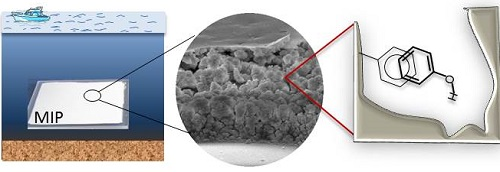
Development of Molecularly Imprinted Polymer in Porous Film Format for Binding of Phenol and Alkylphenols from Water

Abstract
:
1. Introduction
2. Results and Discussion
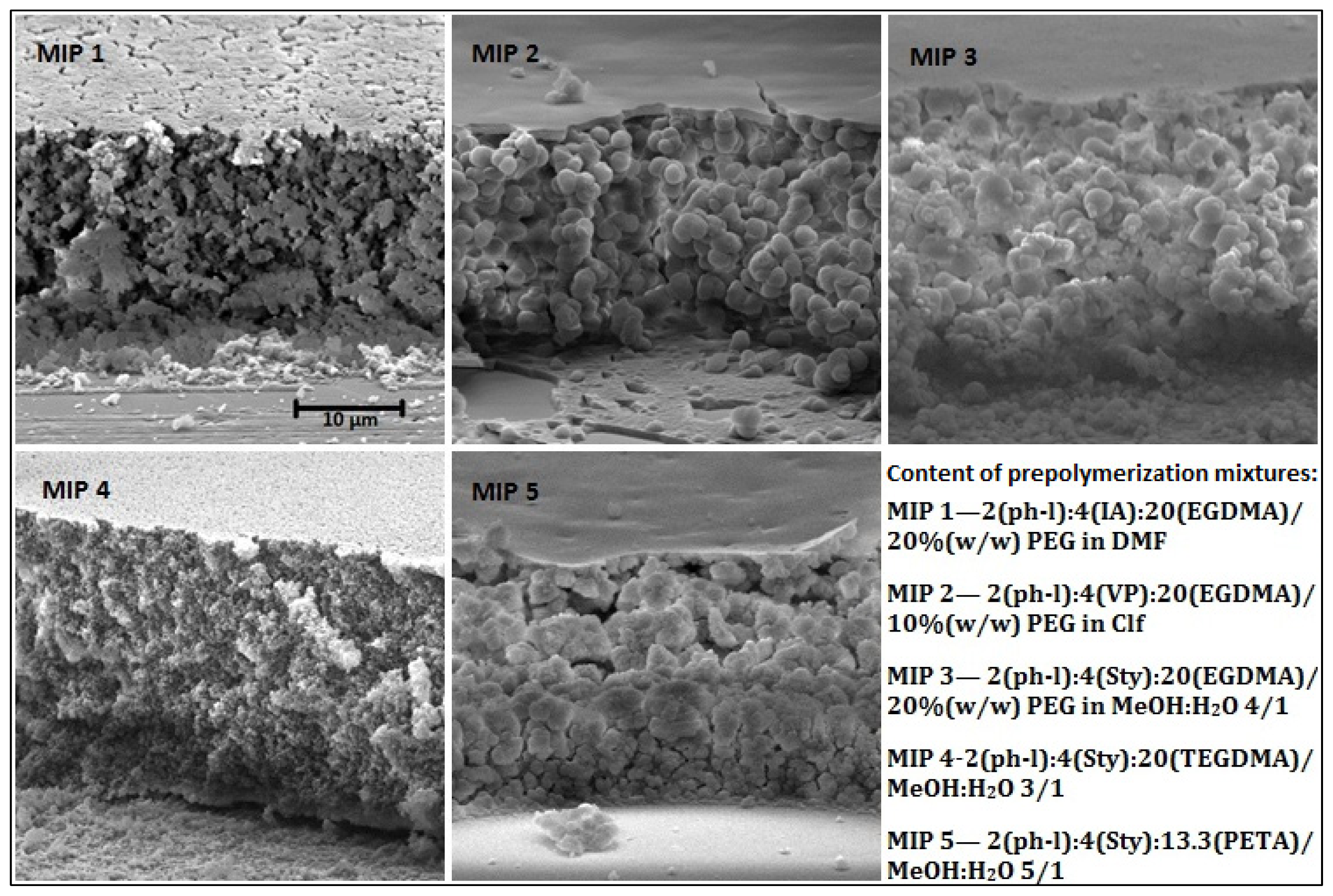
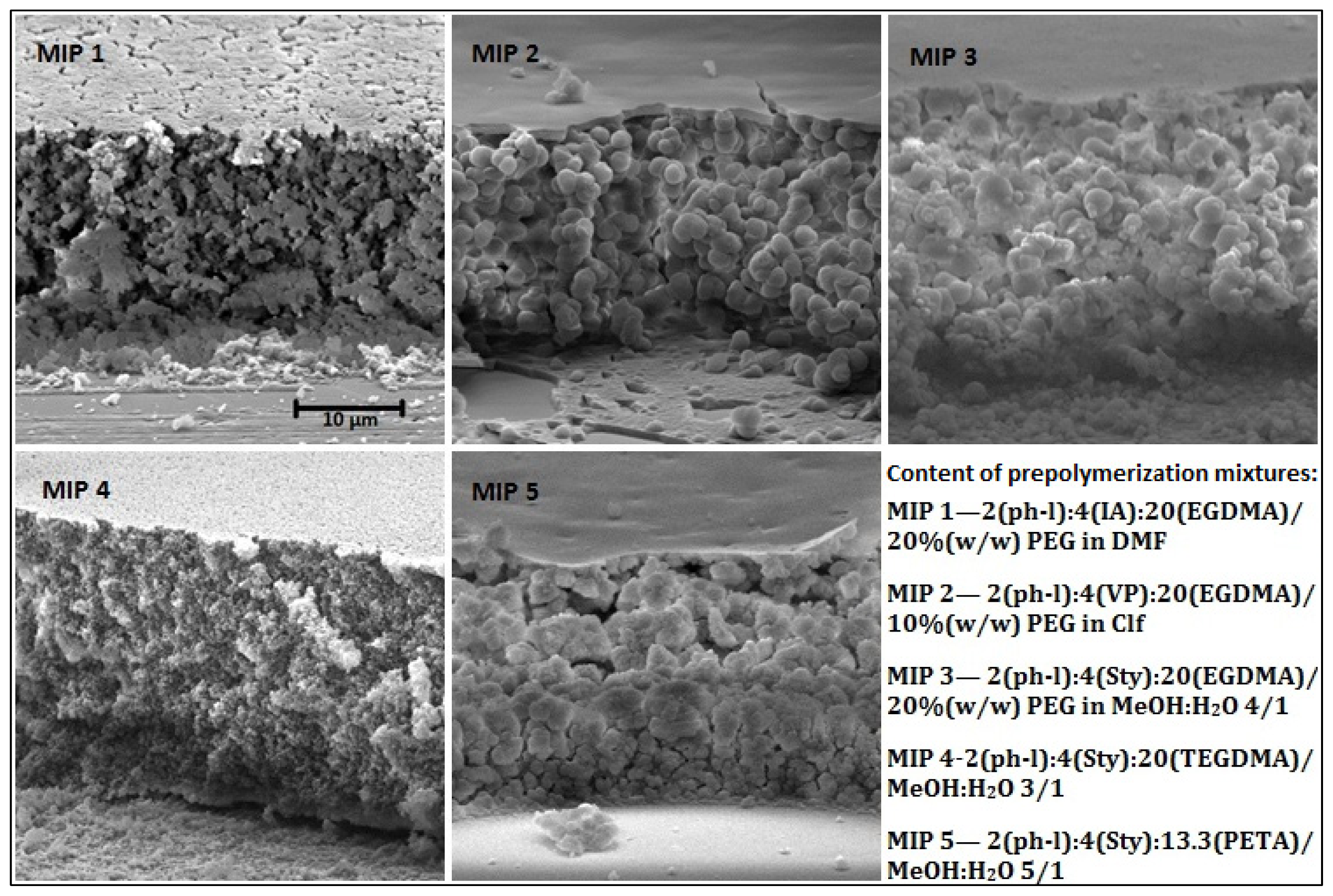
2.1. MIP Films Prepared by “Sandwich” Technique
2.2. Choice of Monomer and Solvent
2.2.1. Phenol Binding Studies in Water for MIPs Prepared on Selected Monomers and Solvents
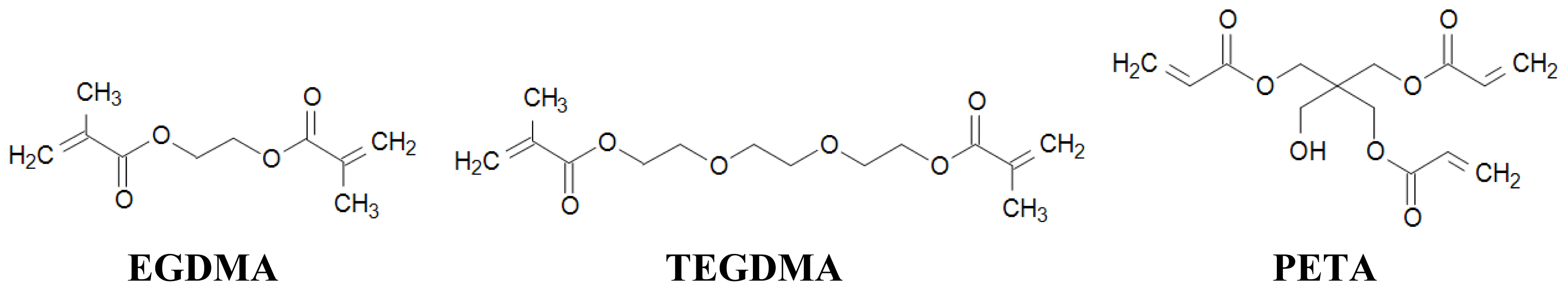
2.3. Choice of Cross-Linker
2.4. Characterization of Styrene/PETA MIP (MIP 5)
2.4.1. Binding Properties Study
2.4.2. Cross-Reactivity Study
3. Experimental Section
3.1. Materials
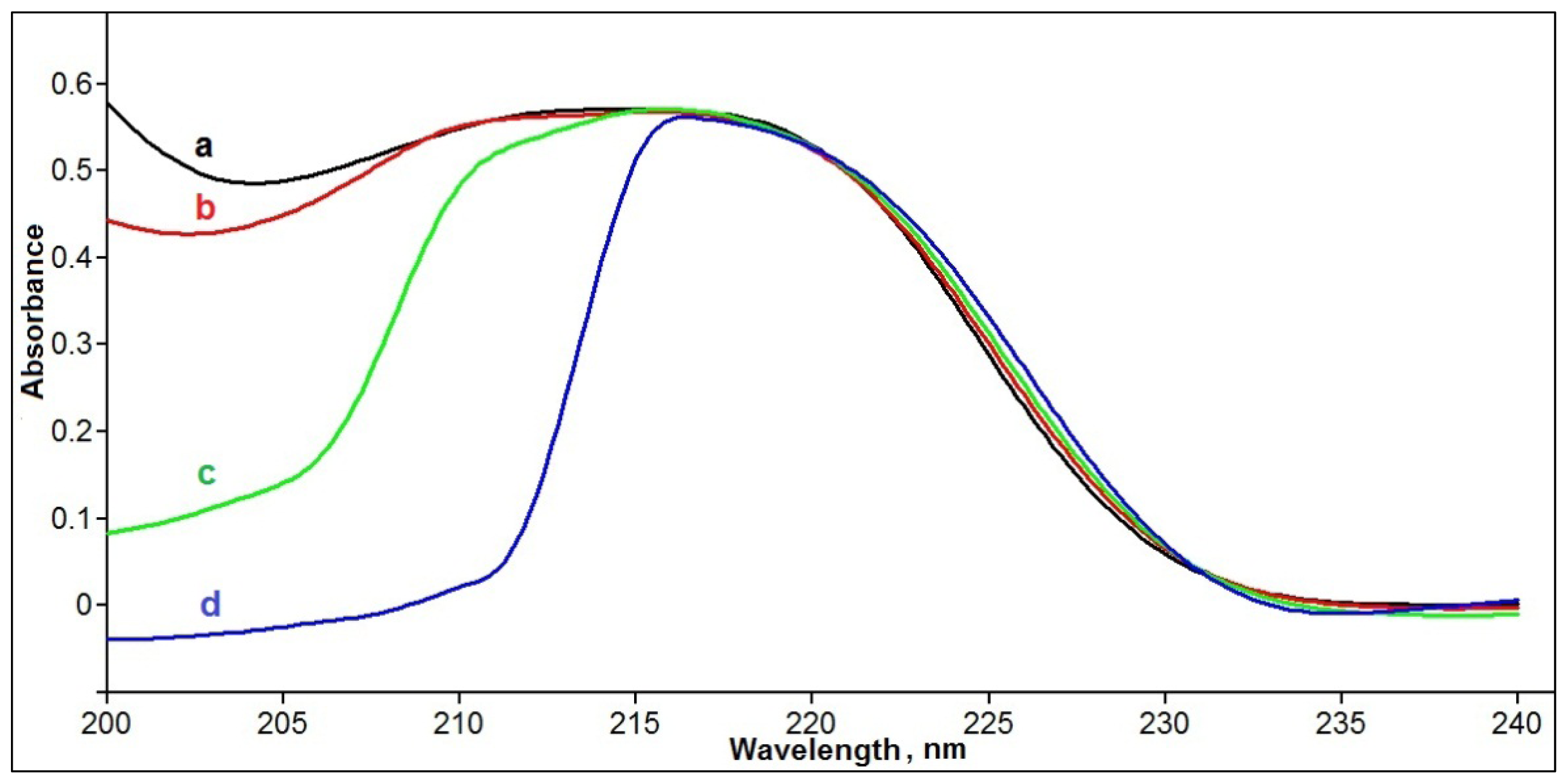
