1. Introduction
During vascular development and pathological angiogenesis, the maintenance of blood vessel homeostasis and its functional execution depend on the integrity of vascular endothelium [
1], which is affected by proliferation, migration and apoptosis of endothelial cells. Recovery of injured endothelial cells through regulated endothelial cell proliferation plays significant roles in thrombosis disease, such as late stent thrombosis after drug eluting stent placement in percutaneous coronary intervention [
2]. Therefore, intensive efforts have been directed to identify more active factors that promote endothelial growth, which have not yielded encouraging results, so far.
Cellular repressor of E1A-stimulated genes (CREG), originally reported as an antagonist of transcriptional activation and cellular transformation by E1A in 1998 [
3], was recently identified as a secreted glycoprotein [
4,
5] and a lysosomal protein [
6]. It is widely expressed in adult tissues, but much less so in undifferentiated cells [
4]. CREG has been proven to enhance differentiation [
4] and inhibit growth of the human teratocarcinoma cell line NTERA-2 through its interaction with the insulin-link growth factor II receptor (IGFIIR) [
7]. It can inhibit cardiac cell growth and attenuate cardiac hypertrophy and fibrosis by downregulating the expression of ERK1/2 [
8]. Our previous study demonstrated that CREG was expressed abundantly in the adult vascular endothelium and reduced dramatically in endothelial cells in atherosclerotic lesions [
9]. At the cellular level, forced CREG overexpression led to endothelial cell activation associated with increased cell motility and permeability of endothelial monolayers
in vitro [
10]. Further studies showed that CREG can induce endothelial cell migration by activating the ILK/Akt/mTOR/vascular endothelial growth factor (VEGF) 165 signaling pathway [
10] and attenuate atherosclerotic endothelium apoptosis via the VEGF/PI3K/Akt pathway [
9]. These series of observations suggest that CREG may play an important role in adult neovascularization and endothelial homeostasis. However, until now, there has been no direct evidence of a CREG effect on the proliferation of endothelial cells. In this study, we investigated the effect of CREG on vascular endothelial cell cycle dynamics and the possible molecular mediators. Our study identifies CREG as a novel mitogen that can promote cell cycle progression and subsequent proliferation of human umbilical vein endothelial cells (HUVEC).
3. Discussion
In the present study, we identified that CREG can serve as a potent proliferative factor for HUVEC and may, therefore, be represented as a novel modulator of vascular barrier functions and the pathogenesis of vascular lesions. Since proliferation of endothelial cells is a key protective mechanism to sustain endothelium homeostasis [
15], recognition of CREG as a potent factor that promotes the proliferation of endothelial cells will shed new light on the identification of therapeutic targets for both the prevention and treatment of various vascular diseases, especially atherosclerosis and ischemic cardiovascular diseases [
16,
17].
CREG is a recently discovered secreted glycoprotein expressed in mature tissues and cells and involved in homeostatic modulation [
18]. Our previous studies confirmed that overexpression of CREG inhibited the pathological apoptosis of human vascular smooth muscle cells (SMCs) [
19] and rat mesenchymal stem cells [
20], supporting its potential for antagonizing apoptosis and sustaining cellular homeostasis. Another recent study demonstrated that CREG was expressed abundantly in the adult vascular endothelium and was reduced dramatically in endothelial cells in atherosclerotic lesions [
9], which indicated that CREG might play a role in the regulation of endothelium homeostasis. Our series of studies have identified CREG as an active participant in the regulation of apoptosis, inflammation and wound healing of vascular endothelial cells [
9,
15]. Although a growing body of evidence revealed CREG as an inhibitory factor to the proliferation of undifferentiated tissues and cells [
3,
7], a recent study found that CREG was highly expressed in gastric cancer (GC) tissues, and downregulation of CREG expression inhibited the growth of GC cells [
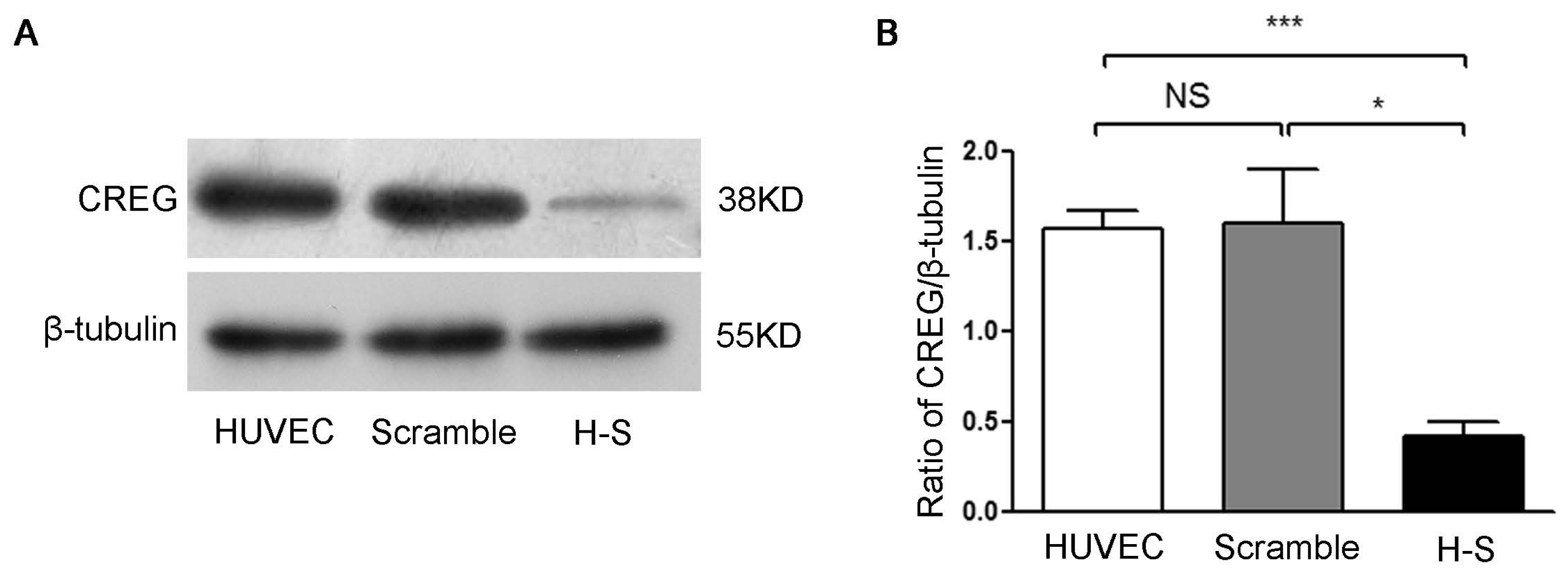
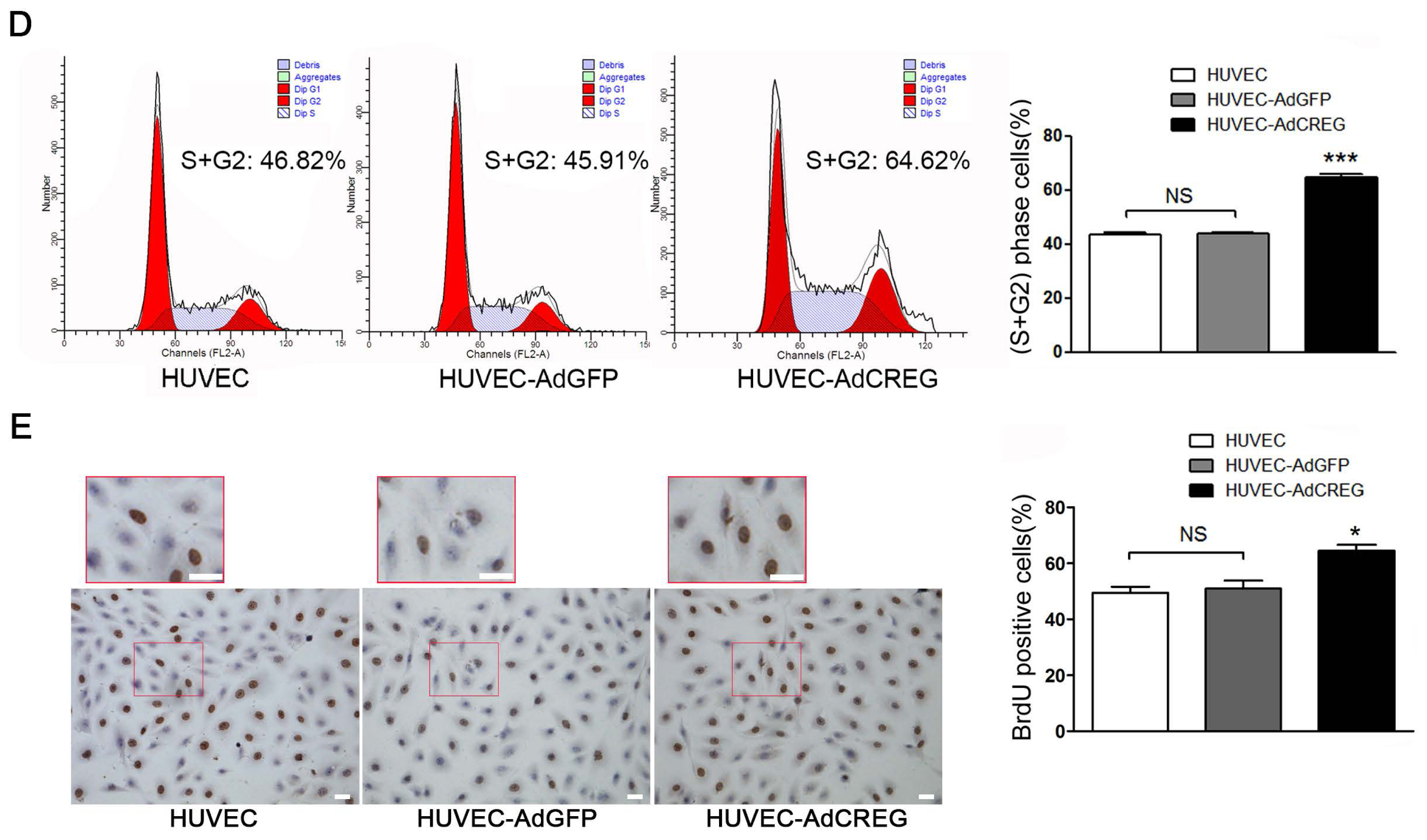
21], indicating that CREG exerted a pro-proliferative effect on GC cells. These results suggest that CREG may play either anti- or pro-proliferative roles in the differentiation of different cell types. The effect of CREG on endothelium growth and proliferation remains unknown. Therefore, in this study, we try to identify the effect of CREG overexpression on the proliferation of vascular endothelial cells. HUVEC with CREG overexpression was obtained via adenovirus infection. Our study identified that CREG can promote the proliferation of HUVEC, as determined by BrdU incorporation assay and the cell cycle analysis by FCM. In addition, the effect of CREG on endothelial proliferation was further confirmed by loss-of-function studies via silencing of CREG expression with shRNA. It is thus implied that the level of CREG expression might be an important determinant for the proliferative potential of endothelial cells and has a role in the regulation of vascular structure and functioning.
