Membrane Trafficking of Death Receptors: Implications on Signalling

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Death Receptors: General Aspects and Essential Structural Characteristics
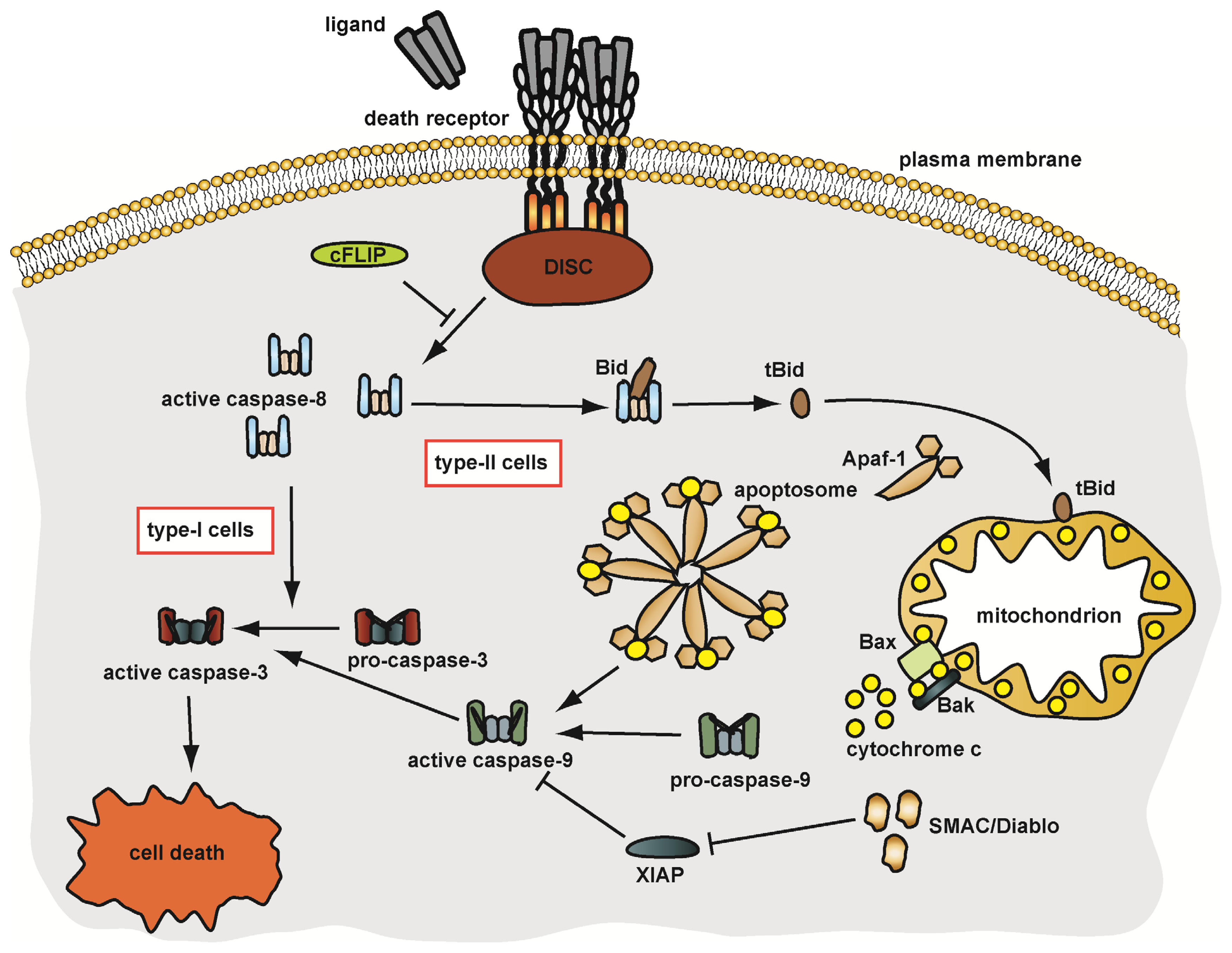
3. Death Receptor Signalling: A Double-Edged Sword
4. Structure, Physiology and Pathophysiology of TNFR1, TRAIL-R1/2, Fas and DR3
4.1. The TNF/TNFR1 System
4.1.1. Structure, Physiological and Pathophysiological Roles of the TNF/TNFR1 System
4.1.1.1. Structure
4.1.1.2. Physiological and Pathophysiological Roles of the TNF/TNFR1 System
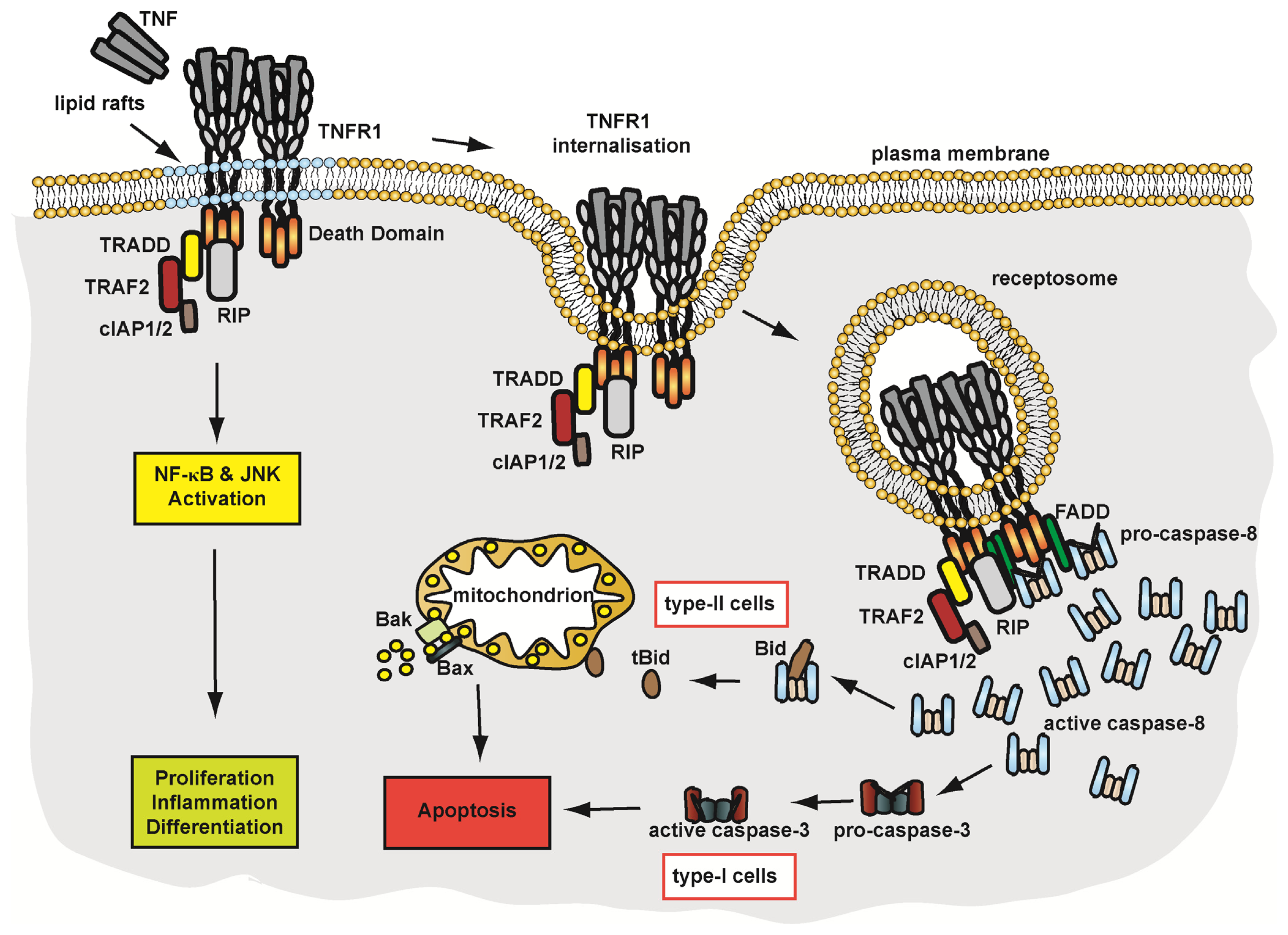
4.1.2. Pathways of TNFR1 Signalling
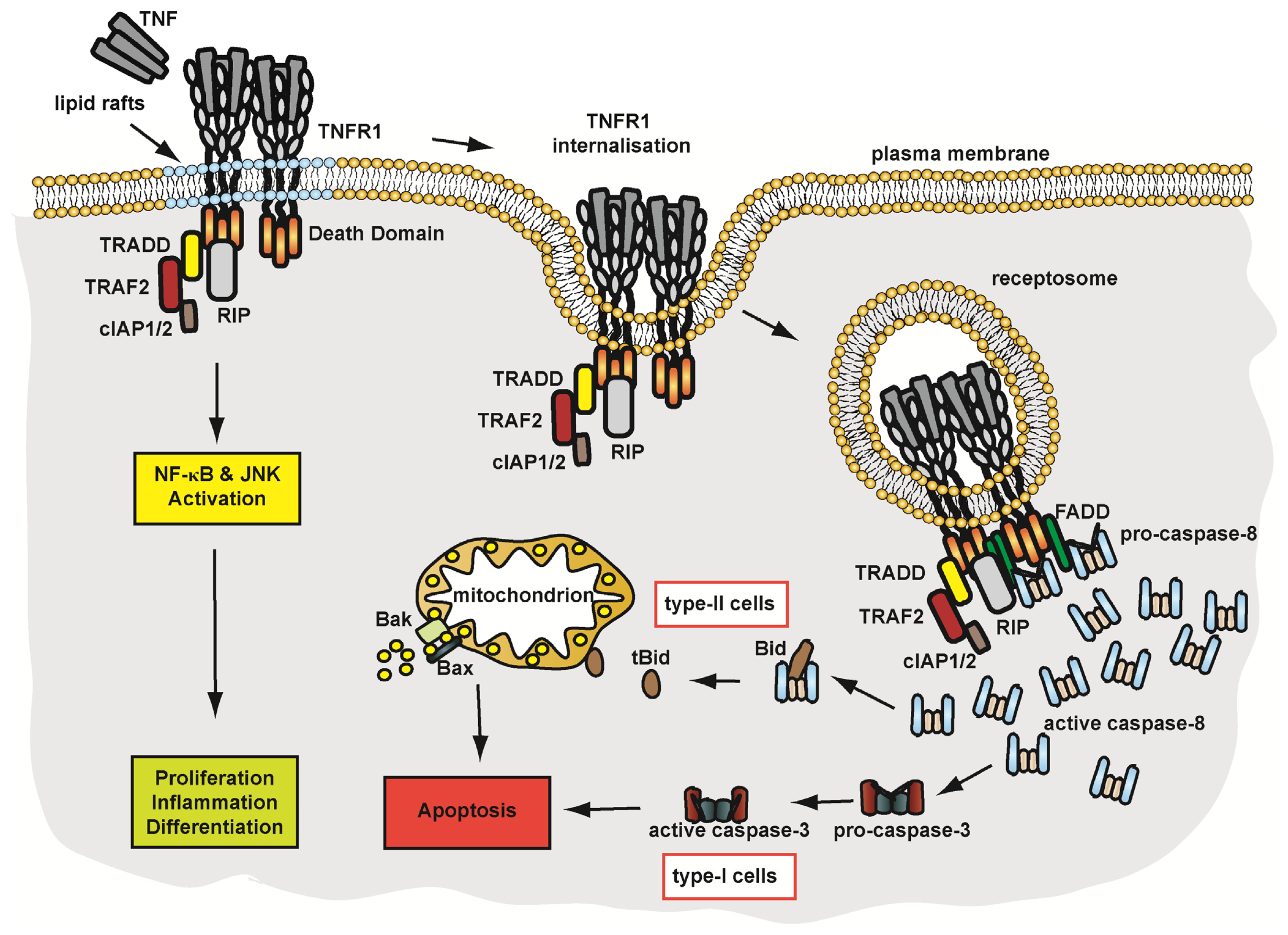
4.1.3. Membrane Trafficking and Implications on TNFR1 Signalling
4.2. The TRAIL/TRAIL-Receptor System
4.2.1. Structure, Physiological and Pathophysiological Roles of the TRAIL/TRAIL-Receptor System
4.2.1.1. Structure
4.2.1.2. Physiological and Pathophysiological Roles of the TRAIL/TRAIL-Receptor System
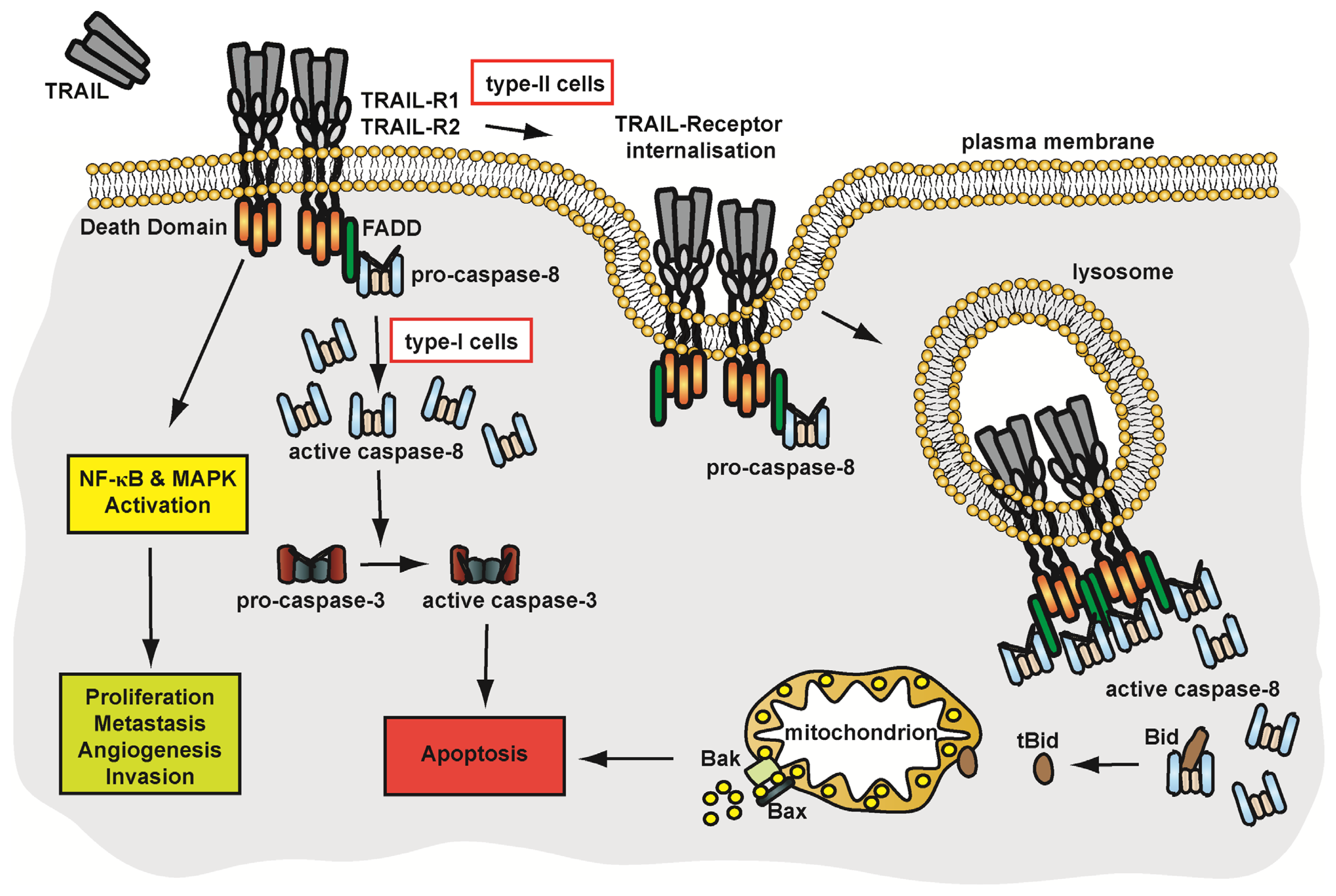
4.2.2. Pathways of TRAIL-Receptor Signalling
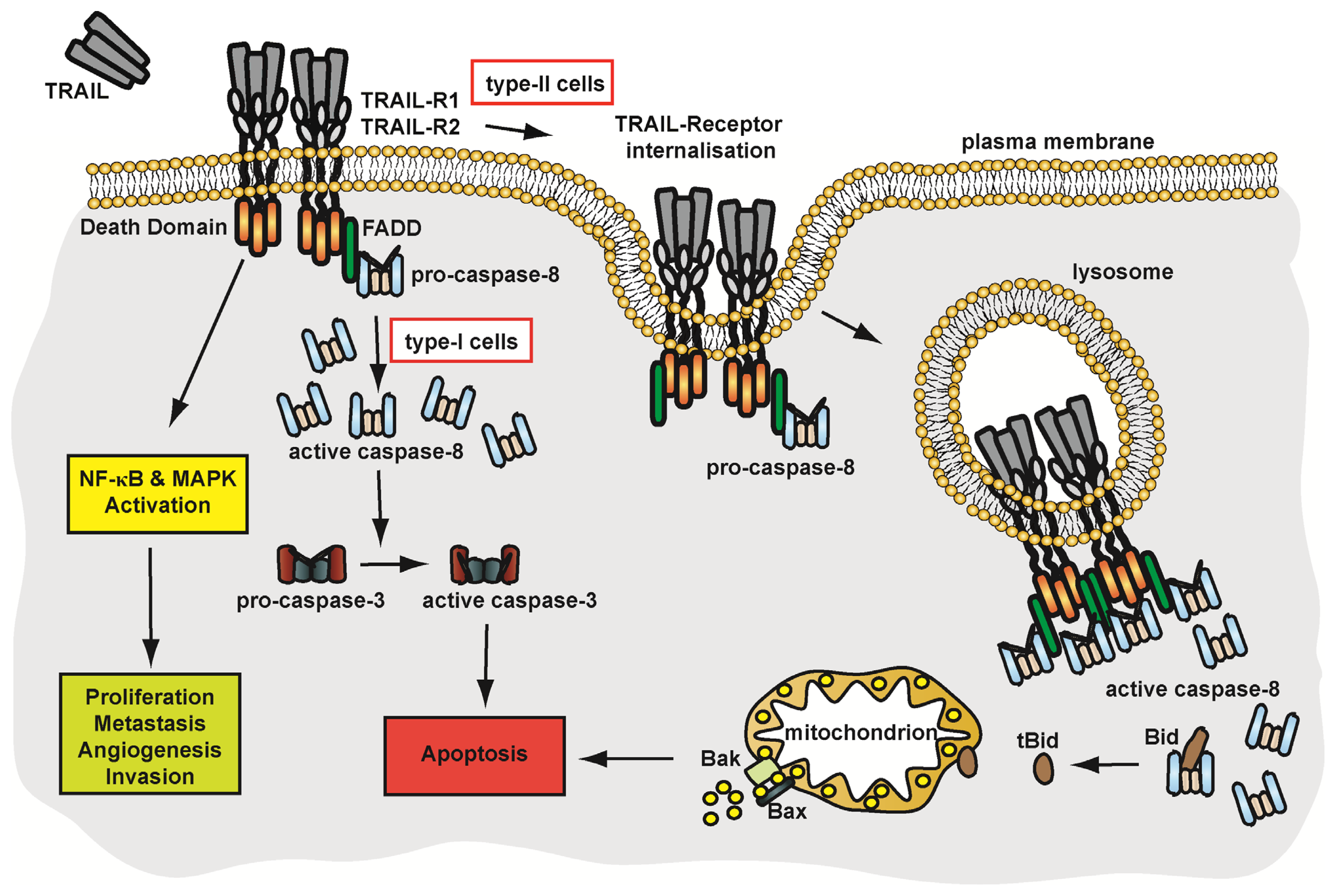
4.2.3. Membrane Trafficking and Implications on TRAIL-Receptor Signalling
4.3. The FasL/Fas System
4.3.1. Structure, Physiological and Pathophysiological Roles of the FasL/Fas System
4.3.1.1. Structure
4.3.1.2. Physiological and Pathophysiological Roles of the FasL/Fas System
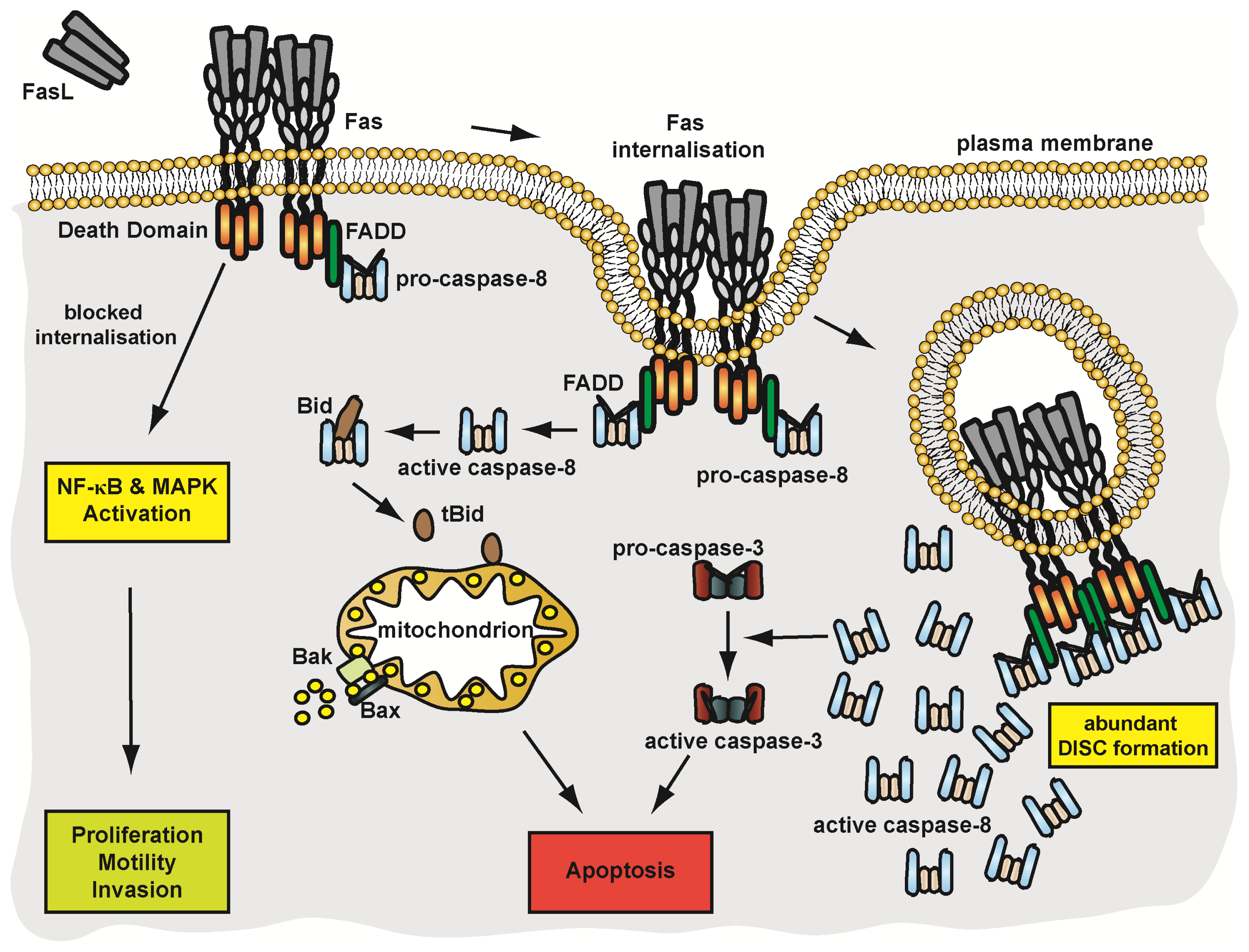
4.3.2. Pathways of Fas Signalling
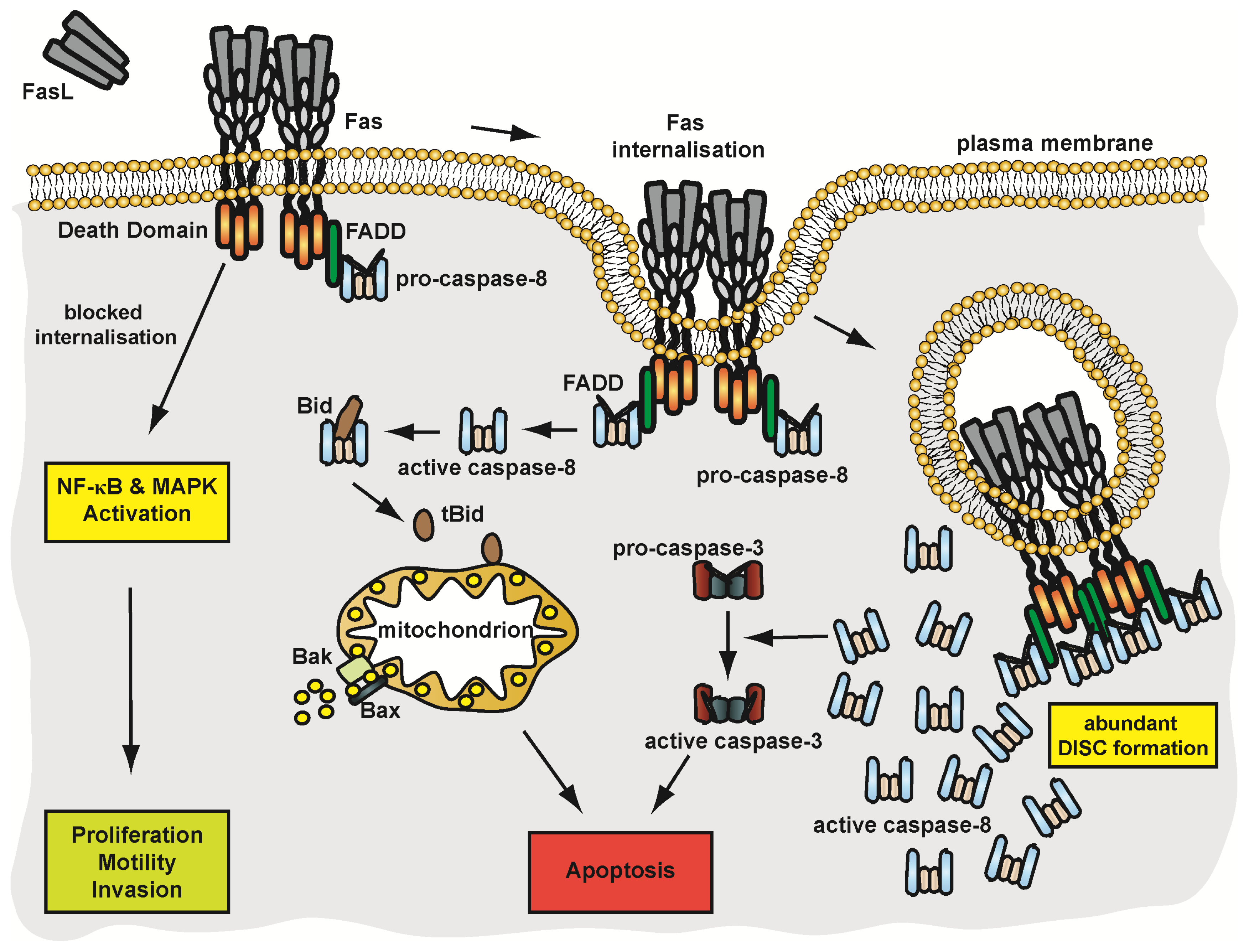
4.3.3. Membrane Trafficking and Implications on Fas Signalling
4.4. The TL1A/DR3 System
4.4.1. Structure, Physiological and Pathophysiological Roles of the TL1A/DR3 System
4.4.1.1. Structure
4.4.1.2. Physiological and Pathophysiological Roles of the TL1A/DR3 System
4.4.2. Pathways of DR3 Signalling
4.4.3. Membrane Trafficking and Implications on DR3 Signalling
5. Conclusions and Perspectives
Acknowledgments
Conflict of Interest
References
- Croft, M.; Duan, W.; Choi, H.; Eun, S.Y.; Madireddi, S.; Mehta, A. TNF superfamily in inflammatory disease: translating basic insights. Trends Immunol 2012, 33, 144–152. [Google Scholar]
- Bodmer, J.L.; Schneider, P.; Tschopp, J. The molecular architecture of the TNF superfamily. Trends Biochem. Sci 2002, 27, 19–26. [Google Scholar]
- Ware, C.F. The TNF superfamily. Cytokine Growth Factor Rev 2003, 14, 181–184. [Google Scholar]