3.2. Study of Phenol-Styrene Interactions by UV Absorbance Spectrometry
3.3. Fabrication of MIP Films by “Sandwich” Technique
3.4. Gravimetrical Analysis of Porosity
3.5. SEM Imaging and Thickness Measurements
3.6. Adsorbate Binding Studies
4. Conclusions
5. Future Prospects
Supporting Information

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ci(phenol), mg·L−1 | 15 | 40 | 100 | 150 | 200 | 250 | 300 |
|---|---|---|---|---|---|---|---|
| MIP 3 (EGDMA) | |||||||
| Q(MIP), mg·g−1 | 2.60 | 6.30 | 13.0 | 17.0 | 21.7 | 23.7 | 26.2 |
| Q(NIP), mg·g−1 | 2.60 | 6.51 | 13.7 | 17.3 | 22.4 | 24.6 | 27.2 |
| IF | 1.00 | 1.03 | 1.05 | 1.02 | 1.03 | 1.04 | 1.04 |
| MIP 4 (TEGDMA) | |||||||
| Q(MIP), mg·g−1 | 1.79 | 4.91 | 10.1 | 14.9 | 18.4 | 22.3 | 27.1 |
| Q(NIP), mg·g−1 | 1.72 | 4.76 | 9.7 | 14.2 | 17.4 | 21.2 | 25.6 |
| IF | 1.04 | 1.03 | 1.04 | 1.05 | 1.06 | 1.05 | 1.06 |
| MIP 5 (PETA) | |||||||
| Q(MIP), mg·g−1 | 2.52 | 5.41 | 11.5 | 15.4 | 18.2 | 21.9 | 24.6 |
| Q(NIP), mg·g−1 | 2.31 | 5.20 | 10.7 | 14.2 | 16.9 | 19.8 | 22.1 |
| IF | 1.09 | 1.04 | 1.07 | 1.09 | 1.08 | 1.11 | 1.12 |
| Ci(phenol), mg·L−1 | 0.1 | 0.5 | 1 | 5 | 15 | 25 | 40 |
|---|---|---|---|---|---|---|---|
| Q(NIP), mg·g−1 | 0.0168 (0.0043) | 0.0865 (0.0008) | 0.192 (0.009) | 0.882 (0.007) | 2.31 (0.011) | 3.45 (0.13) | 5.20 (0.28) |
| Q(MIP), mg·g−1 | 0.0202 (0.0051) | 0.1004 (0.0011) | 0.212 (0.0001) | 0.957 (0.025) | 2.52 (0.020) | 3.70 (0.11) | 5.41 (0.32) |
| IF | 1.20 (0.023) | 1.16 (0.016) | 1.11 (0.0044) | 1.09 (0.0200) | 1.09 (0.0037) | 1.07 (0.0060) | 1.04 (0.0080) |
