Genetic Deletion of Rheb1 in the Brain Reduces Food Intake and Causes Hypoglycemia with Altered Peripheral Metabolism

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
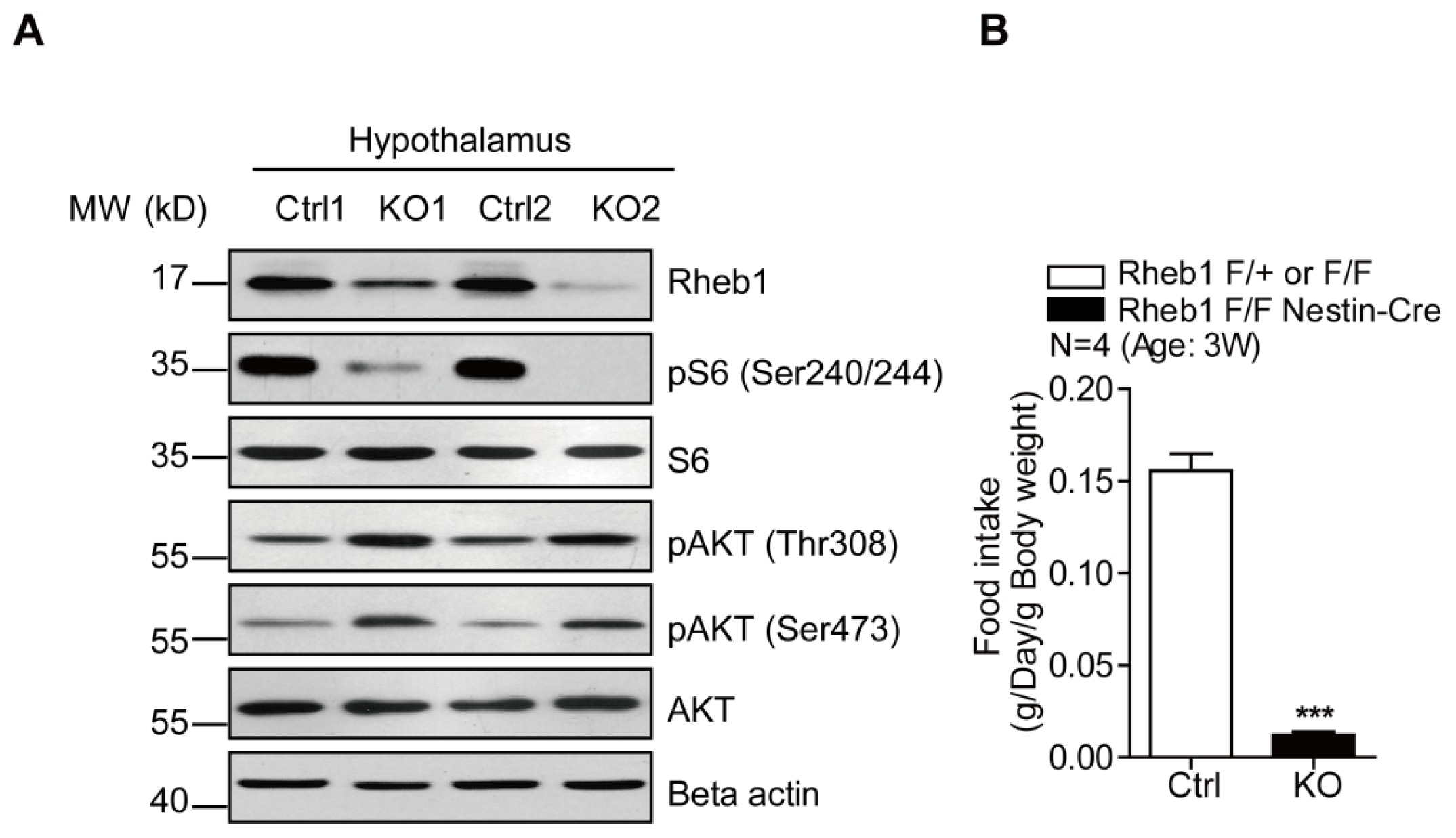
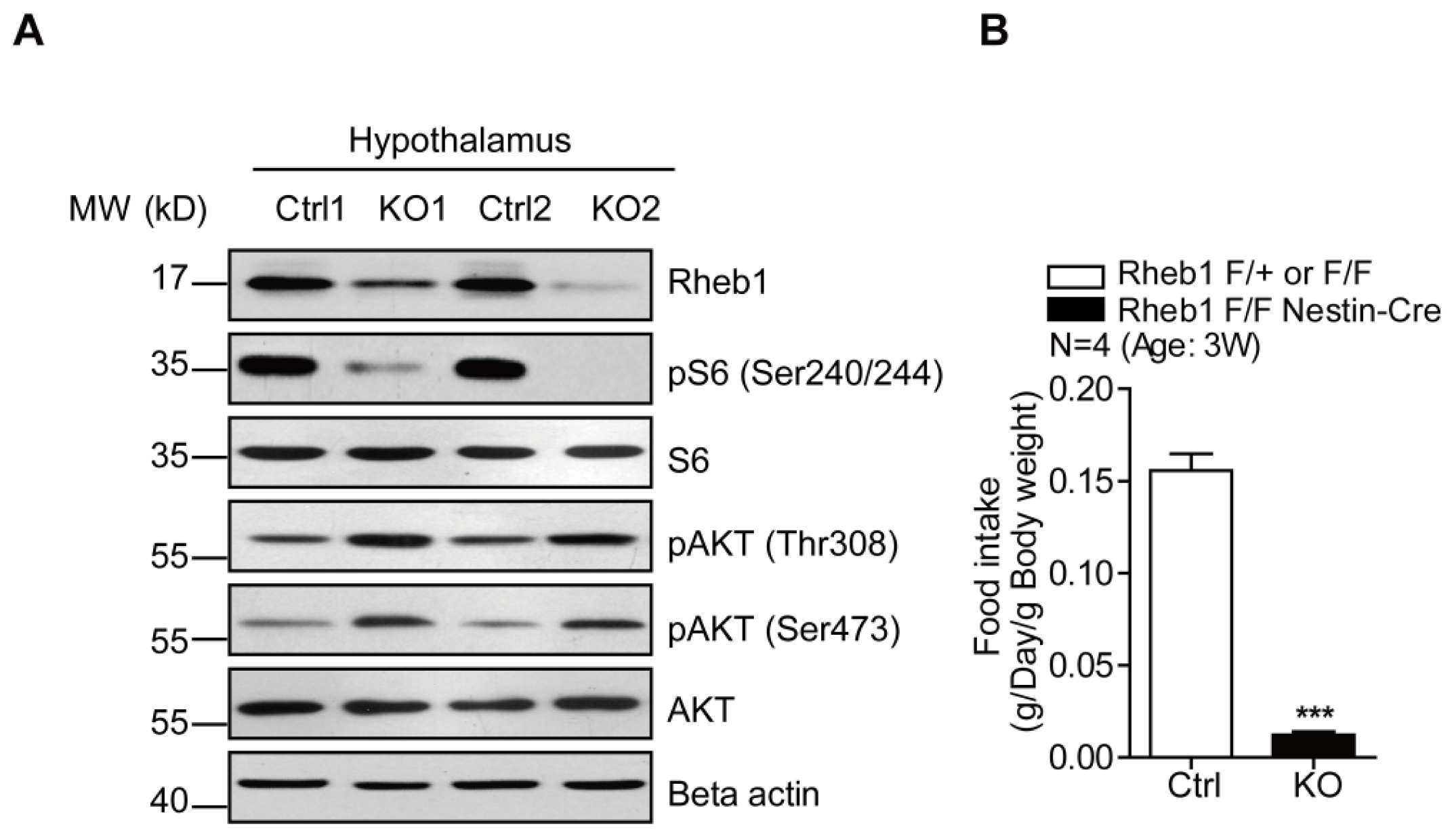
2.1. Genetic Deletion of Rheb1 in the Brain Disrupts Food Intake