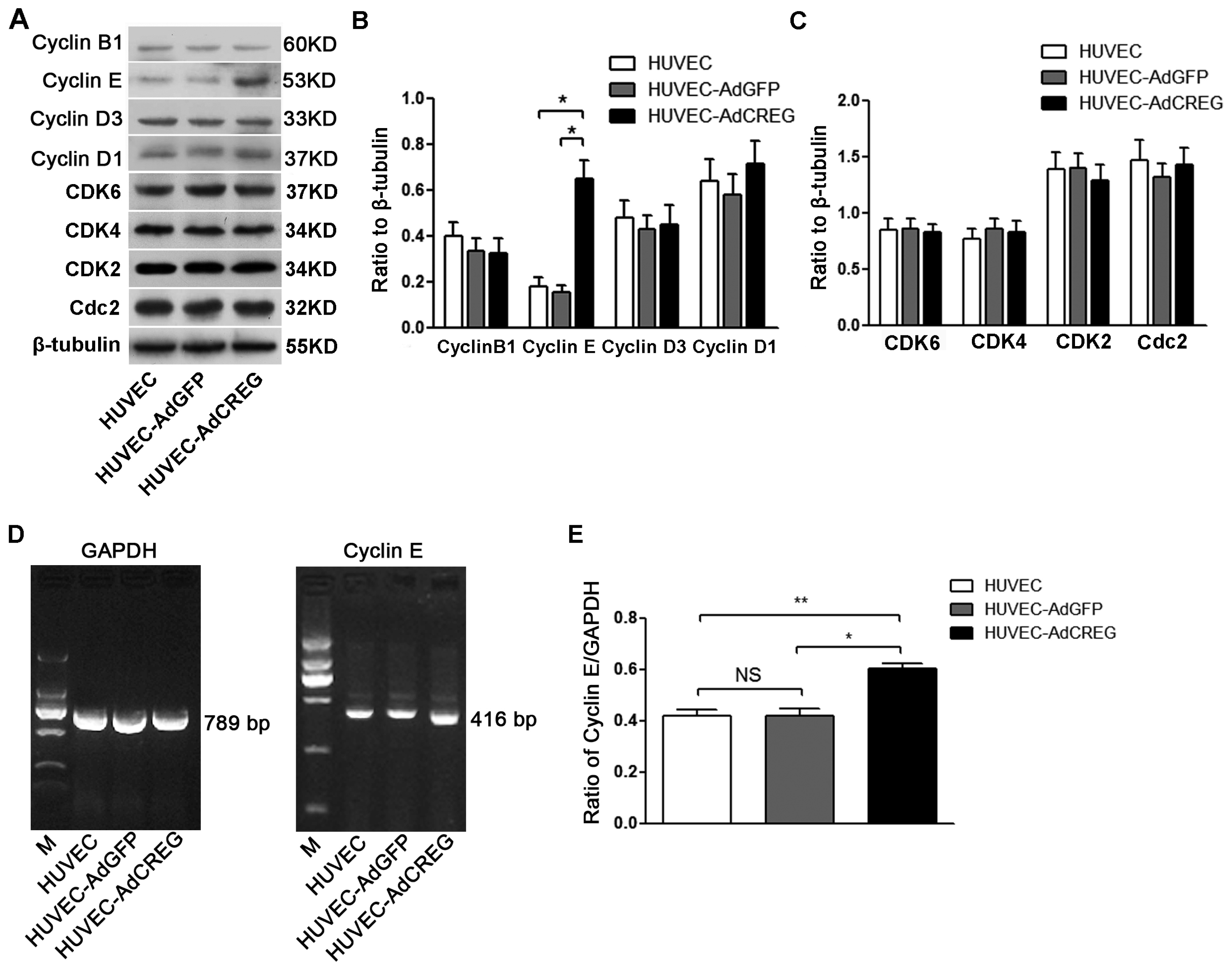
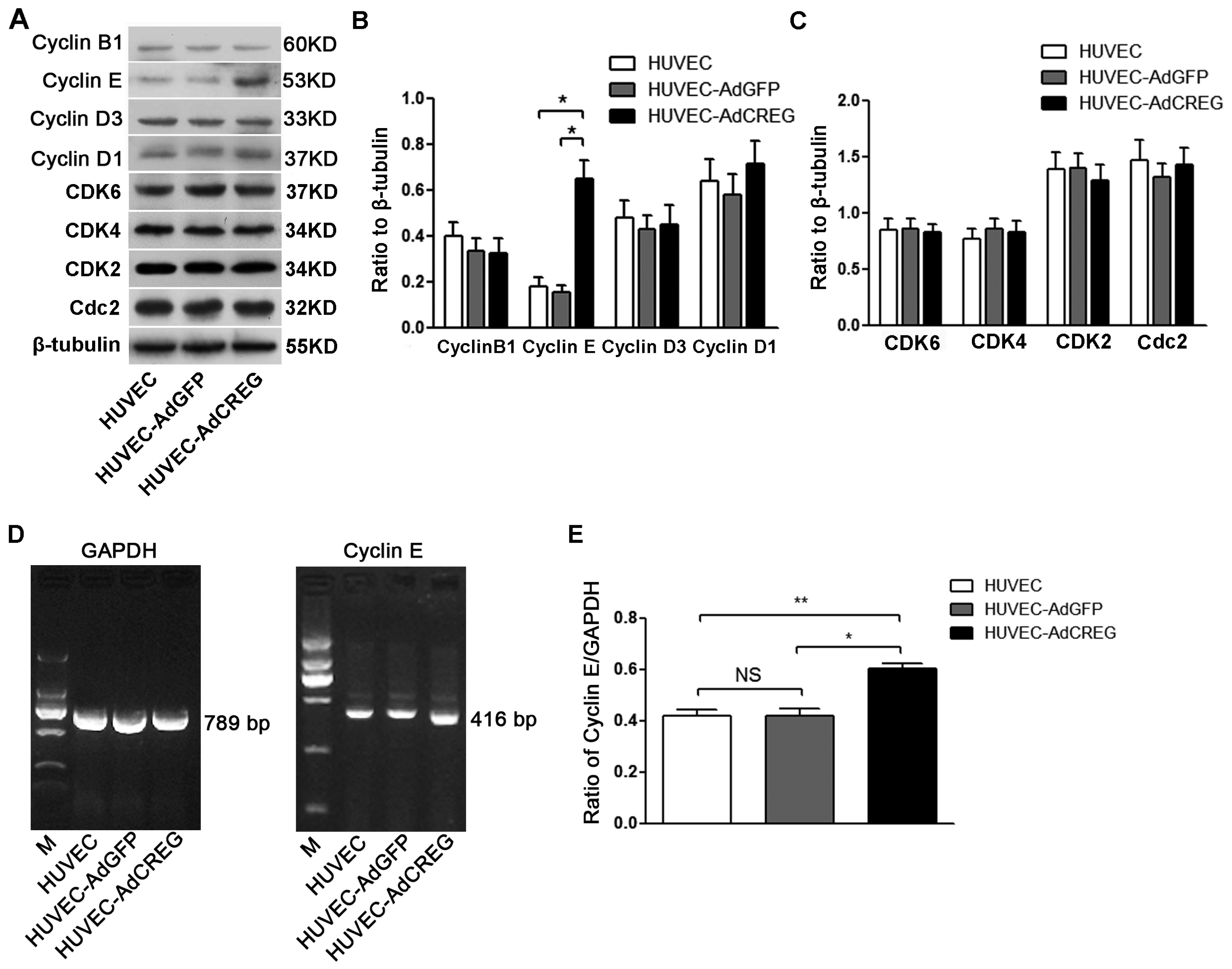
Cell cycle progression requires well-balanced and coordinated expression of both positive and negative regulators, whose expression fluctuates in a manner that tailors to cell cycle directionality. As a key step for cell cycle progression, cyclin members, including B, D1, D3 and E, are synthesized in response to mitogenic stimuli and form active kinase complexes with CDKs to initiate proliferation [
22,
23]. In the present study, the expressions of cyclins and CDKs were detected, and our results showed that CREG can increase the expression of cyclin E, both in protein and mRNA levels, but failed to alter the protein expression levels of other modulators. Therefore, CREG might control G1/S phase progression in the proliferation of HUVEC by regulating the expression of cyclin E, which is central to the mechanisms that underpin the CREG mitogenic effect on HUVEC.
It is normally recognized that the G1/S-phase stage transition is collectively regulated by cyclins and their dependent kinases [
24]. Mitogenic signals have been documented to promote the sequential assembly and activation of cyclin D/CDK4, 6 and cyclin E/CDK2, respectively, in the early and late G1 phase. Then, members of the retinoblastoma (Rb) protein family were hyper-phosphorylated, and E2F transcription factors were released to propel the G1-S transition [
25]. CDK2 activity is commonly induced by E-type cyclins [
26], but in the present study, induction of cyclin E by CREG did not result in a significant overexpression of CDK2. This could be conceivably explained by the fact that the activities of the CDKs are also regulated by two families of cyclin-dependent kinase inhibitors (CKIs), the Cip/Kip family and the INK4 family [
24]. Since studies on single, as well as combined gene deletion models have confirmed the overlapping and complexity of functions of cell cycle regulators during cell lineage and developmental timing [
27], we did not comprehensively examine the effect of CREG overexpression on the activation of these specific inhibitors; however, analysis of cell lines deficient of specific cell cycle regulators may be helpful to finally decipher the molecular mechanisms underlying CREG modulation of cell cycle progression.