{kind=link}
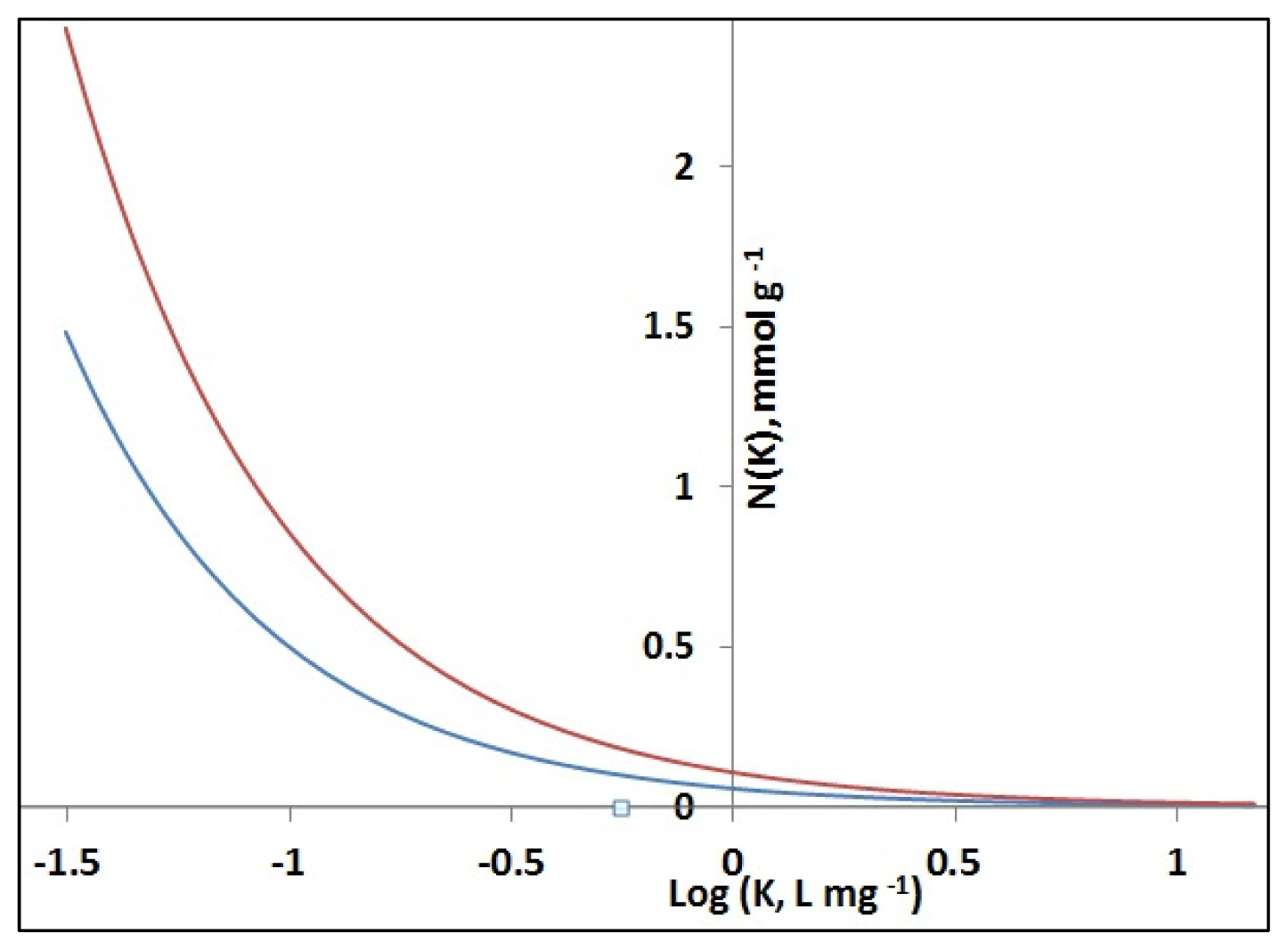
Affinity distributions corresponding to Freundlich Isotherm binding model [31] for MIP 5 on PETA (
 ) and corresponding NIP (
) and corresponding NIP (
 ) calculated based on binding parameters (Formula S3).
) calculated based on binding parameters (Formula S3).
{kind=link}
Acknowledgments
Conflicts of Interest
References
- Michałowicz, J.; Duda, W. Phenols—Sources and toxicity. Pol. J. Environ. Stud 2007, 16, 16. [Google Scholar]
- Toxilogical Profile for Phenol; U.S. Department of Health and Human Services: Atlanta, GA, USA, 2008; p. 27.
- Ambient Water Quality Criteria for Phenol; U.S. Environmental Protection Agency: Washington, DC, USA, 1980; EPA440/5-80-066.