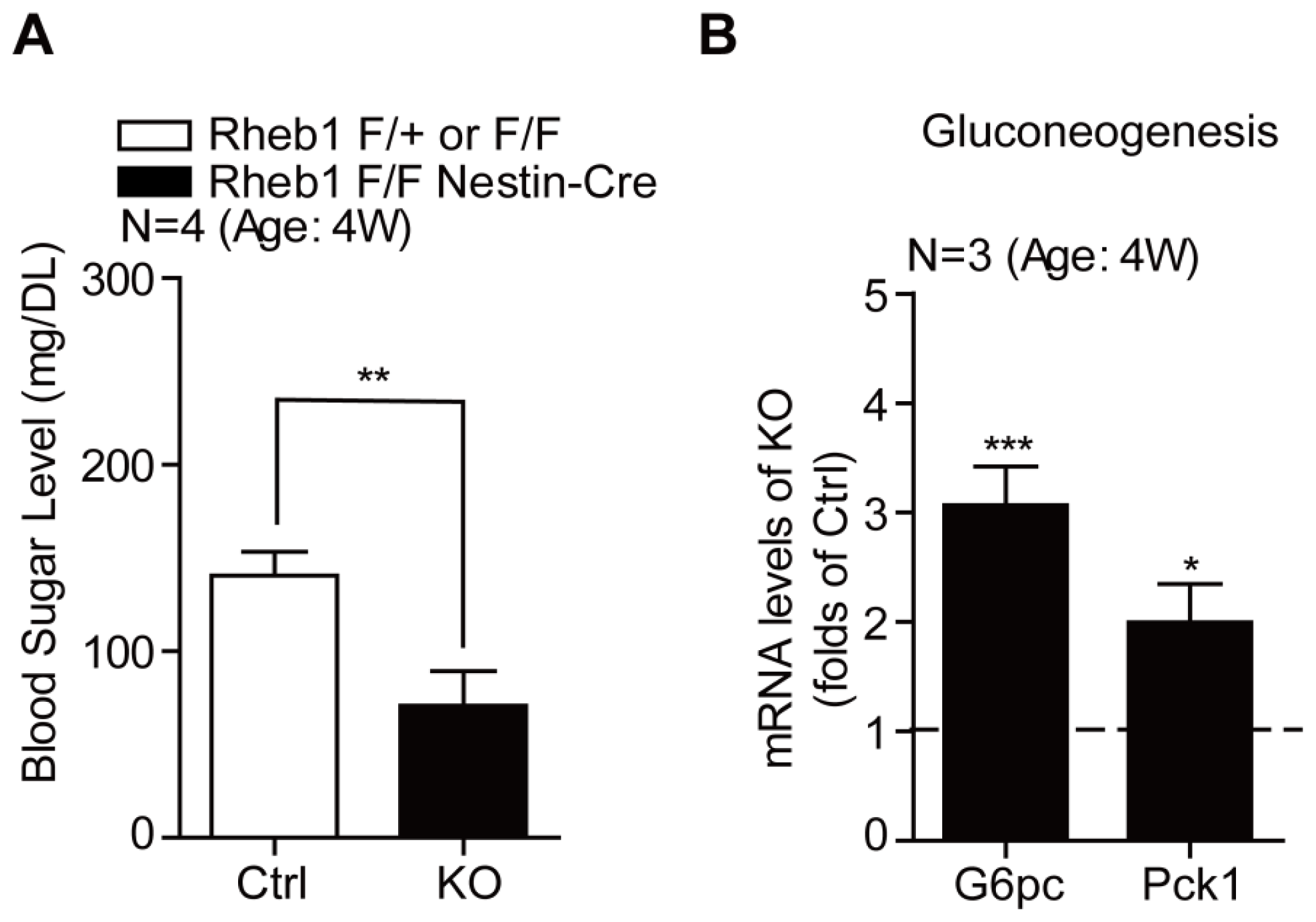
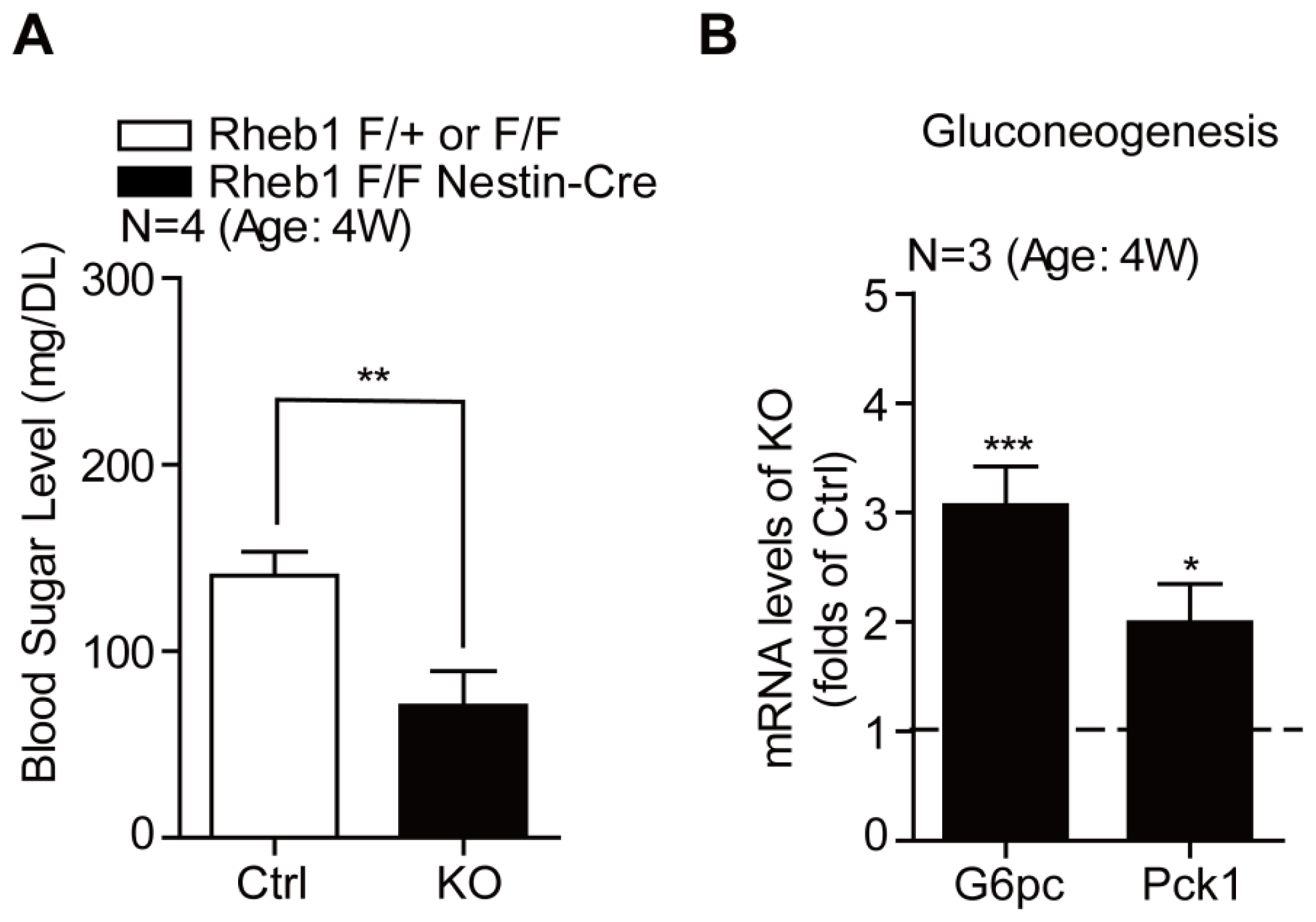
2.2. Genetic Deletion of Rheb1 in the Brain Leads to Hypoglycemia
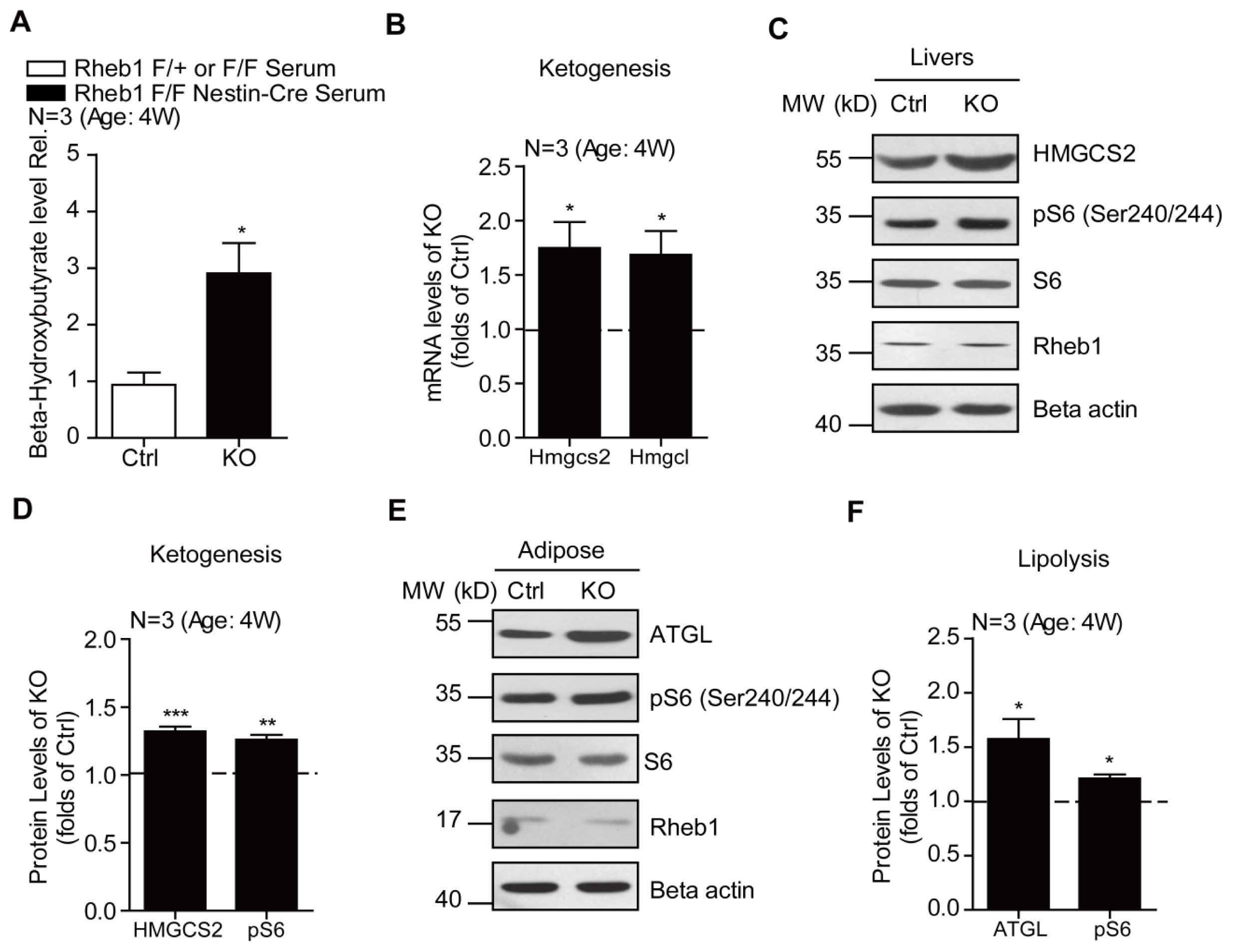
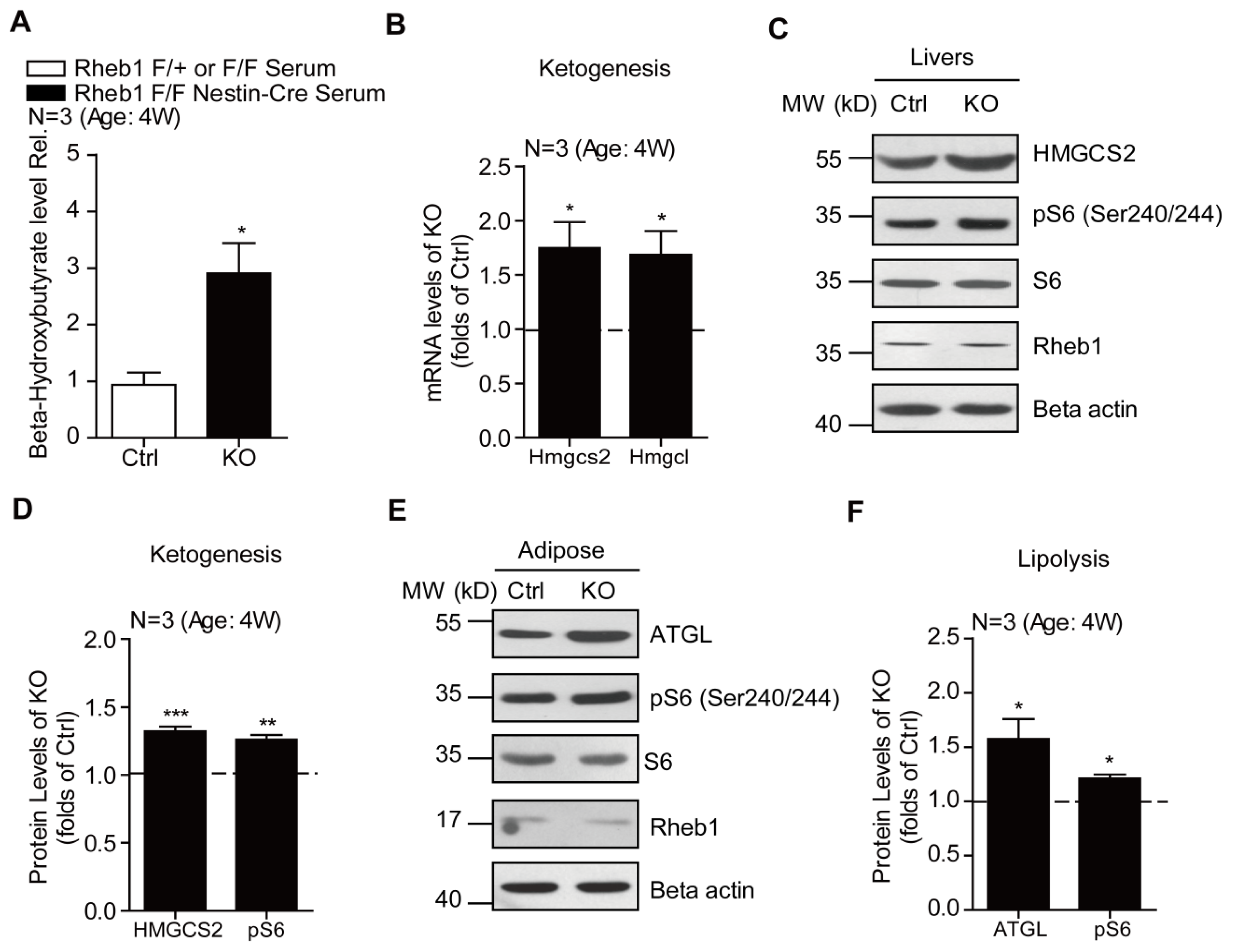
2.3. Genetic Deletion of Rheb1 in the Brain Induces Ketogenesis in the Liver