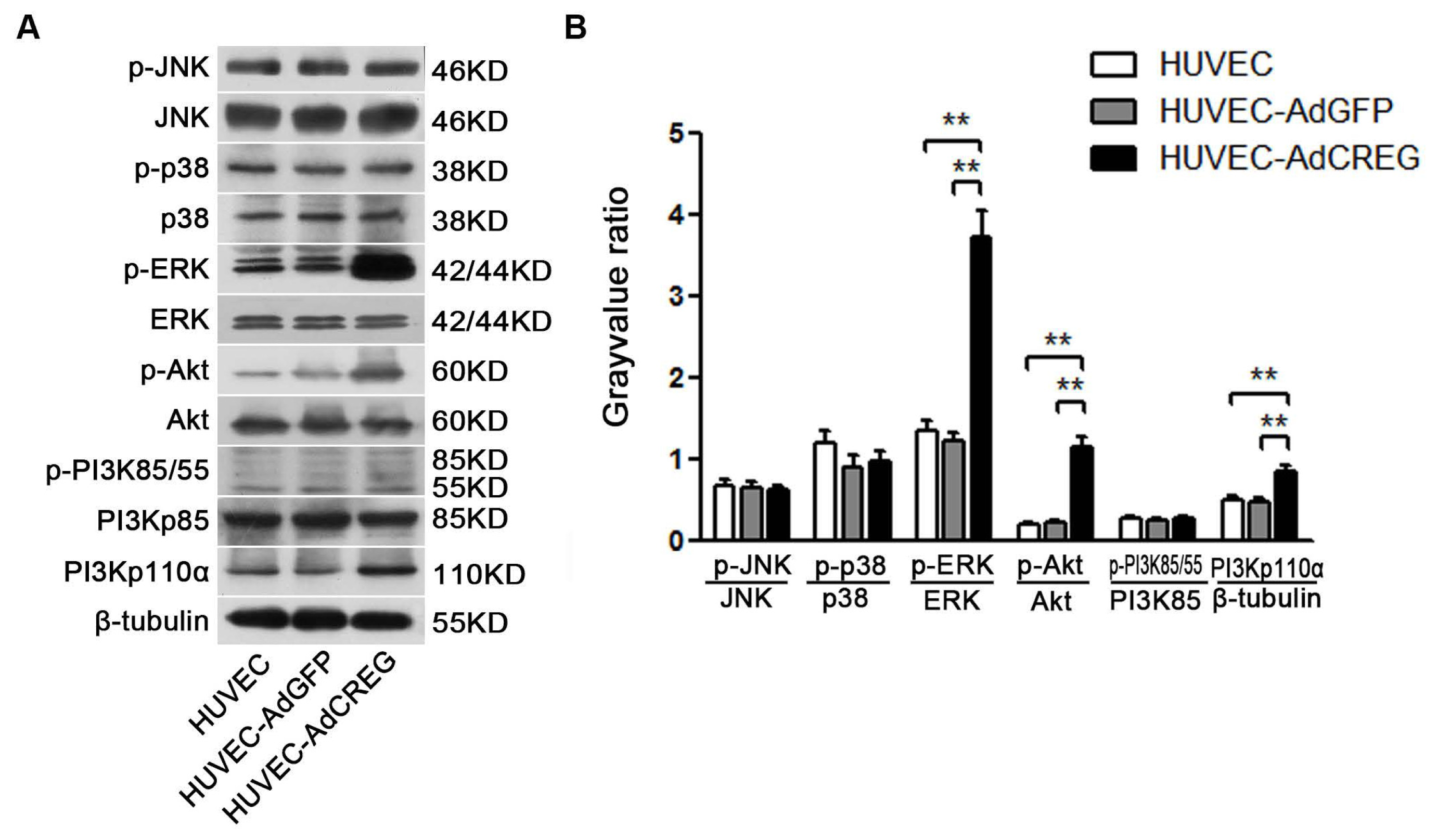
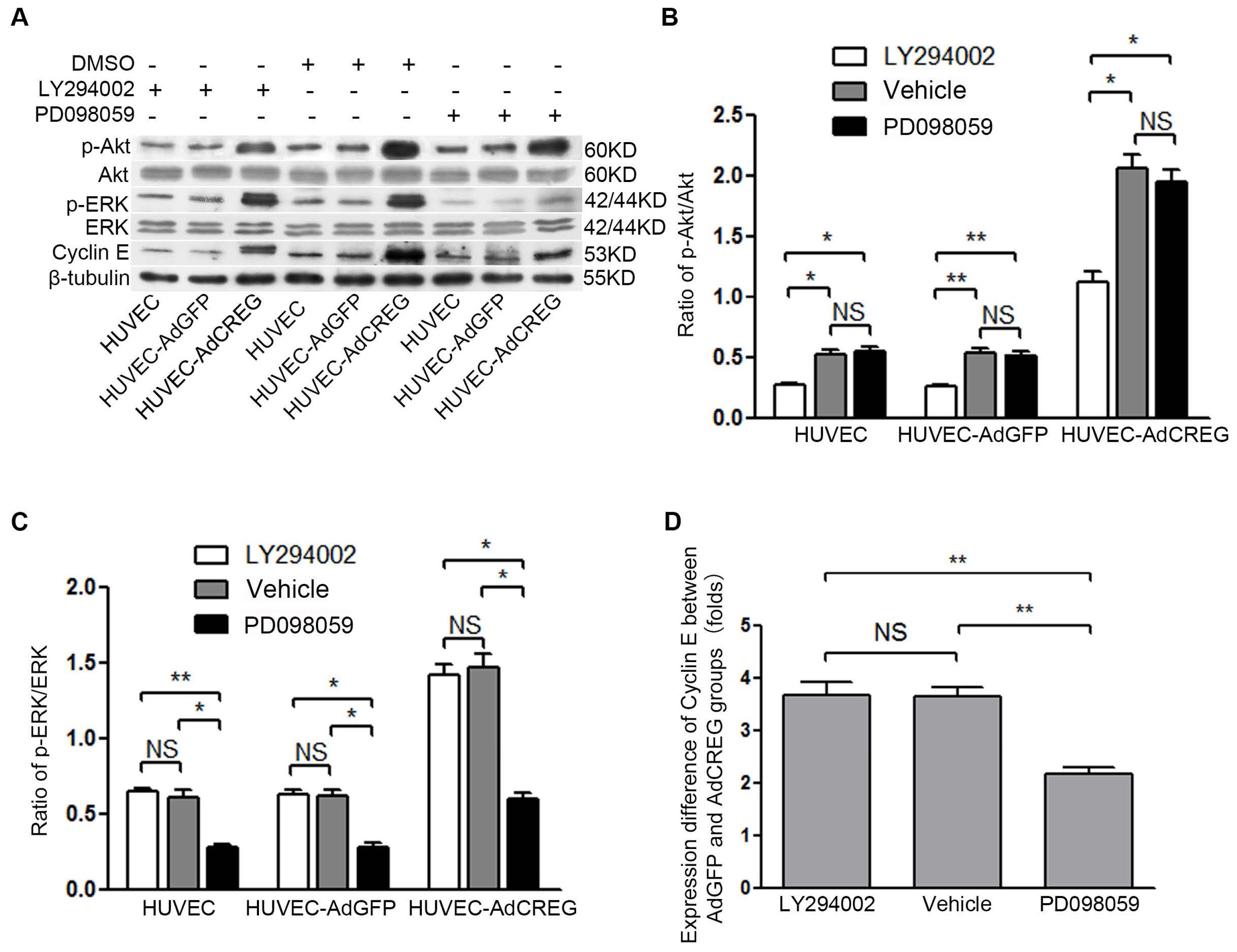
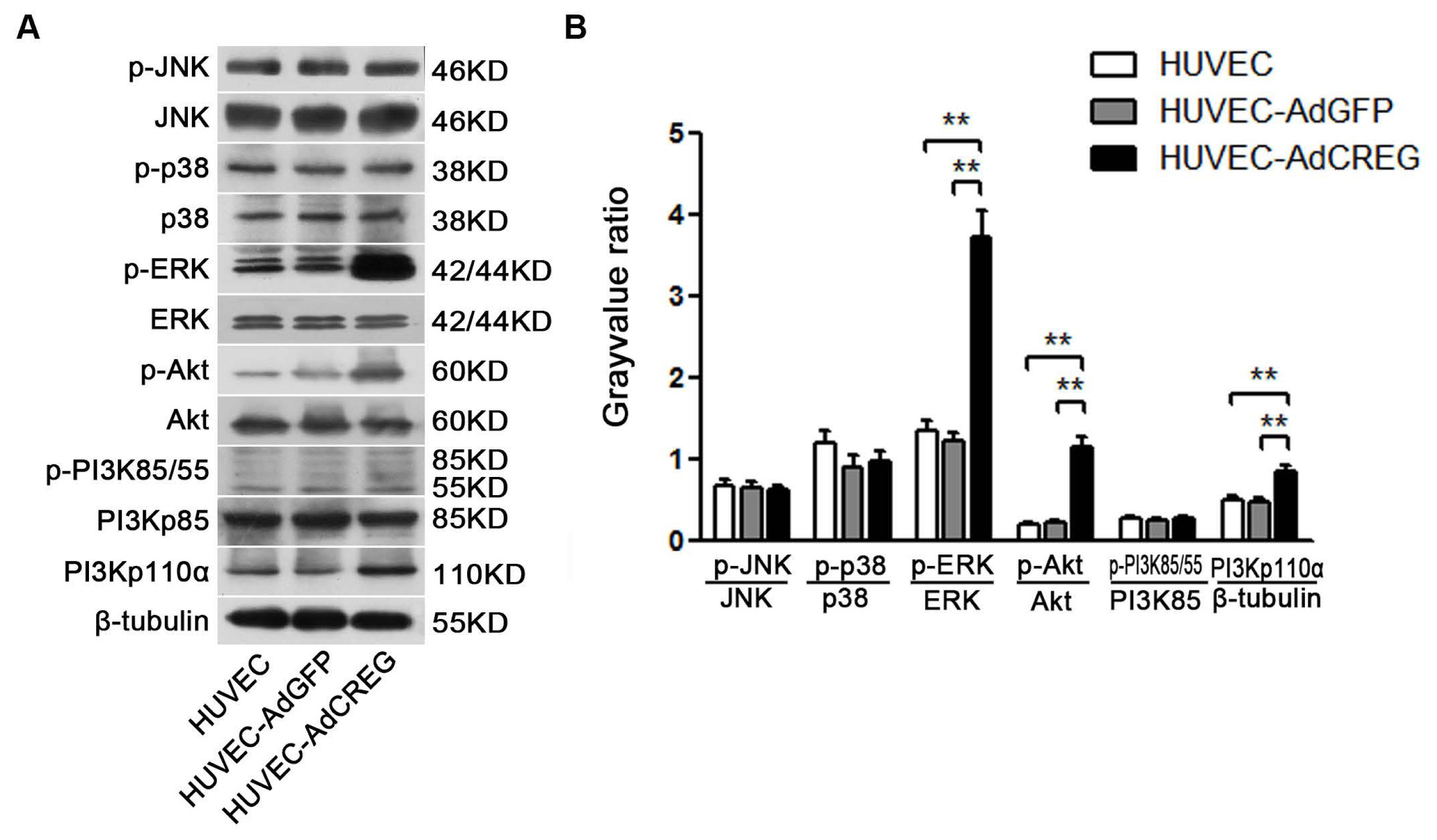
We further aimed to explore the mechanism of CREG regulation of cyclin E expression. Two pathways, ERK and PI3K/Akt signaling, were chosen for their established roles in the regulation of cell proliferation by affecting cyclins/CDKs expression [
11,
12]. Moreover, our and others’ previous studies have reported that both the PI3K/Akt and ERK pathways can be activated by CREG to modulate varied biofunctions in different cells [
9,
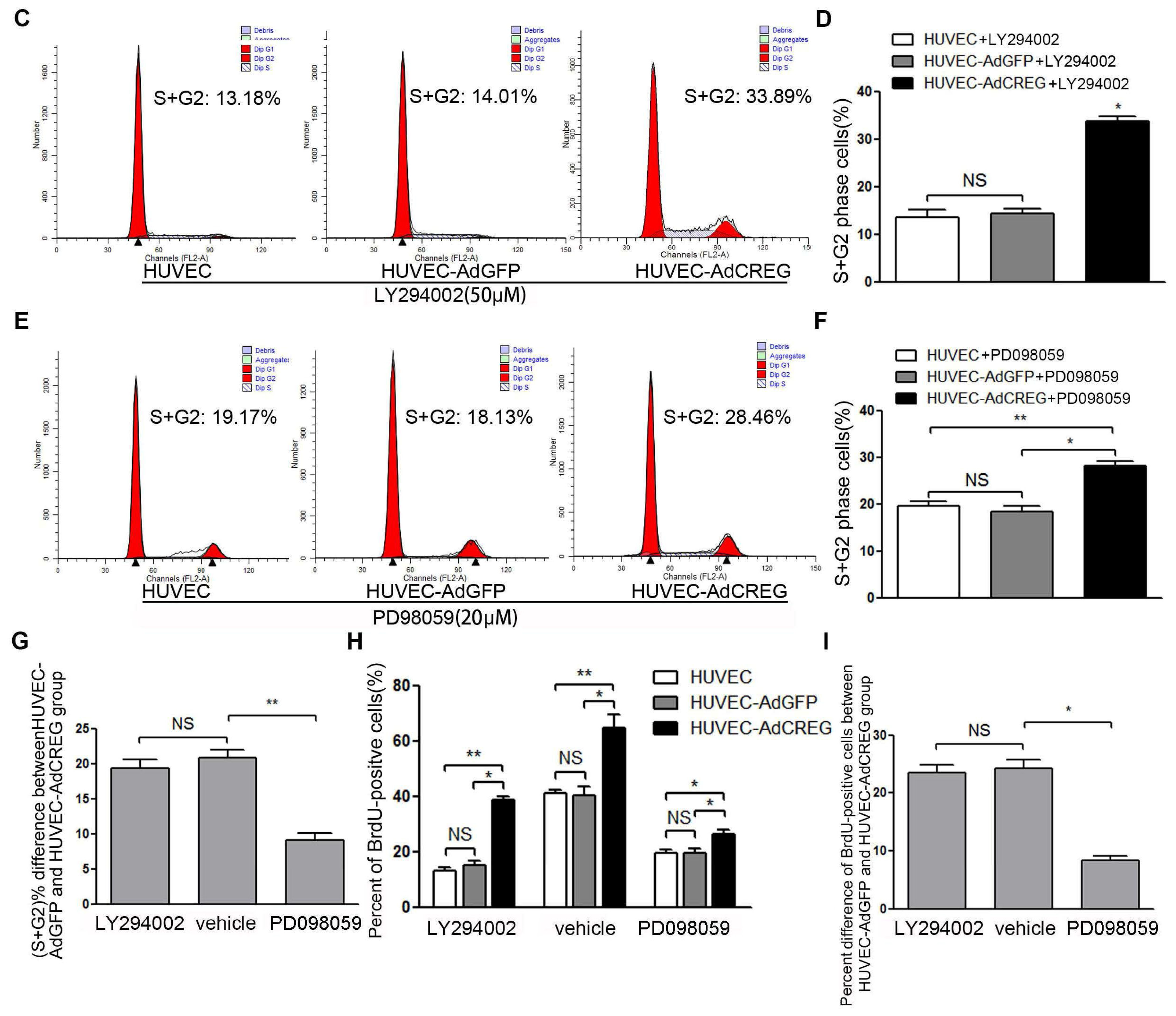