- Shiraishi, Y.; Suzuki, T.; Hirai, T. Selective photooxidation of chlorophenols with molecularly imprinted polymers containing a photosensitizer. N. J. Chem 2010, 34, 714–717. [Google Scholar]
- Caro, E.; Marcé, R.M.; Cormack, P.A.G.; Sherrington, D.C.; Borrull, F. On-line solid-phase extraction with molecularly imprinted polymers to selectively extract substituted 4-chlorophenols and 4-nitrophenol from water. J. Chromatogr. A 2003, 995, 233–238. [Google Scholar]
- Lakshmi, D.; Whitcombe, M.J.; Davis, F.; Chianella, I.; Piletska, E.V.; Guerreiro, A.; Subrahmanyam, S.; Brito, P.S.; Fowler, S.A.; Piletsky, S.A. Chimeric polymers formed from a monomer capable of free radical, oxidative and electrochemical polymerisation. Chem. Commun 2009, 2759–2761. [Google Scholar]
- Mayes, A.G.; Whitcombe, M.J. Synthetic strategies for the generation of molecularly imprinted organic polymers. Adv. Drug Delivery Rev 2005, 57, 1742–1778. [Google Scholar]
- Feng, Q.; Zhao, L.; Lin, J.M. Molecularly imprinted polymer as micro-solid phase extraction combined with high performance liquid chromatography to determine phenolic compounds in environmental water samples. Anal. Chim. Acta 2009, 650, 70–76. [Google Scholar]
- Feng, Q.Z.; Zhao, L.X.; Yan, W.; Ji, F.; Wei, Y.L.; Lin, J.M. Molecularly imprinted solid-phase extraction and flow-injection chemiluminescence for trace analysis of 2,4-dichlorophenol in water samples. Anal. Bioanal. Chem 2008, 391, 1073–1079. [Google Scholar]
- Zakaria, N.D.; Yusof, N.A.; Haron, J.; Abdullah, A.H. Synthesis and evaluation of a molecularly imprinted polymer for 2,4-dinitrophenol. Int. J. Mol. Sci 2009, 10, 354–365. [Google Scholar]
- An, F.Q.; Gao, B.J.; Feng, X.Q. Binding and recognition ability of molecularly imprinted polymer toward p-nitrophenol. J. Appl. Polym. Sci 2012, 125, 2549–2555. [Google Scholar]
- Kan, X.; Zhao, Q.; Zhang, Z.; Wang, Z.; Zhu, J.J. Molecularly imprinted polymers microsphere prepared by precipitation polymerization for hydroquinone recognition. Talanta 2008, 75, 22–26. [Google Scholar]
- Guerreiro, A.; Soares, A.; Piletska, E.; Mattiasson, B.; Piletsky, S. Preliminary evaluation of new polymer matrix for solid-phase extraction of nonylphenol from water samples. Anal. Chim. Acta 2008, 612, 99–104. [Google Scholar]
- Cela-Perez, M.C.; Castro-Lopez, M.M.; Lasagabaster-Latorre, A.; Lopez-Vilarino, J.M.; Gonzalez-Rodriguez, M.V.; Barral-Losada, L.F. Synthesis and characterization of bisphenol-a imprinted polymer as a selective recognition receptor. Anal. Chim. Acta 2011, 706, 275–284. [Google Scholar]
- Lv, Y.-Q.; Lin, Z.; Feng, W.; Tan, T. Evaluation of the polymerization and recognition mechanism for phenol imprinting SPE. Chromatographia 2007, 66, 339–347. [Google Scholar]
- An, F.; Gao, B. Adsorption of phenol on a novel adsorption material PEI/Sio2. J. Hazard. Mater 2008, 152, 1186–1191. [Google Scholar]
- Sergeyeva, T.A.; Gorbach, L.A.; Slinchenko, O.A.; Goncharova, L.A.; Piletska, O.V.; Brovko, O.O.; Sergeeva, L.M.; Elska, G.V. Towards development of colorimetric test-systems for phenols detection based on computationally-designed molecularly imprinted polymer membranes. Mater. Sci. Eng. C 2010, 30, 431–436. [Google Scholar]