- Wiens, G.D.; Glenney, G.W. Origin and evolution of TNF and TNF receptor superfamilies. Dev. Comp. Immunol 2011, 35, 1324–1335. [Google Scholar]
- Locksley, R.M.; Killeen, N.; Lenardo, M.J. The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell 2001, 104, 487–501. [Google Scholar]
- Wertz, I.E.; Dixit, V.M. Regulation of death receptor signaling by the ubiquitin system. Cell Death Differ 2010, 17, 14–24. [Google Scholar]
- Fesik, S.W. Insights into programmed cell death through structural biology. Cell 2000, 103, 273–282. [Google Scholar]
- Bossen, C.; Cachero, T.G.; Tardivel, A.; Ingold, K.; Willen, L.; Dobles, M.; Scott, M.L.; Maquelin, A.; Belnoue, E.; Siegrist, C. A.; et al. TACI, unlike BAFF-R, is solely activated by oligomeric BAFF and APRIL to support survival of activated B cells and plasmablasts. Blood 2008, 111, 1004–1012. [Google Scholar]
- Kreuz, S.; Siegmund, D.; Rumpf, J.J.; Samel, D.; Leverkus, M.; Janssen, O.; Hacker, G.; Dittrich-Breiholz, O.; Kracht, M.; Scheurich, P.; et al. NFkappaB activation by Fas is mediated through FADD, caspase-8, and RIP and is inhibited by FLIP. J. Cell Biol 2004, 166, 369–380. [Google Scholar]
- Trauzold, A.; Wermann, H.; Arlt, A.; Schutze, S.; Schafer, H.; Oestern, S.; Roder, C.; Ungefroren, H.; Lampe, E.; Heinrich, M.; et al. CD95 and TRAIL receptor-mediated activation of protein kinase C and NF-kappaB contributes to apoptosis resistance in ductal pancreatic adenocarcinoma cells. Oncogene 2001, 20, 4258–4269. [Google Scholar]
- Varfolomeev, E.; Vucic, D. (Un)expected roles of c-IAPs in apoptotic and NFkappaB signaling pathways. Cell Cycle 2008, 7, 1511–1521. [Google Scholar]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-kappaB signaling. Cell 2008, 132, 344–362. [Google Scholar]
- Wertz, I.E.; O’Rourke, K.M.; Zhou, H.; Eby, M.; Aravind, L.; Seshagiri, S.; Wu, P.; Wiesmann, C.; Baker, R.; Boone, D.L.; et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature 2004, 430, 694–699. [Google Scholar]
- De Valck, D.; Jin, D.Y.; Heyninck, K.; van de Craen, M.; Contreras, R.; Fiers, W.; Jeang, K.T.; Beyaert, R. The zinc finger protein A20 interacts with a novel anti-apoptotic protein which is cleaved by specific caspases. Oncogene 1999, 18, 4182–4190. [Google Scholar]
