GRP78 in cancer. Oncogene 2013, 32, 4694–4701. [Google Scholar]
- Huang, G.; Zeng, Y.; Liang, P.; Zhou, C.; Zhao, S.; Huang, X.; Wu, L.; He, X. Indoleamine 2,3-Dioxygenase (IDO) Downregulates the Cell Surface Expression of the CD4 Molecule. Int. J. Mol. Sci 2012, 13, 10863–10879. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer name | Primer sequence |
|---|---|
| GRP78- sense | 5′-CCTGGTACTGCTTGATGTAT-3′ |
| GRP78-antisense | 5′-TTCTGCTGTATCCTCTTCAC-3′ |
| CHOP-sense | 5′-CTTCATACATCACCACACCT-3′ |
| CHOP-antisense | 5′-GTAGTCAGTAGCCACTTCTG-3′ |
| caspase-3-sense | 5′-CTATTGTGAGGCGGTTGT-3′ |
| caspase-3-antisense | 5′-TCCAGAGTCCATTGATTCG-3′ |
| caspase-4-sense | 5′-TGAACTGGAAGGAAGAGGAA-3′ |
| caspase-4-antisense | 5′-GCGGTTGTTGAATATCTGGA-3′ |
| 18s rRNA-sense | 5′-TGGTGGAGCGATTTGTCTG-3′ |
| 18s rRNA-antisense | 5′-AATGGGGTTCAACGGGTTAC-3′ |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wu, L.-F.; Guo, Y.-T.; Zhang, Q.-H.; Xiang, M.-Q.; Deng, W.; Ye, Y.-Q.; Pu, Z.-J.; Feng, J.-L.; Huang, G.-Y. Enhanced Antitumor Effects of Adenoviral-Mediated siRNA against GRP78 Gene on Adenosine-Induced Apoptosis in Human Hepatoma HepG2 Cells. Int. J. Mol. Sci. 2014, 15, 525-544. https://doi.org/10.3390/ijms15010525
Wu L-F, Guo Y-T, Zhang Q-H, Xiang M-Q, Deng W, Ye Y-Q, Pu Z-J, Feng J-L, Huang G-Y. Enhanced Antitumor Effects of Adenoviral-Mediated siRNA against GRP78 Gene on Adenosine-Induced Apoptosis in Human Hepatoma HepG2 Cells. International Journal of Molecular Sciences. 2014; 15(1):525-544. https://doi.org/10.3390/ijms15010525
Chicago/Turabian StyleWu, Ling-Fei, Yi-Tian Guo, Qing-Hua Zhang, Meng-Qi Xiang, Wei Deng, Yan-Qing Ye, Ze-Jin Pu, Jia-Lin Feng, and Guan-You Huang. 2014. "Enhanced Antitumor Effects of Adenoviral-Mediated siRNA against GRP78 Gene on Adenosine-Induced Apoptosis in Human Hepatoma HepG2 Cells" International Journal of Molecular Sciences 15, no. 1: 525-544. https://doi.org/10.3390/ijms15010525