- Harz, S.; Schimmelpfennig, M.; Tse, Sum; Bui, B.; Marchyk, N.; Haupt, K.; Feller, K.-H. Fluorescence optical spectrally resolved sensor based on molecularly imprinted polymers and microfluidics. Eng. Life Sci 2011, 11, 559–565. [Google Scholar]
- Van Biesen, G.; Wiseman, J.M.; Li, J.; Bottaro, C.S. Desorption electrospray ionization-mass spectrometry for the detection of analytes extracted by thin-film molecularly imprinted polymers. Analyst 2010, 135, 2237–2240. [Google Scholar]
- Pérez-Moral, N.; Mayes, A. Novel mip formats. Bioseparation 2001, 10, 287–299. [Google Scholar]
- Schmidt, R.H.; Haupt, K. Molecularly imprinted polymer films with binding properties enhanced by the reaction-induced phase separation of a sacrificial polymeric porogen. Chem. Mater 2005, 17, 1007–1016. [Google Scholar]
- Ulbricht, M. Molecularly imprinted polymer films and membranes. In Molecularly Imprinted Materials: Science and Technology; Yan, M., Ramström, O., Eds.; Marcel Dekker: New York, NY, USA, 2005; pp. 455–491. [Google Scholar]
- Jakusch, M.; Janotta, M.; Mizaikoff, B.; Mosbach, K.; Haupt, K. Molecularly imprinted polymers and infrared evanescent wave spectroscopy. A chemical sensors approach. Anal. Chem 1999, 71, 4786–4791. [Google Scholar]
- Yan, H.; Row, K. Characteristic and synthetic approach of molecularly imprinted polymer. Int. J. Mol. Sci 2006, 7, 155–178. [Google Scholar]
- Lulinski, P.; Maciejewska, D. Examination of imprinting process with molsidomine as a template. Molecules 2009, 14, 2212–2225. [Google Scholar]
- Spivak, D.A. Selectivity in molecularly imprinted matrices. In Molecularly Imprinted Materials: Science and Technology; Yan, M., Ramström, O., Eds.; Marcel Dekker: New York, NY, USA, 2005; p. 413. [Google Scholar]
- Manesiotis, P.; Borrelli, C.; Aureliano, C.S.A.; Svensson, C.; Sellergren, B. Water-compatible imprinted polymers for selective depletion of riboflavine from beverages. J. Mater. Chem 2009, 19, 6185–6193. [Google Scholar]
- Turner, N.W.; Holdsworth, C.I.; Donne, S.W.; McCluskey, A.; Bowyer, M.C. Microwave induced mip synthesis: Comparative analysis of thermal and microwave induced polymerisation of caffeine imprinted polymers. N. J. Chem 2010, 34, 686–692. [Google Scholar]
- Corton, E.; García-Calzón, J.A.; Díaz-García, M.E. Kinetics and binding properties of cloramphenicol imprinted polymers. J. Non-Cryst. Solids 2007, 353, 974–980. [Google Scholar]
- Umpleby, R.J.; Baxter, S.C.; Chen, Y.; Shah, R.N.; Shimizu, K.D. Characterization of molecularly imprinted polymers with the langmuir–freundlich isotherm. Anal. Chem 2001, 73, 4584–4591. [Google Scholar]
- Rampey, A.M.; Umpleby, R.J.; Rushton, G.T.; Iseman, J.C.; Shah, R.N.; Shimizu, K.D. Characterization of the imprint effect and the influence of imprinting conditions on affinity, capacity, and heterogeneity in molecularly imprinted polymers using the Freundlich isotherm-affinity distribution analysis. Anal. Chem 2004, 76, 1123–1133. [Google Scholar]
- Haupt, K.; Dzgoev, A.; Mosbach, K. Assay system for the herbicide 2,4-dichlorophenoxyacetic acid using a molecularly imprinted polymer as an artificial recognition element. Anal. Chem 1998, 70, 628–631. [Google Scholar]
- The Handbook of Chemistry and Physics. Available online: http://www.hbcpnetbase.com (accessed on 7 October 2013).

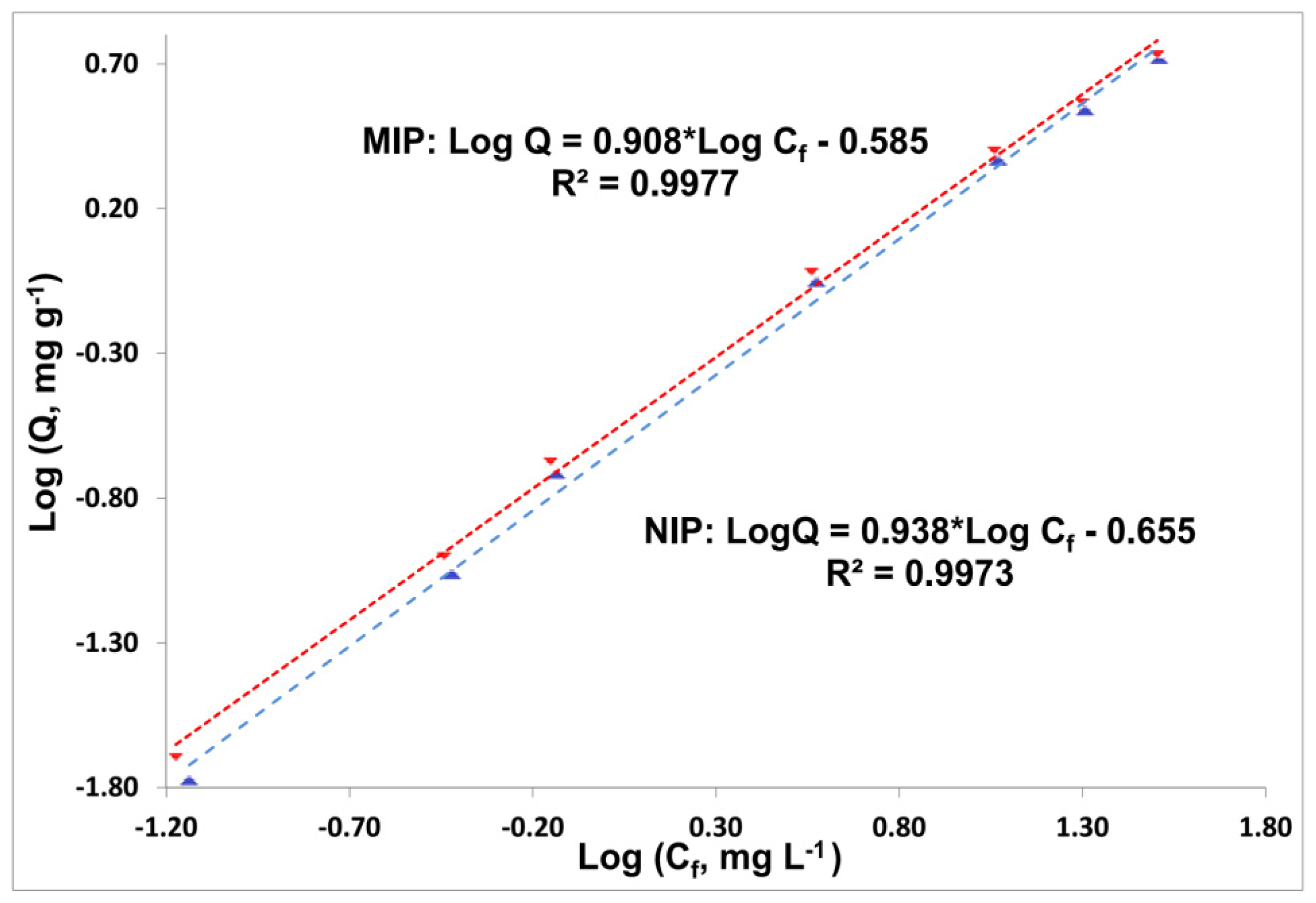
 —MIP,
—MIP,
 —NIP. Note: Cf—phenol concentration at binding equilibrium.