- Shembade, N.; Harhaj, N.S.; Parvatiyar, K.; Copeland, N.G.; Jenkins, N.A.; Matesic, L.E.; Harhaj, E.W. The E3 ligase Itch negatively regulates inflammatory signaling pathways by controlling the function of the ubiquitin-editing enzyme A20. Nat. Immunol 2008, 9, 254–262. [Google Scholar]
- Evans, P.C.; Taylor, E.R.; Coadwell, J.; Heyninck, K.; Beyaert, R.; Kilshaw, P.J. Isolation and characterization of two novel A20-like proteins. Biochem. J 2001, 357, 617–623. [Google Scholar]
- Wright, A.; Reiley, W.W.; Chang, M.; Jin, W.; Lee, A.J.; Zhang, M.; Sun, S.C. Regulation of early wave of germ cell apoptosis and spermatogenesis by deubiquitinating enzyme CYLD. Dev. Cell 2007, 13, 705–716. [Google Scholar]
- Peter, M.E.; Krammer, P.H. The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ 2003, 10, 26–35. [Google Scholar]
- Budd, R.C.; Yeh, W.C.; Tschopp, J. cFLIP regulation of lymphocyte activation and development. Nat. Rev. Immunol 2006, 6, 196–204. [Google Scholar]
- Meng, X.W.; Peterson, K.L.; Dai, H.; Schneider, P.; Lee, S.H.; Zhang, J.S.; Koenig, A.; Bronk, S.; Billadeau, D.D.; Gores, G.J.; et al. High cell surface death receptor expression determines type I versus type II signaling. J. Biol. Chem 2011, 286, 35823–35833. [Google Scholar]
- Jost, P.J.; Grabow, S.; Gray, D.; McKenzie, M.D.; Nachbur, U.; Huang, D.C.; Bouillet, P.; Thomas, H.E.; Borner, C.; Silke, J.; et al. XIAP discriminates between type I and type II FAS-induced apoptosis. Nature 2009, 460, 1035–1039. [Google Scholar]
- Kurita, S.; Mott, J.L.; Cazanave, S.C.; Fingas, C.D.; Guicciardi, M.E.; Bronk, S.F.; Roberts, L.R.; Fernandez-Zapico, M.E.; Gores, G.J. Hedgehog inhibition promotes a switch from Type II to Type I cell death receptor signaling in cancer cells. PLoS One 2011, 6, e18330. [Google Scholar]
- Barnhart, B.C.; Alappat, E.C.; Peter, M.E. The CD95 type I/type II model. Cell Death Differ 2003, 15, 185–193. [Google Scholar]
- Vaux, D.L.; Silke, J. Mammalian mitochondrial IAP binding proteins. Biochem. Biophys. Res. Commun 2003, 304, 499–504. [Google Scholar]
- Van de Walle, L.; Lamkanfi, M.; Vandenabeele, P. The mitochondrial serine protease HtrA2/Omi: An overview. Cell Death Differ 2008, 15, 453–460. [Google Scholar]
- Wurstle, M.L.; Laussmann, M.A.; Rehm, M. The central role of initiator caspase-9 in apoptosis signal transduction and the regulation of its activation and activity on the apoptosome. Exp. Cell. Res 2012, 318, 1213–1220. [Google Scholar]