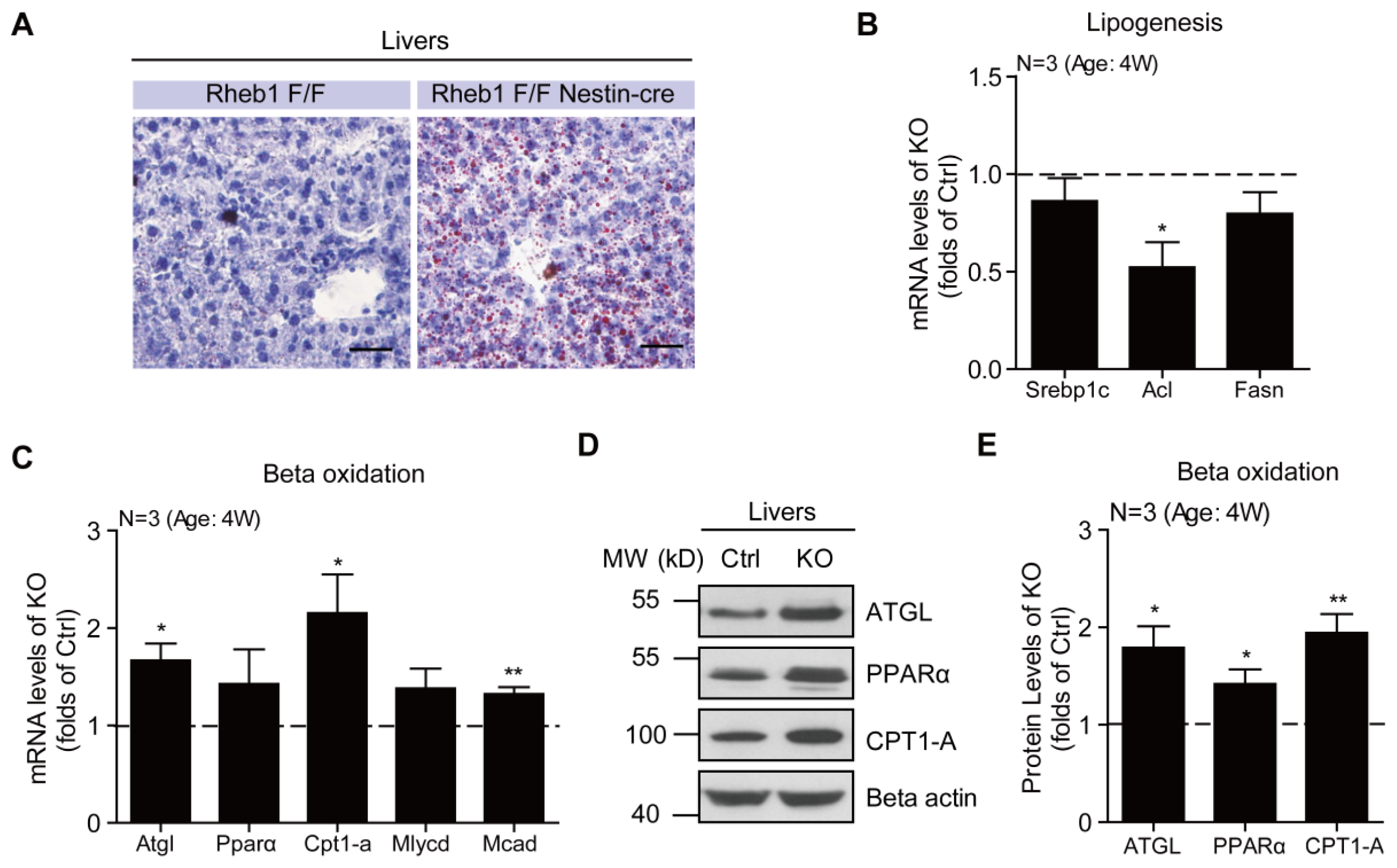
2.4. Genetic Deletion of Rheb1 in the Brain Increases Liver Lipid Droplets and Beta Oxidation
3. Experimental Section
3.1. Reagents and Antibodies
3.2. Animals
3.3. Blood Glucose Measurement
3.4. Western Blotting
3.5. RNA Extraction and Real-Time PCR Assay