—MIP,
—NIP. Note: Cf—phenol concentration at binding equilibrium.
—NIP. Note: Cf—phenol concentration at binding equilibrium.
—MIP,
—NIP. Note: Cf—phenol concentration at binding equilibrium.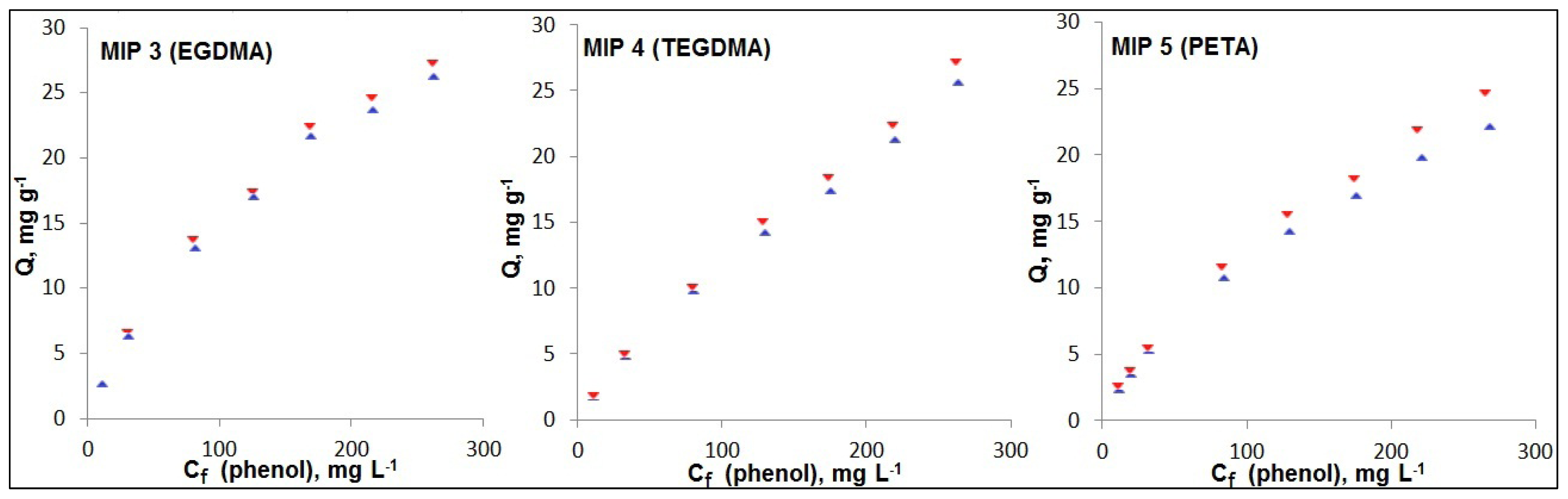 MIP,
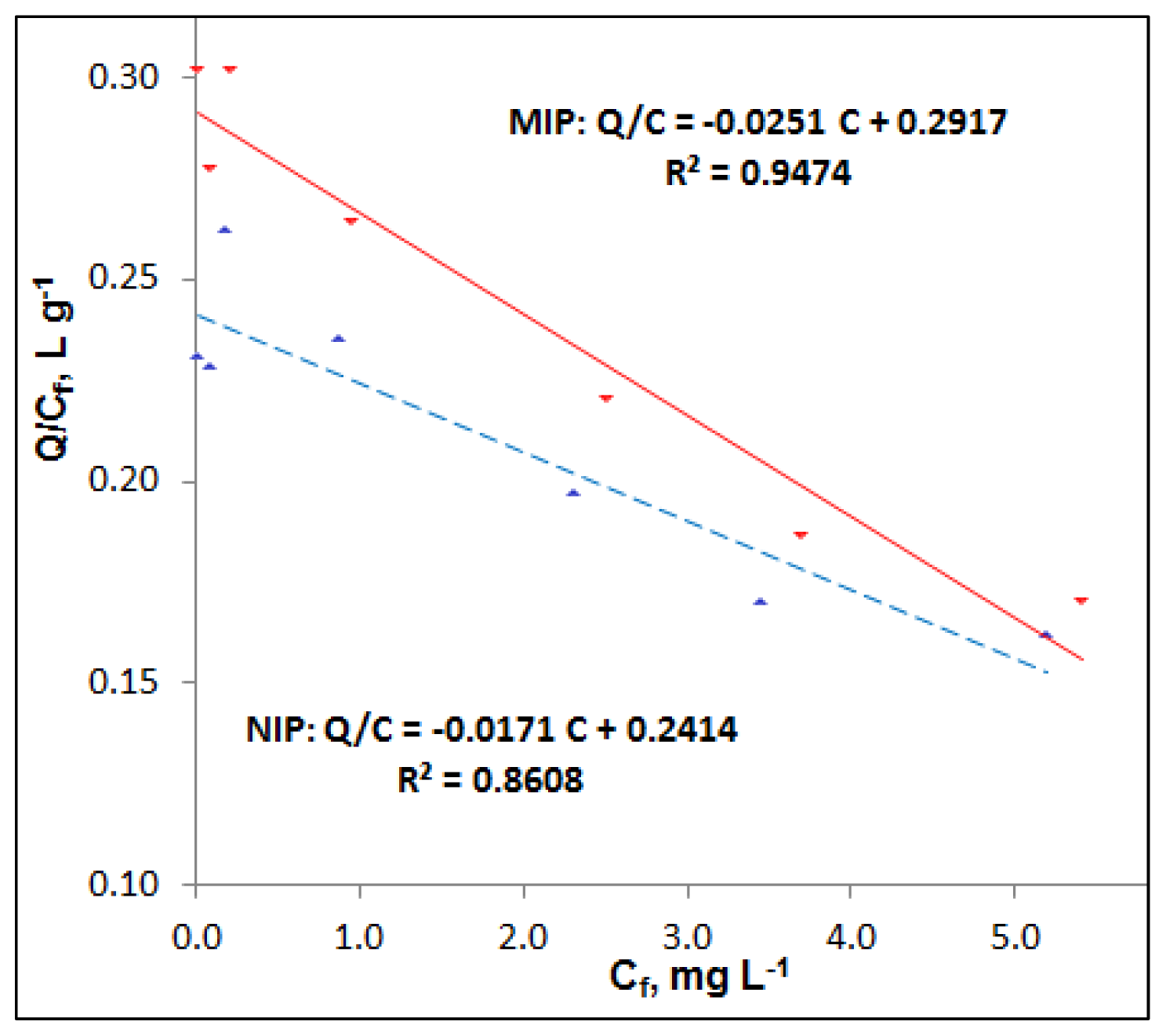
NIP.