- Kriegler, M.; Perez, C.; DeFay, K.; Albert, I.; Lu, S.D. A novel form of TNF/cachectin is a cell surface cytotoxic transmembrane protein: Ramifications for the complex physiology of TNF. Cell 1988, 53, 45–53. [Google Scholar]
- Tang, P.; Hung, M.C.; Klostergaard, J. Human pro-tumor necrosis factor is a homotrimer. Biochemistry 1996, 35, 8216–8225. [Google Scholar]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 1997, 385, 729–733. [Google Scholar]
- Dembic, Z.; Loetscher, H.; Gubler, U.; Pan, Y.C.; Lahm, H.W.; Gentz, R.; Brockhaus, M.; Lesslauer, W. Two human TNF receptors have similar extracellular, but distinct intracellular, domain sequences. Cytokine 1990, 2, 231–237. [Google Scholar]
- Wajant, H.; Pfizenmaier, K.; Scheurich, P. Tumor necrosis factor signaling. Cell Death Differ 2003, 10, 45–65. [Google Scholar]
- Schneider-Brachert, W.; Tchikov, V.; Neumeyer, J.; Jakob, M.; Winoto-Morbach, S.; Held-Feindt, J.; Heinrich, M.; Merkel, O.; Ehrenschwender, M.; Adam, D.; et al. Compartmentalization of TNF receptor 1 signaling: Internalized TNF receptosomes as death signaling vesicles. Immunity 2004, 21, 415–428. [Google Scholar]
- Adam, D.; Wiegmann, K.; Adam-Klages, S.; Ruff, A.; Kronke, M. A novel cytoplasmic domain of the p55 tumor necrosis factor receptor initiates the neutral sphingomyelinase pathway. J. Biol. Chem 1996, 271, 14617–14622. [Google Scholar]
- Sethi, G.; Sung, B.; Kunnumakkara, A.B.; Aggarwal, B.B. Targeting TNF for treatment of cancer and autoimmunity. Adv. Exp. Med. Biol 2009, 647, 37–51. [Google Scholar]
- Van Hauwermeiren, F.; Vandenbroucke, R.E.; Libert, C. Treatment of TNF mediated diseases by selective inhibition of soluble TNF or TNFR1. Cytokine Growth Factor Rev 2011, 22, 311–319. [Google Scholar]
- Leibovich, S.J.; Polverini, P.J.; Shepard, H.M.; Wiseman, D.M.; Shively, V.; Nuseir, N. Macrophage-induced angiogenesis is mediated by tumour necrosis factor-alpha. Nature 1987, 329, 630–632. [Google Scholar]
- Moore, R.J.; Owens, D.M.; Stamp, G.; Arnott, C.; Burke, F.; East, N.; Holdsworth, H.; Turner, L.; Rollins, B.; Pasparakis, M.; et al. Mice deficient in tumor necrosis factor-alpha are resistant to skin carcinogenesis. Nat. Med 1999, 5, 828–831. [Google Scholar]
- Arnott, C.H.; Scott, K.A.; Moore, R.J.; Robinson, S.C.; Thompson, R.G.; Balkwill, F.R. Expression of both TNF-alpha receptor subtypes is essential for optimal skin tumour development. Oncogene 2004, 23, 1902–1910. [Google Scholar]