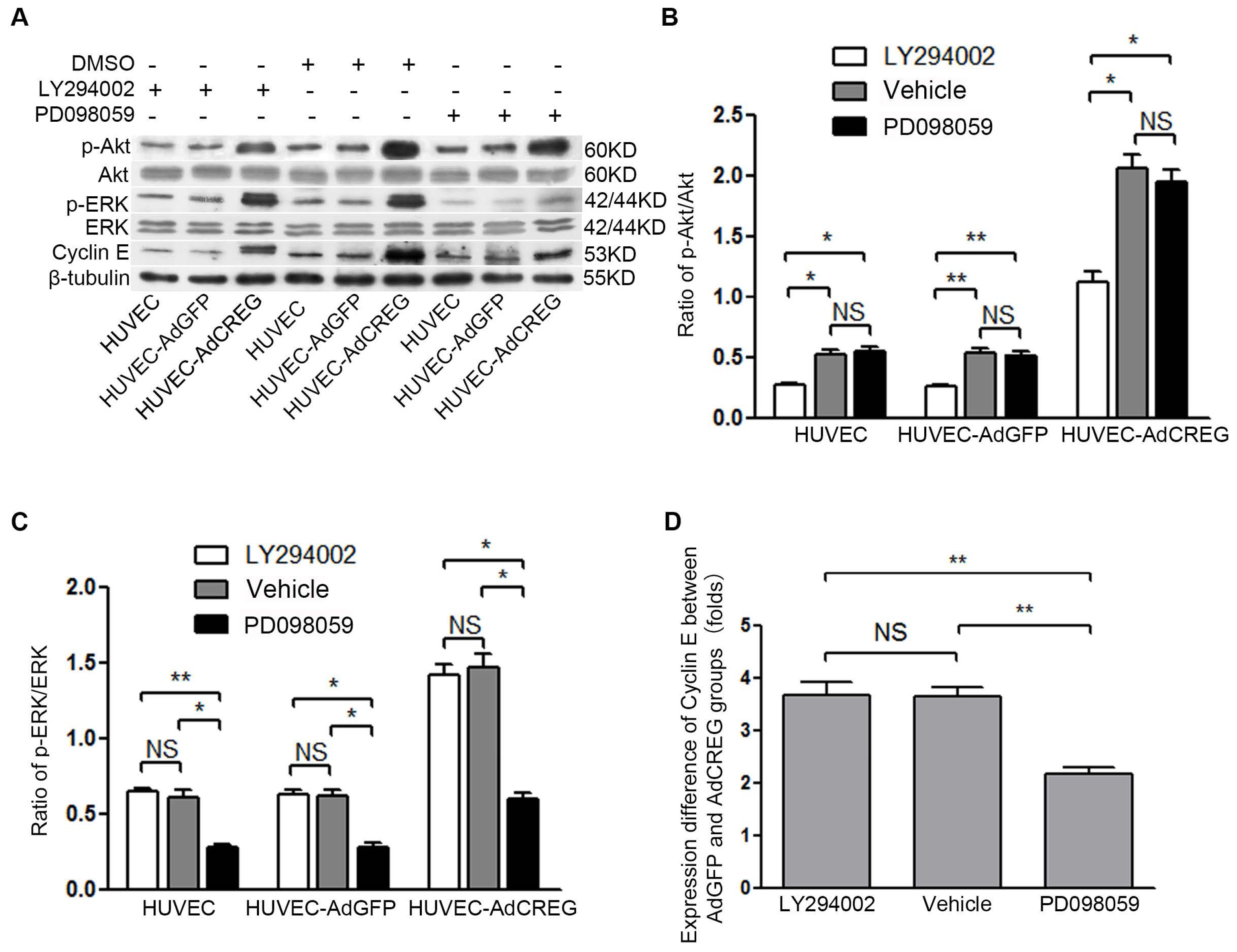
18]. In the current study, we found that CREG overexpression can markedly activate both PI3K/Akt and ERK pathways, which is consistent with our previous knowledge. Further, a blockage study identified that ERK is a mediator of the CREG effect with respect to enhanced cyclin E expression and proliferation of HUVEC. On the other hand, although the PI3K/Akt pathway has also been a well-established modulator of endothelial growth, it does not seem to be directly involved in the CREG effect on endothelial proliferation.