- G6pc: 5′-GTGTCCGTGATCGCAGACC-3′ and 5′-GACGAGGTTGAGCCAGTCTC-3′;
- Pck1: 5′-T TGAGAAAGCGTTCAATGCCA-3′ and 5′-CACGTAGGGTGAATCCGTCAG-3′;
- Hmgcs2: 5′-ATA CCACCAACGCCTGTTATGG-3′ and 5′-CAATGTCACCACAGACCACCAG-3′;
- Hmgcl: 5′-ACTA CCCAGTCCTGACTCCAA-3′ and 5′-TAGAGCAGTTCGCGTTCTTCC-3′;
- Srebp1c: 5′-GGAGCC ATGGATTGCACATT-3′ and 5′-GCTTCCAGAGAGGAGGCCAG-3′;
- Acl: 5′-GAAGCTGACCTT GCTGAACC-3′ and 5′-CTGCCTCCAATGATGAGGAT-3′;
- Fasn: 5′-TGGGTTCTAGCCAGCAGA GT-3′ and 5′-ACCACCAGAGACCGTTATGC-3′;
- Atgl: 5′-TGTGGCCTCATTCCTCCTAC-3′ and 5′-TGCTGGATGTTGGTGGAGCT-3′;
- Pparα: 5′-TGTTTGTGGCTGCTATAATTTGC-3′ and 5′-GCAACTTCTCAATGTAGCCTATGTTT-3′;
- Cpt1-a: 5′-GGAGAGAATTTCATCCACTTCCA-3′ and 5′-CTTCCCAAAGCGGTGTGAGT-3′;
- Mlycd: 5′-GCACGTCCGGGAAATG AAC-3′ and 5′-GCCTCACACTCGCTGATCTT-3′;
- Mcad: 5′-TTTCGAAGACGTCAGAGTGC-3′ and 5′-TGCGACTGTAGGTCTGGTTC-3′;
- Beta-actin: 5′-GAGACCTTCAACACCCCAGC-3′ and 5′-ATGTCACGCACGATTTCCC-3′.