- Tomita, Y.; Yang, X.; Ishida, Y.; Nemoto-Sasaki, Y.; Kondo, T.; Oda, M.; Watanabe, G.; Chaldakov, G.N.; Fujii, C.; Mukaida, N. Spontaneous regression of lung metastasis in the absence of tumor necrosis factor receptor p55. Int. J. Cancer 2004, 112, 927–933. [Google Scholar]
- Kitakata, H.; Nemoto-Sasaki, Y.; Takahashi, Y.; Kondo, T.; Mai, M.; Mukaida, N. Essential roles of tumor necrosis factor receptor p55 in liver metastasis of intrasplenic administration of colon 26 cells. Cancer Res 2002, 62, 6682–6687. [Google Scholar]
- Malik, S.T.; Naylor, M.S.; East, N.; Oliff, A.; Balkwill, F.R. Cells secreting tumour necrosis factor show enhanced metastasis in nude mice. Eur. J. Cancer 1990, 26, 1031–1034. [Google Scholar]
- Orosz, P.; Echtenacher, B.; Falk, W.; Ruschoff, J.; Weber, D.; Mannel, D.N. Enhancement of experimental metastasis by tumor necrosis factor. J. Exp. Med 1993, 177, 1391–1398. [Google Scholar]
- Popivanova, B.K.; Kitamura, K.; Wu, Y.; Kondo, T.; Kagaya, T.; Kaneko, S.; Oshima, M.; Fujii, C.; Mukaida, N. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J. Clin. Invest 2008, 118, 560–570. [Google Scholar]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar]
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004, 118, 285–296. [Google Scholar]
- Rothe, J.; Lesslauer, W.; Lotscher, H.; Lang, Y.; Koebel, P.; Kontgen, F.; Althage, A.; Zinkernagel, R.; Steinmetz, M.; Bluethmann, H. Mice lacking the tumour necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature 1993, 364, 798–802. [Google Scholar]
- Flynn, J.L.; Goldstein, M.M.; Chan, J.; Triebold, K.J.; Pfeffer, K.; Lowenstein, C.J.; Schreiber, R.; Mak, T.W.; Bloom, B.R. Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity 1995, 2, 561–572. [Google Scholar]
- Silke, J. The regulation of TNF signalling: What a tangled web we weave. Curr. Opin. Immunol 2011, 23, 620–626. [Google Scholar]
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar]
- Harper, N.; Hughes, M.; MacFarlane, M.; Cohen, G.M. Fas-associated death domain protein and caspase-8 are not recruited to the tumor necrosis factor receptor 1 signaling complex during tumor necrosis factor-induced apoptosis. J. Biol. Chem 2003, 278, 25534–25541. [Google Scholar]