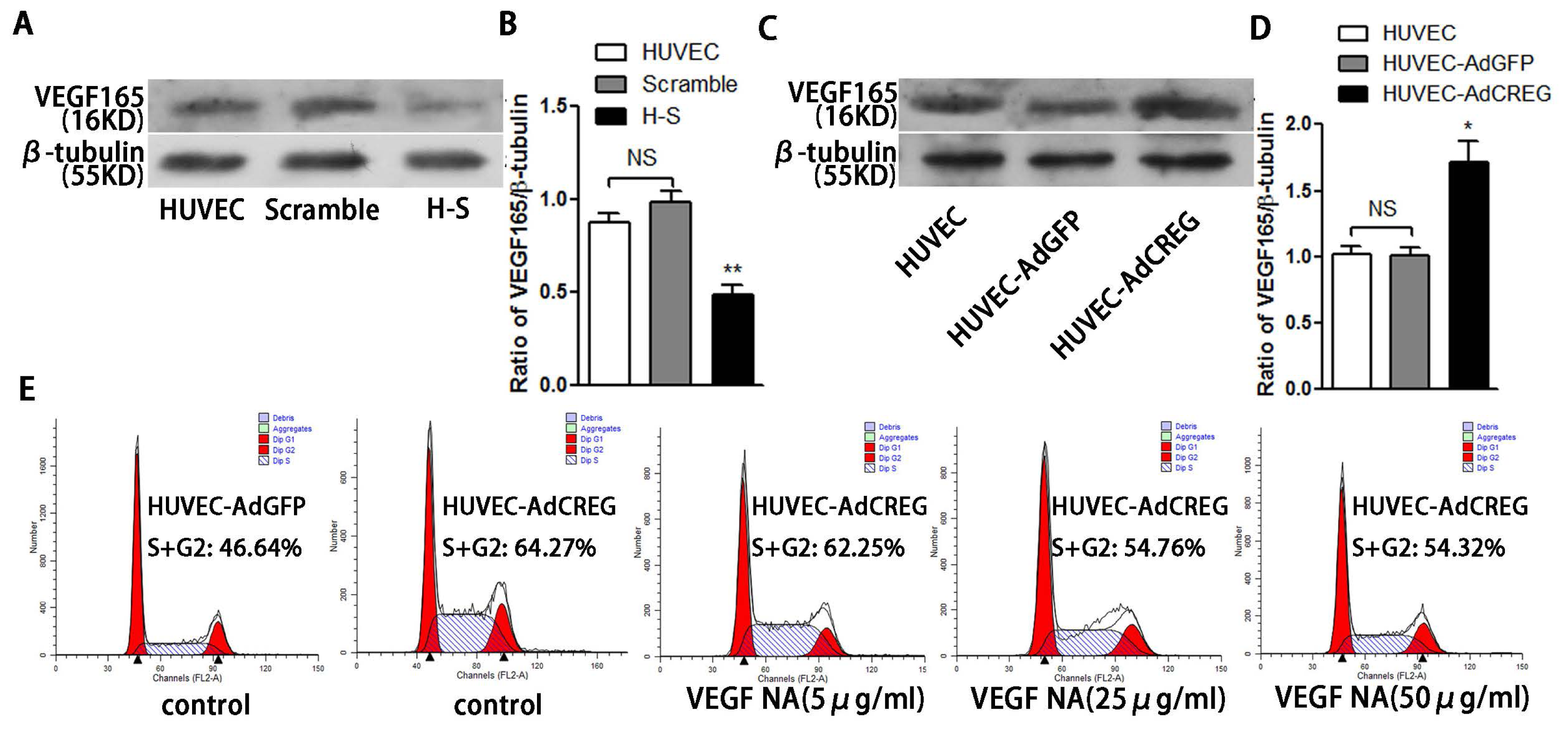
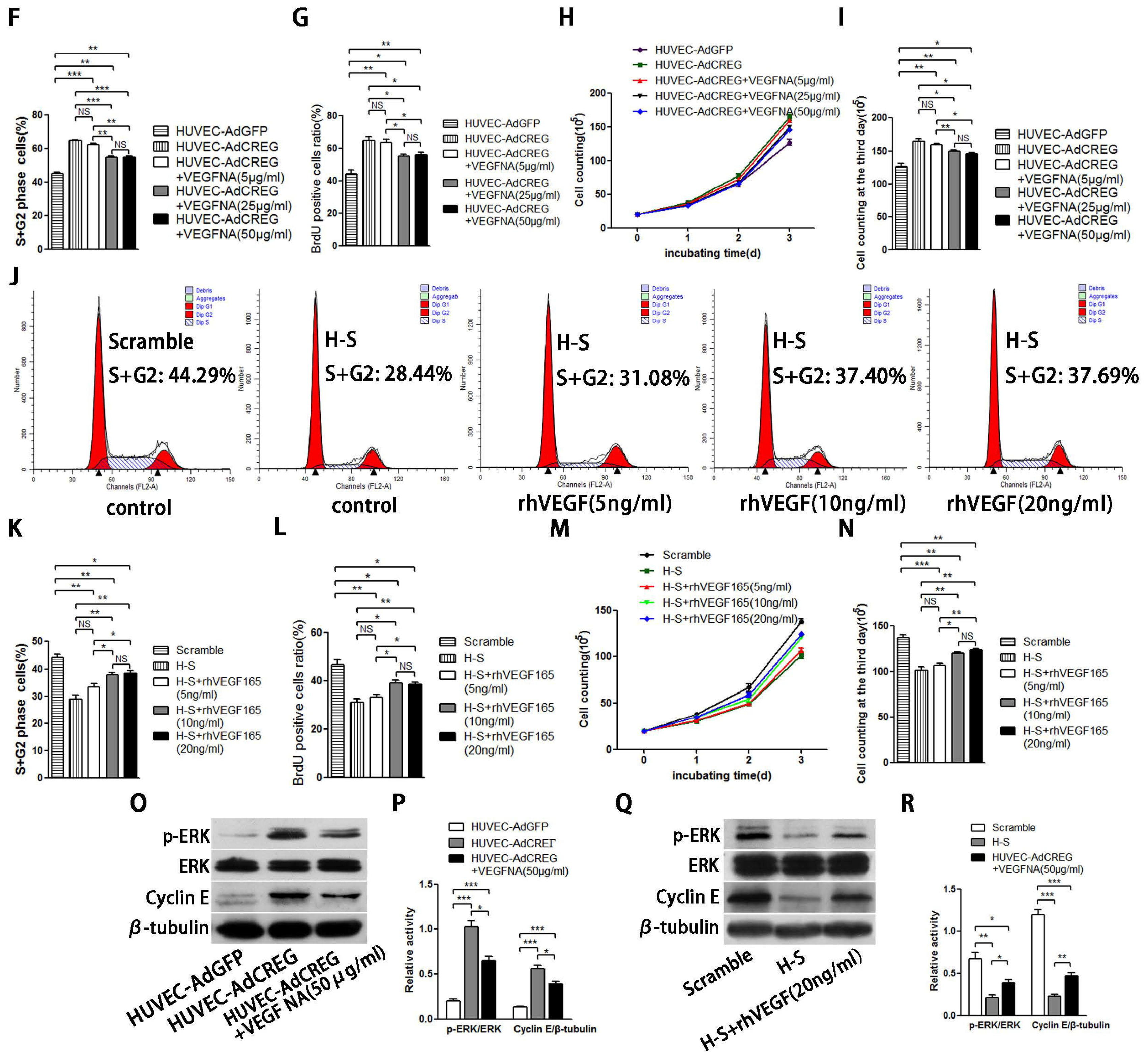
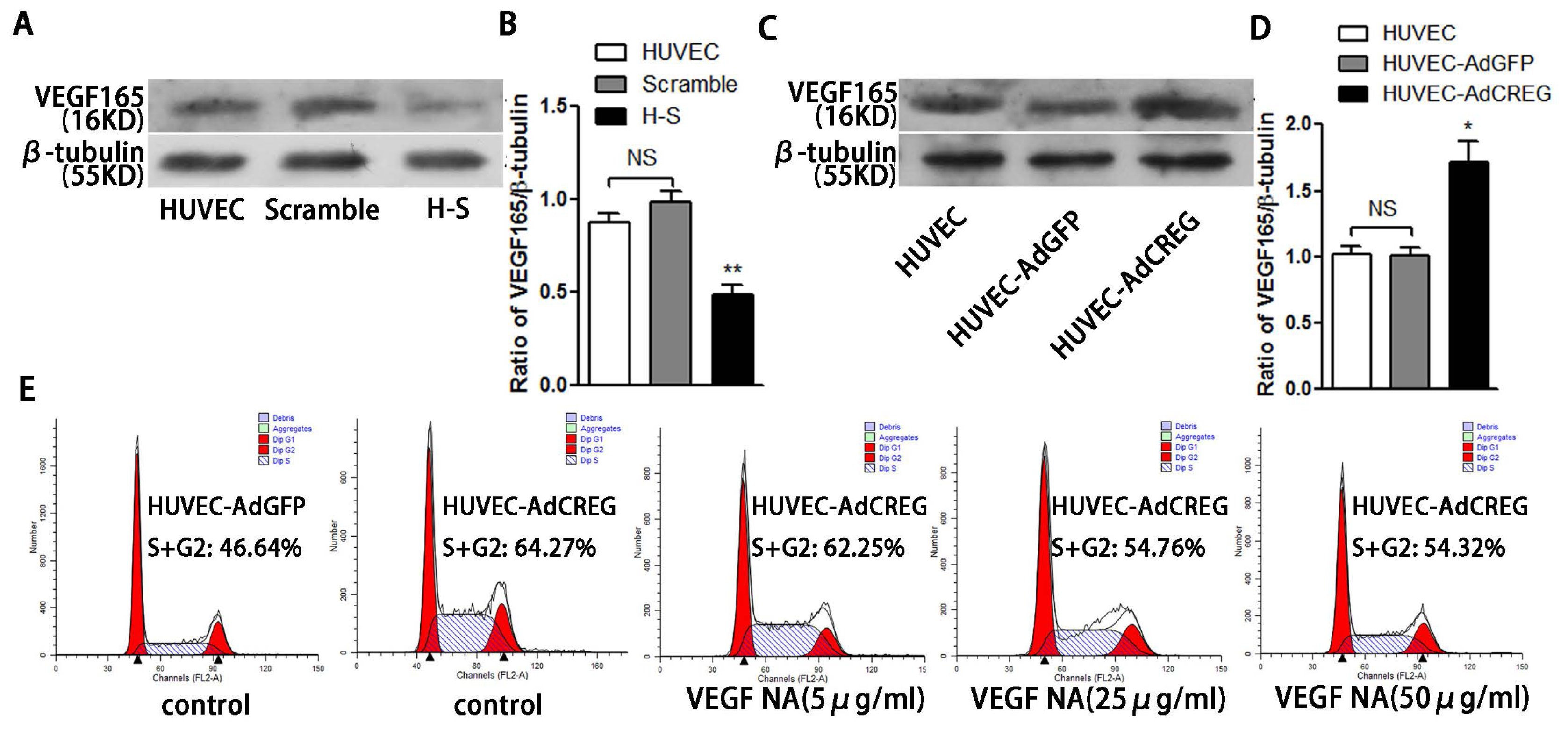
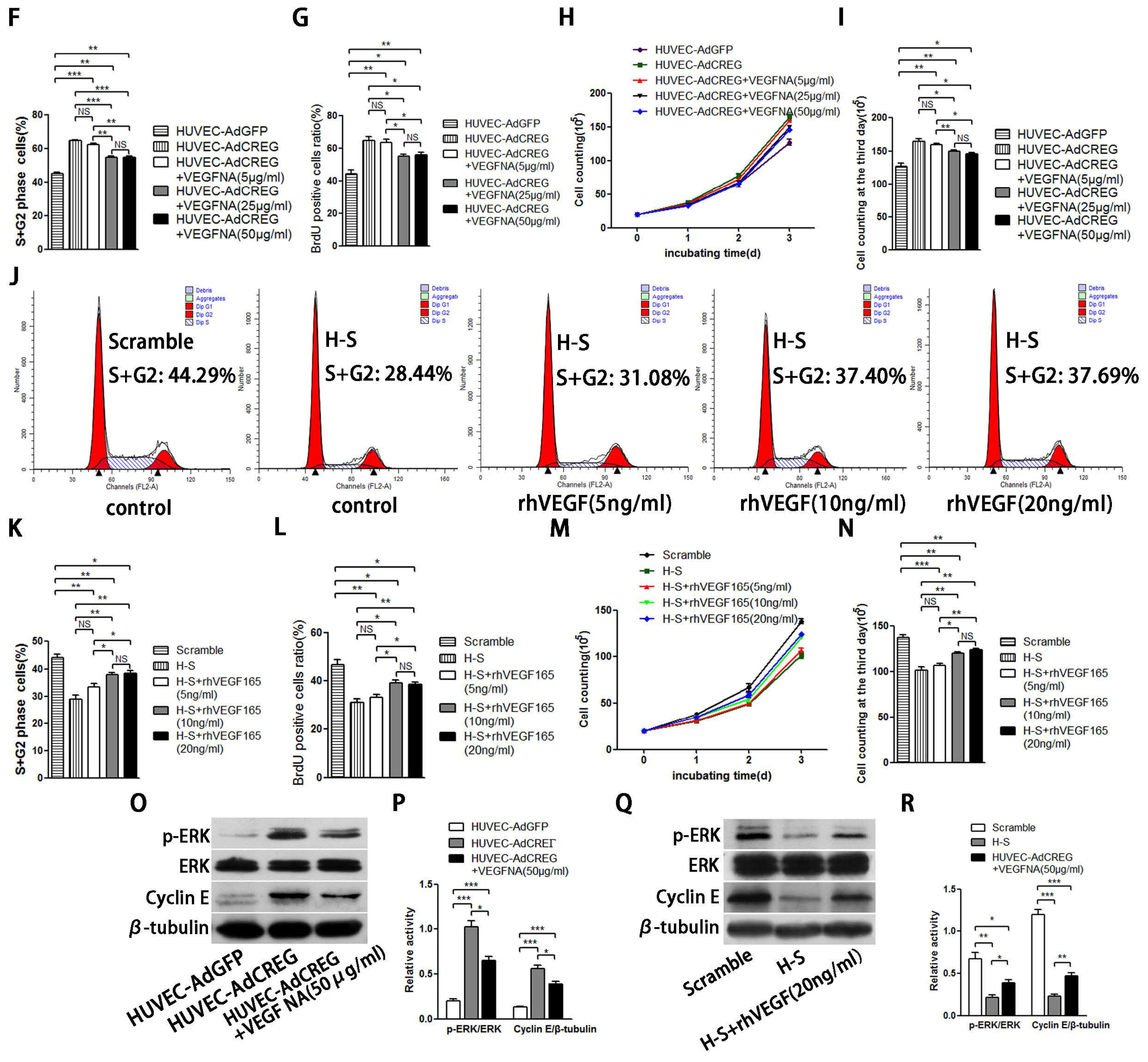
Endothelial cell proliferation and migration are tightly regulated by angiogenic factors, such as VEGF-A, which have a central role in the process of angiogenesis. We have previously reported that CREG can induce VEGF production and regulate endothelial cell migration and apoptosis [
9,
10]. Moreover, the VEGF has also been reported to activate cyclin E/Cdk2 through either ERK or PI3K/Akt signaling to modulate centrosome over-duplication in tumor endothelial cells [
28]. In the present study, we found that the pro-proliferative effect of CREG on HUVEC was only partially attributed to upregulation of VEGF165, implying that other underlying mechanism may be involved in this process and need to be identified in the future. Nevertheless, our recent study evaluating the efficacy of a nanoporous CREG-eluting stent in inhibiting neointimal formation in a porcine coronary model has shown an advantage of CREG-eluting stents over the widely used sirolimus-eluting stents or bare metal stents, as evidenced by accelerated re-endothelialization in the presence of CREG [
29].
Taken together, our data indicate that CREG promotes endothelial proliferation, and this function is partially mediated by VEGF-induced ERK/cyclin E activation. In addition, our identification of CREG and its downstream effectors in this important modulatory process might provide new targets for the intervention of vascular disorders associated with endothelial cell injury and cell loss.
4. Material and Methods
4.1. Reagents