MIP,
NIP.
MIP,
NIP.
MIP,
NIP.


| Characteristic determined | MIP | ||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | |
| H (SD, n = 3), μm | 24.1 (2.5) | 23.4 (3.5) | 18.5 (3.1) | 21.5 (5.2) | 20.6 (2.5) |
| ν (SD, n = 4), mL·g−1 | 1.22 (0.04) | 1.08 (0.08) | 1.46 (0.05) | 0.76 (0.07) | 0.91 (0.04) |
| MIP (composition) | Ci (phenol), mg L−1 | |||
|---|---|---|---|---|
| 10 | 15 | 100 | 300 | |
| IF (SD, n = 2) | ||||
| MIP 1 (IA/DMF) | 1.04 (0.006) | 1.04 (0.013) | 1.01 (0.007) | 0.99 (0.030) |
| MIP 2 (VP/CHCl3) | 1.02 (0.013) | 0.96 (0.170) | 0.99 (0.022) | 1.00 (0.015) |
| MIP 3 (Sty/(MeOH:H2O)] | 1.01 (0.020) | 1.00 (0.016) | 1.05 (0.025) | 1.04 (0.010) |
| Adsorbent | R2 | a, mg g−1 (mg L−1)−m | m | NK1–K2, mmol g−1 | KK1–K2, L mg−1 |
|---|---|---|---|---|---|
| MIP 5 | 0.9977 | 0.260 (0.012) | 0.908 (0.020) | 0.0112 (0.0018) | 0.237 (0.011) |
| NIP 5 | 0.9973 | 0.221 (0.012) | 0.938 (0.022) | 0.0073 (0.0022) | 0.221 (0.010) |
| Polymer component | MIP 1 | MIP 2 | MIP 3 | MIP 4 | MIP 5 |
|---|---|---|---|---|---|
| template | phenol 0.4 mmol (37.6 mg) | ||||
| monomer | IA 0.8 mmol (104 mg) | VP 0.8 mmol (85.4 μL) | Sty 0.8 mmol (92.0 μL) | Sty 0.8 mmol (92.0 μL) | Sty 0.8 mmol (92.0 μL) |
| cross-linker | EGDMA 4 mmol 755 μL | EGDMA 4 mmol 755 μL | EGDMA 4 mmol 755 μL | TEGDMA 4 mmol 1049 μL | PETA 2.67 mmol 674 μL |
| photoinitiator | DMPA 0.06 mmol 15.4 mg | ||||
| solvent (1000 μL) | 15% (w/w) PEG in DMF | 10% (w/w) PVA in CHCl3 | 20% (w/w) PEG in MeOH:H2O 4:1 | MeOH:H2O 3:1 | MeOH:H2O 5:1 |
| Analyzed species | Mobile phase (v/v) | Detection wavelength, nm | |
|---|---|---|---|
| CH3CN | H2O with 5% CH3CN (v/v) | ||
| phenol | 55 | 45 | 195 *; 216 *; 272 |
| 4-methylphenol | 55 | 45 | 279 |
| resorcinol | 35 | 65 | 276 |
| 2,4-dimethylphenol | 65 | 35 | 280 |
| 4-propylphenol | 65 | 35 | 278 |
| 3-octanone | 85 | 15 | 279 |
© 2014 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gryshchenko, A.O.; Bottaro, C.S. Development of Molecularly Imprinted Polymer in Porous Film Format for Binding of Phenol and Alkylphenols from Water. Int. J. Mol. Sci. 2014, 15, 1338-1357. https://doi.org/10.3390/ijms15011338
Gryshchenko AO, Bottaro CS. Development of Molecularly Imprinted Polymer in Porous Film Format for Binding of Phenol and Alkylphenols from Water. International Journal of Molecular Sciences. 2014; 15(1):1338-1357. https://doi.org/10.3390/ijms15011338
Chicago/Turabian StyleGryshchenko, Andriy O., and Christina S. Bottaro. 2014. "Development of Molecularly Imprinted Polymer in Porous Film Format for Binding of Phenol and Alkylphenols from Water" International Journal of Molecular Sciences 15, no. 1: 1338-1357. https://doi.org/10.3390/ijms15011338