- Legler, D.F.; Micheau, O.; Doucey, M.A.; Tschopp, J.; Bron, C. Recruitment of TNF receptor 1 to lipid rafts is essential for TNFalpha-mediated NF-kappaB activation. Immunity 2003, 18, 655–664. [Google Scholar]
- Li, H.; Kobayashi, M.; Blonska, M.; You, Y.; Lin, X. Ubiquitination of RIP is required for tumor necrosis factor alpha-induced NF-kappaB activation. J. Biol. Chem 2006, 281, 13636–13643. [Google Scholar]
- Ea, C.K.; Deng, L.; Xia, Z.P.; Pineda, G.; Chen, Z.J. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol. Cell 2006, 22, 245–257. [Google Scholar]
- Tang, G.; Minemoto, Y.; Dibling, B.; Purcell, N.H.; Li, Z.; Karin, M.; Lin, A. Inhibition of JNK activation through NF-kappaB target genes. Nature 2001, 414, 313–317. [Google Scholar]
- Bradley, J.R.; Johnson, D.R.; Pober, J.S. Four different classes of inhibitors of receptor-mediated endocytosis decrease tumor necrosis factor-induced gene expression in human endothelial cells. J. Immunol 1993, 150, 5544–5555. [Google Scholar]
- Schutze, S.; Machleidt, T.; Adam, D.; Schwandner, R.; Wiegmann, K.; Kruse, M.L.; Heinrich, M.; Wickel, M.; Kronke, M. Inhibition of receptor internalization by monodansylcadaverine selectively blocks p55 tumor necrosis factor receptor death domain signaling. J. Biol. Chem 1999, 274, 10203–10212. [Google Scholar]
- Schneider-Brachert, W.; Tchikov, V.; Merkel, O.; Jakob, M.; Hallas, C.; Kruse, M.L.; Groitl, P.; Lehn, A.; Hildt, E.; Held-Feindt, J.; et al. Inhibition of TNF receptor 1 internalization by adenovirus 14.7K as a novel immune escape mechanism. J. Clin. Invest 2006, 116, 2901–2913. [Google Scholar]
- Mahul-Mellier, A.L.; Strappazzon, F.; Petiot, A.; Chatellard-Causse, C.; Torch, S.; Blot, B.; Freeman, K.; Kuhn, L.; Garin, J.; Verna, J.M.; et al. Alix and ALG-2 are involved in tumor necrosis factor receptor 1-induced cell death. J. Biol. Chem 2008, 283, 34954–34965. [Google Scholar]
- Edelmann, B.; Bertsch, U.; Tchikov, V.; Winoto-Morbach, S.; Perrotta, C.; Jakob, M.; Adam-Klages, S.; Kabelitz, D.; Schutze, S. Caspase-8 and caspase-7 sequentially mediate proteolytic activation of acid sphingomyelinase in TNF-R1 receptosomes. EMBO J 2011, 30, 379–394. [Google Scholar]
- Heinrich, M.; Neumeyer, J.; Jakob, M.; Hallas, C.; Tchikov, V.; Winoto-Morbach, S.; Wickel, M.; Schneider-Brachert, W.; Trauzold, A.; Hethke, A.; et al. Cathepsin D links TNF-induced acid sphingomyelinase to Bid-mediated caspase-9 and -3 activation. Cell Death Differ 2004, 11, 550–563. [Google Scholar]