Mouse monoclonal antibodies against CREG (MAB2380), CD31 (BBA7), E-cadherin (AF648), ERK (AF1576) and p-ERK (MAB1018) were from R & D Systems (Minneapolis, MN, USA). Mouse monoclonal anti-β-tubulin (ab6046) was from Abcam (Hong Kong, China). P38 (sc-7149), p-p38 (sc-101758), JNK (sc-572), p-JNK (sc-6254), cyclin-dependent kinases 2 (CDK2) (sc-163), CDK4 (sc-260), CDK6 (sc-177), Cdc2 (sc-54), cyclin D1 (sc-753), cyclin D3 (sc-182), cyclin B1 (sc-245) and cyclin E (sc-481) antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies against Akt (#4691), p-Akt (Ser473, #4060), PI3K p85 (#4292), p-PI3K p85/p55 (#4228) and p110α (#4249) were purchased from Cell Signaling (Monsey, NY, USA). Secondary antibodies conjugated to Horseradish Peroxidase (HRP) were obtained from Santa Cruz Biotechnology. The ERK phosphorylation inhibitor, PD098059 (2′-amino-3′-methoxyflavone, 513000), and the PI3K/Akt inhibitor, LY294002 [2-(4-morholinyl)-8-phenyl-4H-1-benzopyran-4-one, 440202], were purchased from Calbiochem (San Diego, CA, USA). Anti-vascular endothelial growth factor (VEGF) antibody and neutralizing antibody (NA) were obtained from BD biotechnology (Franklin Lakes, NJ, USA), and recombinant human VEGF165 were obtained from PeproTech (Rocky Hill, NJ, USA). TRIzol reagent, PrimeScript™ RT reagent kit and SYBR® Premis Ex Taq™ were from TaKaRa Biotechnology Co. (Liaoning, China). The primers were synthesized by TaKaRa Biotechnology Co. (Liaoning, China).
4.2. Culture of Primary HUVEC
HUVEC were isolated from human umbilical cords using collagenase and cultured in Medium 199 (Invitrogen, Carlsbad, CA, USA) containing 10% (v/v) fetal bovine serum (FBS, HyClone, UT, USA) and conditioned supplement (recombinant human (rh) VEGF, 5 ng/mL; rhEGF, 5 ng/mL; rhFGF basic, 5 ng/mL; rhIGF-1, 15 ng/mL) at 37 °C in an atmosphere of 5% CO2 and 95% air. Endothelial cell identity was confirmed by immunostaining for CD31 and VE-cadherin. HUVEC were used at passages 2–6 in all experiments.