3.6. β-Hydroxybutyrate Assay
3.7. Oil Red O Staining
3.8. Food Intake Assay
3.9. Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| mTOR | mammalian target of rapamycin |
| mTORC1 | mammalian target of rapamycin complex 1 |
| mTORC2 | mammalian target of rapamycin complex 2 |
| Rheb1 | Ras homolog enriched in brain 1 |
| ARC | arcuate |
| POMC | proopiomelanocortin |
| DN | dominant negative |
| CA | constitutively active |
| Rheb1 ko | Rheb1 f/f Nestin-cre |
| HMG-CoA | hydroxy-methyl-glutaryl coenzyme A |
| HMGCS2 | HMG-CoA synthase 2 |
| HMGCL | HMG-CoA lyase |
| ATGL | adipose triglyceride lipase |
References
- Coupe, B.; Grit, I.; Hulin, P.; Randuineau, G.; Parnet, P. Postnatal growth after intrauterine growth restriction alters central leptin signal and energy homeostasis. PLoS One 2012, 7, e30616. [Google Scholar]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol 2012, 13, 251–262. [Google Scholar]
- Pimentel, G.D.; Ropelle, E.R.; Rocha, G.Z.; Carvalheira, J.B. The role of neuronal AMPK as a mediator of nutritional regulation of food intake and energy homeostasis. Metab. Clin. Exp 2013, 62, 171–178. [Google Scholar]
- Cota, D.; Proulx, K.; Smith, K.A.; Kozma, S.C.; Thomas, G.; Woods, S.C.; Seeley, R.J. Hypothalamic mTOR signaling regulates food intake. Science 2006, 312, 927–930. [Google Scholar]
- Wiczer, B.M.; Thomas, G. The role of the mTOR pathway in regulating food intake. Curr. Opin. Drug Discov. Dev 2010, 13, 604–612. [Google Scholar]
- Woods, S.C.; Seeley, R.J.; Cota, D. Regulation of food intake through hypothalamic signaling networks involving mTOR. Ann. Rev. Nutr 2008, 28, 295–311. [Google Scholar]
- Laplante, M.; Sabatini, D.M. An emerging role of mTOR in lipid biosynthesis. Curr. Biol 2009, 19, R1046–R1052. [Google Scholar]
- Yecies, J.L.; Manning, B.D. Transcriptional control of cellular metabolism by mTOR signaling. Cancer Res 2011, 71, 2815–2820. [Google Scholar]
- Braun, S.; Bitton-Worms, K.; LeRoith, D. The link between the metabolic syndrome and cancer. Int. J. Biol. Sci 2011, 7, 1003–1015. [Google Scholar]
- Jacinto, E.; Facchinetti, V.; Liu, D.; Soto, N.; Wei, S.; Jung, S.Y.; Huang, Q.; Qin, J.; Su, B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 2006, 127, 125–137. [Google Scholar]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar]
- Nobukuni, T.; Joaquin, M.; Roccio, M.; Dann, S.G.; Kim, S.Y.; Gulati, P.; Byfield, M.P.; Backer, J.M.; Natt, F.; Bos, J.L.; et al. Amino acids mediate mTOR/raptor signaling through activation of class 3 phosphatidylinositol 3OH-kinase. Proc. Natl. Acad. Sci. USA 2005, 102, 14238–14243. [Google Scholar]
- Garami, A.; Zwartkruis, F.J.; Nobukuni, T.; Joaquin, M.; Roccio, M.; Stocker, H.; Kozma, S.C.; Hafen, E.; Bos, J.L.; Thomas, G. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol.Cell 2003, 11, 1457–1466. [Google Scholar]
- Blouet, C.; Ono, H.; Schwartz, G.J. Mediobasal hypothalamic p70 S6 kinase 1 modulates the control of energy homeostasis. Cell Metab 2008, 8, 459–467. [Google Scholar]
- Yang, S.B.; Tien, A.C.; Boddupalli, G.; Xu, A.W.; Jan, Y.N.; Jan, L.Y. Rapamycin ameliorates age-dependent obesity associated with increased mTOR signaling in hypothalamic POMC neurons. Neuron 2012, 75, 425–436. [Google Scholar]
- Zou, J.; Zhou, L.; Du, X.X.; Ji, Y.; Xu, J.; Tian, J.; Jiang, W.; Zou, Y.; Yu, S.; Gan, L.; et al. Rheb1 is required for mTORC1 and myelination in postnatal brain development. Dev. Cell 2011, 20, 97–108. [Google Scholar]
- Tronche, F.; Kellendonk, C.; Kretz, O.; Gass, P.; Anlag, K.; Orban, P.C.; Bock, R.; Klein, R.; Schutz, G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat. Genet 1999, 23, 99–103. [Google Scholar]
- Ozcan, L.; Wong, C.C.; Li, G.; Xu, T.; Pajvani, U.; Park, S.K.; Wronska, A.; Chen, B.X.; Marks, A.R.; Fukamizu, A.; et al. Calcium signaling through CaMKII regulates hepatic glucose production in fasting and obesity. Cell Metab 2012, 15, 739–751. [Google Scholar]
- Kostiuk, M.A.; Keller, B.O.; Berthiaume, L.G. Palmitoylation of ketogenic enzyme HMGCS2 enhances its interaction with PPARalpha and transcription at the Hmgcs2 PPRE. FASEB J 2010, 24, 1914–1924. [Google Scholar]
- Pie, J.; Lopez-Vinas, E.; Puisac, B.; Menao, S.; Pie, A.; Casale, C.; Ramos, F.J.; Hegardt, F.G.; Gomez-Puertas, P.; Casals, N. Molecular genetics of HMG-CoA lyase deficiency. Mol. Genet. Metab 2007, 92, 198–209. [Google Scholar]
- Kolditz, C.I.; Langin, D. Adipose tissue lipolysis. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 377–381. [Google Scholar]