- Yazdanpanah, B.; Wiegmann, K.; Tchikov, V.; Krut, O.; Pongratz, C.; Schramm, M.; Kleinridders, A.; Wunderlich, T.; Kashkar, H.; Utermohlen, O.; et al. Riboflavin kinase couples TNF receptor 1 to NADPH oxidase. Nature 2009, 460, 1159–1163. [Google Scholar]
- Liao, W.; Xiao, Q.; Tchikov, V.; Fujita, K.; Yang, W.; Wincovitch, S.; Garfield, S.; Conze, D.; El-Deiry, W.S.; Schutze, S.; et al. CARP-2 is an endosome-associated ubiquitin ligase for RIP and regulates TNF-induced NF-kappaB activation. Curr. Biol 2008, 18, 641–649. [Google Scholar]
- Wiley, S.R.; Schooley, K.; Smolak, P.J.; Din, W.S.; Huang, C.P.; Nicholl, J.K.; Sutherland, G.R.; Smith, T.D.; Rauch, C.; Smith, C.A.; et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 1995, 3, 673–682. [Google Scholar]
- Falschlehner, C.; Ganten, T.M.; Koschny, R.; Schaefer, U.; Walczak, H. TRAIL and other TRAIL receptor agonists as novel cancer therapeutics. Adv. Exp. Med. Biol 2009, 647, 195–206. [Google Scholar]
- Pan, G.; O’Rourke, K.; Chinnaiyan, A.M.; Gentz, R.; Ebner, R.; Ni, J.; Dixit, V.M. The receptor for the cytotoxic ligand TRAIL. Science 1997, 276, 111–113. [Google Scholar]
- Walczak, H.; Degli-Esposti, M.A.; Johnson, R.S.; Smolak, P.J.; Waugh, J.Y.; Boiani, N.; Timour, M.S.; Gerhart, M.J.; Schooley, K.A.; Smith, C.A.; et al. TRAIL-R2: A novel apoptosis-mediating receptor for TRAIL. EMBO J 1997, 16, 5386–5397. [Google Scholar]
- Neumann, S.; Bidon, T.; Branschadel, M.; Krippner-Heidenreich, A.; Scheurich, P.; Doszczak, M. The transmembrane domains of TNF-related apoptosis-inducing ligand (TRAIL) receptors 1 and 2 co-regulate apoptotic signaling capacity. PLoS One 2012, 7, e42526. [Google Scholar]
- Wu, G.S.; Burns, T.F.; Zhan, Y.; Alnemri, E.S.; El-Deiry, W.S. Molecular cloning and functional analysis of the mouse homologue of the KILLER/DR5 tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) death receptor. Cancer Res 1999, 59, 2770–2775. [Google Scholar]
- Cretney, E.; Takeda, K.; Yagita, H.; Glaccum, M.; Peschon, J.J.; Smyth, M.J. Increased susceptibility to tumor initiation and metastasis in TNF-related apoptosis-inducing ligand-deficient mice. J. Immunol 2002, 168, 1356–1361. [Google Scholar]