4.3. Adenoviral Infection
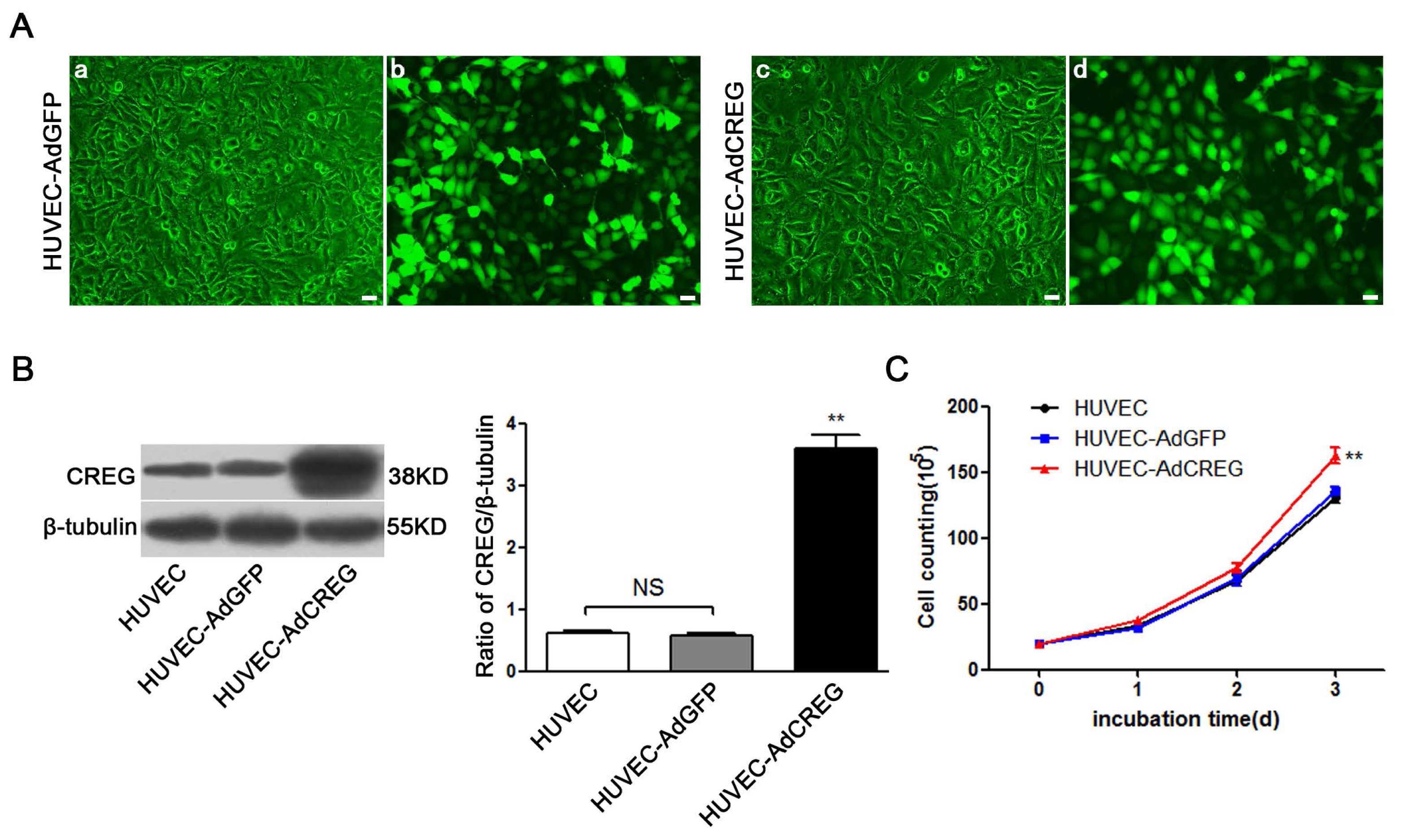
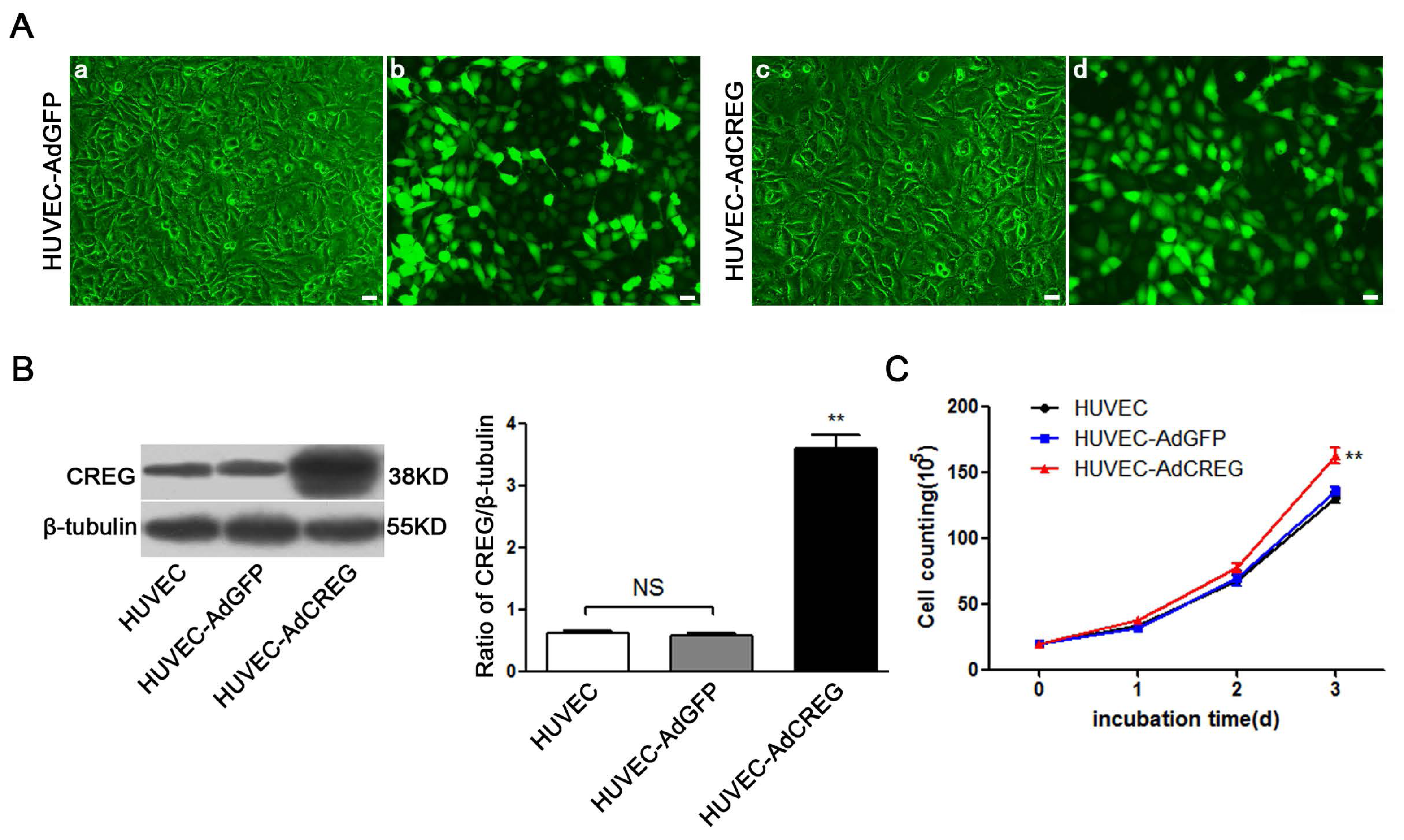
Adenoviral GFP and CREG vectors were created as described previously [
9,
10]. These replication-deficient vectors were propagated in 293 cells using Dulbecco’s Modified Eagle’s medium (DMEM) supplemented with 10% (
v/
v) FBS. The cells were infected with DMEM containing 1 × 10
8 AdGFP or AdCREG adenovirus particles/mL for 2 consecutive days. The expression of CREG was assessed by immunoblotting.
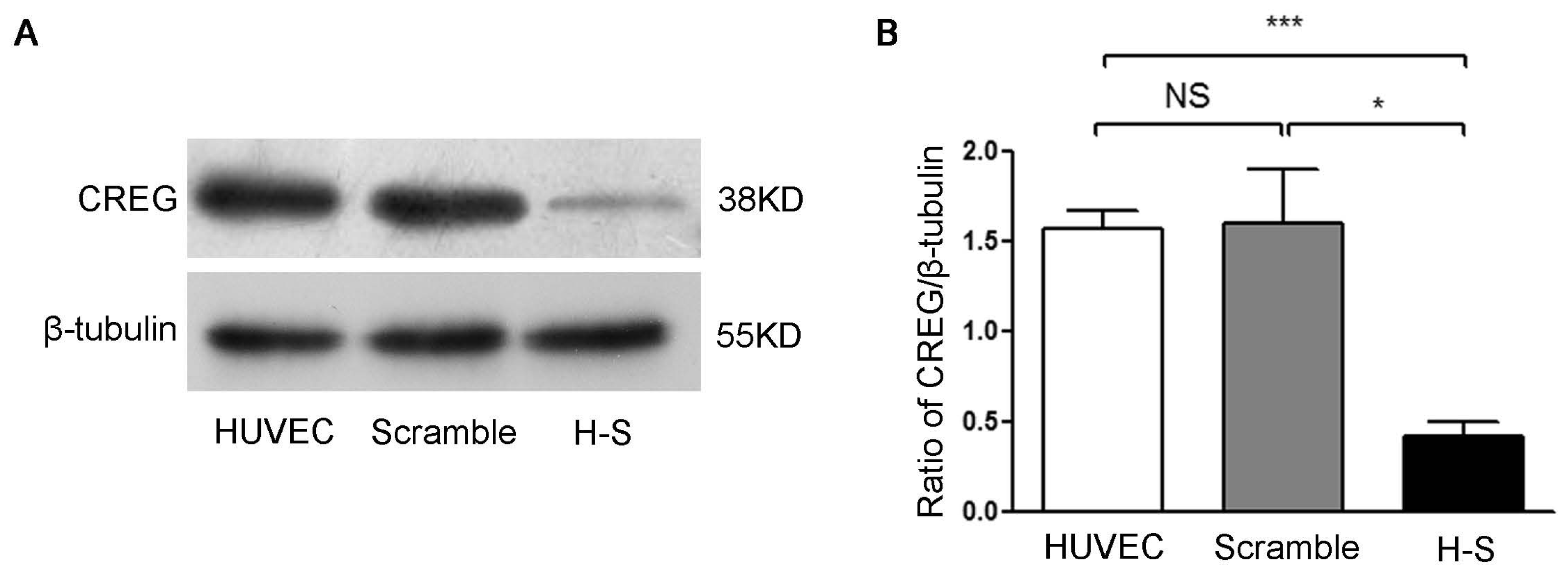
4.4. Generation of CREG Knocked down Endothelial Cell Lines
Retroviral vectors containing either non-effective scramble shRNA cassette (shRNA-scramble) or shRNA targeting human CREG (shRNA-CREG) were purchased from Open Biosystems (Huntsville, AL, USA). To generate infectious retrovirus, 5 μg of the plasmid was transfected into Phoenix amphotropic 293 packaging cells (ATCC, Manassas, VA, USA) by calcium phosphate/DNA co-precipitation. Supernatant containing retrovirus was collected and used to infect HUVEC. Stable HUVEC clones with CREG silenced (H-S) were obtained by selection with puromycin (4 μg/mL) for 2 weeks. The expression of CREG was verified by Western blot analysis. Stable HUVEC clones expressing negative control shRNA-scramble sequence (scramble) were established as a control group.