- Foretz, M.; Ancellin, N.; Andreelli, F.; Saintillan, Y.; Grondin, P.; Kahn, A.; Thorens, B.; Vaulont, S.; Viollet, B. Short-term overexpression of a constitutively active form of AMP-activated protein kinase in the liver leads to mild hypoglycemia and fatty liver. Diabetes 2005, 54, 1331–1339. [Google Scholar]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.L.; Schulze, A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab 2008, 8, 224–236. [Google Scholar]
- Bobrovnikova-Marjon, E.; Hatzivassiliou, G.; Grigoriadou, C.; Romero, M.; Cavener, D.R.; Thompson, C.B.; Diehl, J.A. PERK-dependent regulation of lipogenesis during mouse mammary gland development and adipocyte differentiation. Proc. Natl. Acad. Sci. USA 2008, 105, 16314–16319. [Google Scholar]
- Lopes-Cardozo, M.; van den Bergh, S.G. Ketogenesis in isolated rat liver mitochondria. III. Relationship with the rate of beta-oxidation. Biochim. Biophys. Acta 1974, 357, 53–62. [Google Scholar]
- Serviddio, G.; Giudetti, A.M.; Bellanti, F.; Priore, P.; Rollo, T.; Tamborra, R.; Siculella, L.; Vendemiale, G.; Altomare, E.; Gnoni, G.V. Oxidation of hepatic carnitine palmitoyl transferase-I (CPT-I) impairs fatty acid beta-oxidation in rats fed a methionine-choline deficient diet. PLoS One 2011, 6, e24084. [Google Scholar]
- Chakrabarti, P.; Kandror, K.V. Adipose triglyceride lipase: A new target in the regulation of lipolysis by insulin. Curr. Diabetes Rev 2011, 7, 270–277. [Google Scholar]
- El Kebbaj, Z.; Andreoletti, P.; Mountassif, D.; Kabine, M.; Schohn, H.; Dauca, M.; Latruffe, N.; El Kebbaj, M.S.; Cherkaoui-Malki, M. Differential regulation of peroxisome proliferator-activated receptor (PPAR)-alpha1 and truncated PPARalpha2 as an adaptive response to fasting in the control of hepatic peroxisomal fatty acid beta-oxidation in the hibernating mammal. Endocrinology 2009, 150, 1192–1201. [Google Scholar]
- Gaidhu, M.P.; Fediuc, S.; Ceddia, R.B. 5-Aminoimidazole-4-carboxamide-1-β-d-ribofuranosideinduced AMP-activated protein kinase phosphorylation inhibits basal and insulin-stimulated glucose uptake, lipid synthesis, and fatty acid oxidation in isolated rat adipocytes. J. Biol. Chem 2006, 281, 25956–25964. [Google Scholar]
- Derdak, Z.; Villegas, K.A.; Harb, R.; Wu, A.M.; Sousa, A.; Wands, J.R. Inhibition of p53 attenuates steatosis and liver injury in a mouse model of non-alcoholic fatty liver disease. J. Hepatol 2012, 58, 785–791. [Google Scholar]
- Hanley, P.J.; Gopalan, K.V.; Lareau, R.A.; Srivastava, D.K.; von Meltzer, M.; Daut, J. β-Oxidation of 5-hydroxydecanoate, a putative blocker of mitochondrial ATP-sensitive potassium channels. J. Physiol 2003, 547, 387–393. [Google Scholar]
- Niki, T.; Pekny, M.; Hellemans, K.; Bleser, P.D.; Berg, K.V.; Vaeyens, F.; Quartier, E.; Schuit, F.; Geerts, A. Class VI intermediate filament protein nestin is induced during activation of rat hepatic stellate cells. Hepatology 1999, 29, 520–527. [Google Scholar]
- Fukamauchi, F.; Aihara, O.; Kusakabe, M. Reduced mRNA expression of neuropeptide Y in the limbic system of tenascin gene disrupted mouse brain. Neuropeptides 1998, 32, 265–268. [Google Scholar]
- Tacconi, M.T.; Lligona, L.; Salmona, M.; Pitsikas, N.; Algeri, S. Aging and food restriction: Effect on lipids of cerebral cortex. Neurobiol. Aging 1991, 12, 55–59. [Google Scholar]
- Lassman, D.J.; McKie, S.; Gregory, L.J.; Lal, S.; D’Amato, M.; Steele, I.; Varro, A.; Dockray, G.J.; Williams, S.C.; Thompson, D.G. Defining the role of cholecystokinin in the lipid-induced human brain activation matrix. Gastroenterology 2010, 138, 1514–1524. [Google Scholar]

© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yang, W.; Jiang, W.; Luo, L.; Bu, J.; Pang, D.; Wei, J.; Du, C.; Xia, X.; Cui, Y.; Liu, S.; et al. Genetic Deletion of Rheb1 in the Brain Reduces Food Intake and Causes Hypoglycemia with Altered Peripheral Metabolism. Int. J. Mol. Sci. 2014, 15, 1499-1510. https://doi.org/10.3390/ijms15011499
Yang W, Jiang W, Luo L, Bu J, Pang D, Wei J, Du C, Xia X, Cui Y, Liu S, et al. Genetic Deletion of Rheb1 in the Brain Reduces Food Intake and Causes Hypoglycemia with Altered Peripheral Metabolism. International Journal of Molecular Sciences. 2014; 15(1):1499-1510. https://doi.org/10.3390/ijms15011499
Chicago/Turabian StyleYang, Wanchun, Wanxiang Jiang, Liping Luo, Jicheng Bu, Dejiang Pang, Jing Wei, Chongyangzi Du, Xiaoqiang Xia, Yiyuan Cui, Shuang Liu, and et al. 2014. "Genetic Deletion of Rheb1 in the Brain Reduces Food Intake and Causes Hypoglycemia with Altered Peripheral Metabolism" International Journal of Molecular Sciences 15, no. 1: 1499-1510. https://doi.org/10.3390/ijms15011499