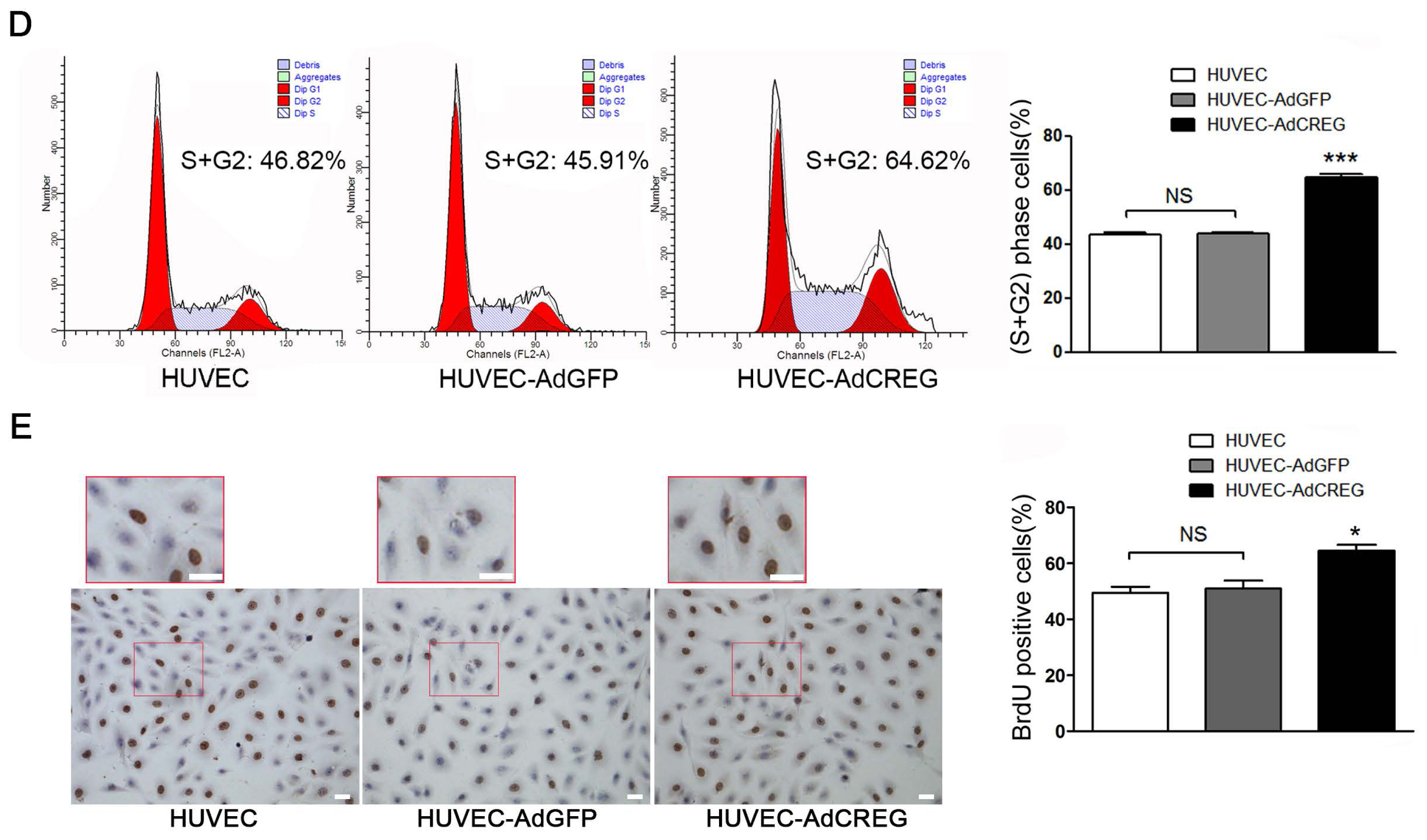
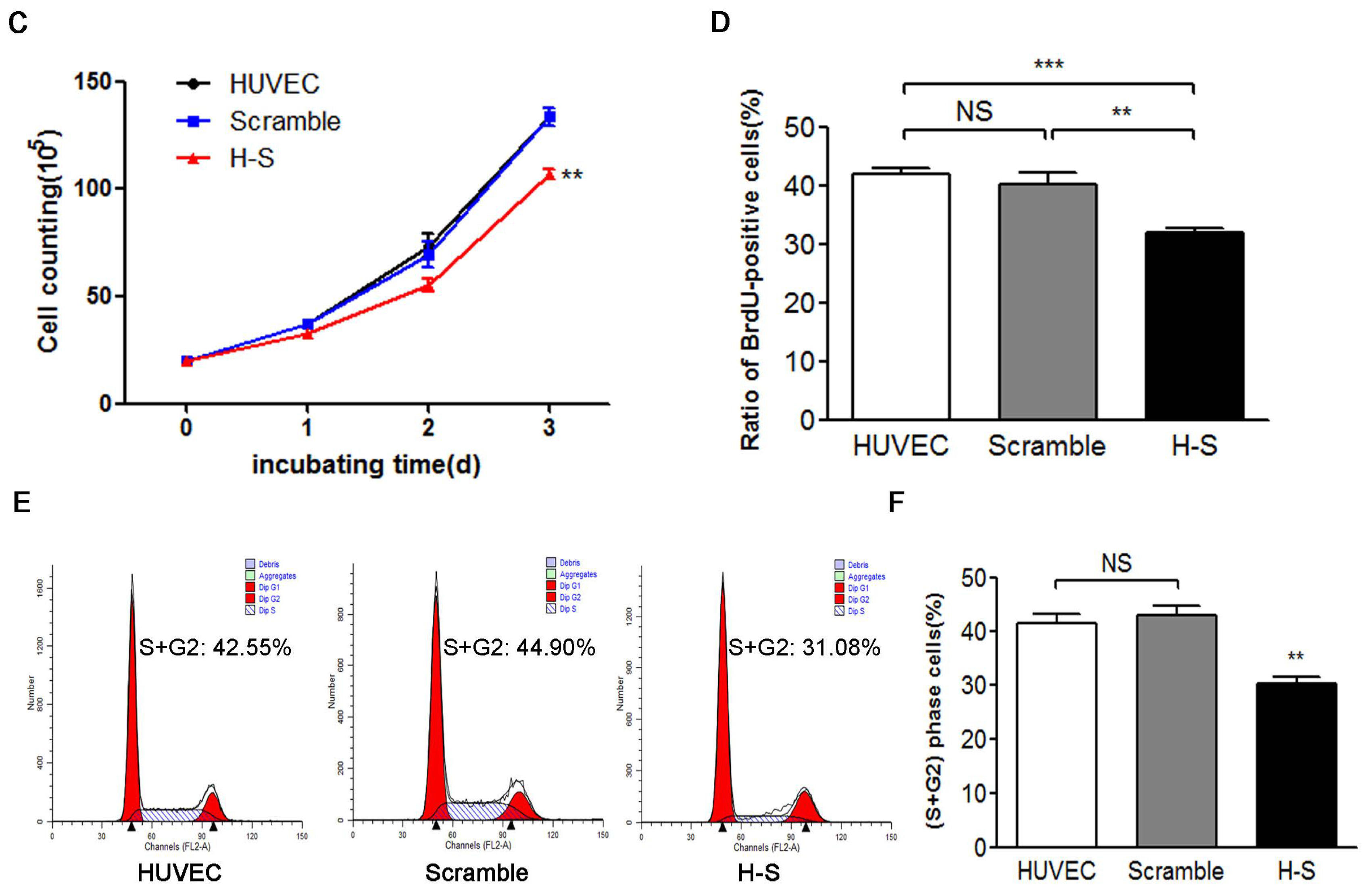
4.5. FCM and 5-bromo-2′-deoxy-uridine (BrdU) Incorporation Assays
Cell cycle analysis was carried out in HUVEC serum-starved for 72 h and then stimulated with medium containing 10% serum for 20 h. After overnight ethanol fixation and propidium iodide staining for 15~30 min, HUVEC with distinct cell cycle distributions were analyzed by FCM. All samples were analyzed on an FACSCalibur flow cytometer (BD Biosciences), and the data were processed using FlowJo 9 software (FlowJo, Ashland, OR, USA). The BrdU incorporation assay was performed with a cell proliferation kit (GE Healthcare Life Sciences) in HUVEC serum-starved for 48 h and then stimulated with medium containing 10% serum for 24 h. Cultured HUVEC were incubated with BrdU for 3 h before fixation. Incorporated BrdU was detected immunohistochemically with an anti-BrdU antibody.
4.6. Cell Counting
After the cell clones were selected and expanded, 2 × 105 HUVEC, HUVEC-AdGFP, HUVEC-AdCREG, HUVEC-scramble and HUVEC-shCREG were planted in a 10-cm diameter culture dish with medium containing 10% serum and were counted with a blood counting chamber every 24 h after being digested by trypsin. Data were analyzed to investigate the influence of CREG expression on the proliferation of HUVEC. Experiments were performed in triplicate.
4.7. Western Blot Analysis
Cell lysates were prepared in lysis buffer containing 10 mM Tris-HCl (pH 7.4) and 1% sodium dodecyl sulfate. After centrifugation at 13,000× g for 10 min, the supernatant was collected for Western blot analysis. Total cell protein concentrations were determined using the BCA Protein Assay Kit (Pierce, Rockford, IL, USA). Proteins were resolved in sodium dodecyl sulfate-polyacrylamide gels and transferred to polyvinylidene fluoride membranes. The membranes were incubated with appropriate primary antibodies for 2 h or overnight and, then, washed for 15 min three times with Tris-buffered saline. Then, the membranes were incubated with HRP-conjugated secondary antibodies for 1 h at room temperature. After being washed again for 15 min three times with Tris-buffered saline, specific binding was detected with enhanced chemiluminescence reagents. The blots were quantified by Quantity One analysis software (Bio-Rad Laboratories). The experiments above were performed in triplicate.
4.8. RT-PCR
Cells were harvested, and total RNA was extracted, purified and reversely transcribed to cDNA. The reverse transcription was conducted at 37 °C for 15 min and 85 °C for 5 s. PCR amplification was run using PCR machine (Bio-Rad, Hercules, CA, USA). The RT-PCR program included a denaturation step at 95 °C for 4 min, 30 cycles of two amplification steps (95 °C for 30 s and 58 °C for 30 s) and an extension step at 72 °C for 7 min. The values for cyclin E mRNA expression were normalized using GAPDH as the housekeeping gene. The primers were as follows: Cyclin E: forward primer 5′-GTC CTG GCT GAA TGT ATA CAT GC-3′; reverse primer 5′-CCCTATTTTGTTCAGACAACATGGC-3′; GAPDH: forward primer 5′-ATT CCA TGG CAC CGT CAA GG-3′; reverse primer 5′-AAT TCG TTG TCA TAC CAG GA-3′. The blots were quantified by Quantity One analysis software. Experiments were performed in triplicate.
4.9. Statistical Analysis
Data are expressed as the mean ± standard deviation (SD). All data were analyzed using SPSS 13.0 statistical software (Chicago, IL, USA). Differences between the two groups were compared using the unpaired Student’s t-test. Differences among three or more groups were compared using one-way analysis of variance. Statistical significance was defined as p < 0.05 (two-tailed).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}