- Sedger, L.M.; Glaccum, M.B.; Schuh, J.C.; Kanaly, S.T.; Williamson, E.; Kayagaki, N.; Yun, T.; Smolak, P.; Le, T.; Goodwin, R.; et al. Characterization of the in vivo function of TNF-alpha-related apoptosis-inducing ligand, TRAIL/Apo2L, using TRAIL/Apo2L gene-deficient mice. Eur. J. Immunol 2002, 32, 2246–2254. [Google Scholar]
- Nitsch, R.; Bechmann, I.; Deisz, R.A.; Haas, D.; Lehmann, T.N.; Wendling, U.; Zipp, F. Human brain-cell death induced by tumour-necrosis-factor-related apoptosis-inducing ligand (TRAIL). Lancet 2000, 356, 827–828. [Google Scholar]
- Gerspach, J.; Pfizenmaier, K.; Wajant, H. Therapeutic targeting of CD95 and the TRAIL death receptors. Recent Pat. Anticancer Drug Discov 2011, 6, 294–310. [Google Scholar]
- Trauzold, A.; Siegmund, D.; Schniewind, B.; Sipos, B.; Egberts, J.; Zorenkov, D.; Emme, D.; Roder, C.; Kalthoff, H.; Wajant, H. TRAIL promotes metastasis of human pancreatic ductal adenocarcinoma. Oncogene 2006, 25, 7434–7439. [Google Scholar]
- Ehrenschwender, M.; Siegmund, D.; Wicovsky, A.; Kracht, M.; Dittrich-Breiholz, O.; Spindler, V.; Waschke, J.; Kalthoff, H.; Trauzold, A.; Wajant, H. Mutant PIK3CA licenses TRAIL and CD95L to induce non-apoptotic caspase-8-mediated ROCK activation. Cell Death Differ 2010, 17, 1435–1447. [Google Scholar]
- Steinwede, K.; Henken, S.; Bohling, J.; Maus, R.; Ueberberg, B.; Brumshagen, C.; Brincks, E.L.; Griffith, T.S.; Welte, T.; Maus, U.A. TNF-related apoptosis-inducing ligand (TRAIL) exerts therapeutic efficacy for the treatment of pneumococcal pneumonia in mice. J. Exp. Med 2012, 209, 1937–1952. [Google Scholar]
- Hameed, A.G.; Arnold, N.D.; Chamberlain, J.; Pickworth, J.A.; Paiva, C.; Dawson, S.; Cross, S.; Long, L.; Zhao, L.; Morrell, N.W.; et al. Inhibition of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) reverses experimental pulmonary hypertension. J. Exp. Med 2012, 209, 1919–1935. [Google Scholar]
- Benedict, C.A.; Ware, C.F. TRAIL: Not just for tumors anymore? J. Exp. Med 2012, 209, 1903–1906. [Google Scholar]
- Kelley, R.F.; Totpal, K.; Lindstrom, S.H.; Mathieu, M.; Billeci, K.; Deforge, L.; Pai, R.; Hymowitz, S.G.; Ashkenazi, A. Receptor-selective mutants of apoptosis-inducing ligand 2/tumor necrosis factor-related apoptosis-inducing ligand reveal a greater contribution of death receptor (DR) 5 than DR4 to apoptosis signaling. J. Biol. Chem 2005, 280, 2205–2212. [Google Scholar]
- Lalaoui, N.; Morle, A.; Merino, D.; Jacquemin, G.; Iessi, E.; Morizot, A.; Shirley, S.; Robert, B.; Solary, E.; Garrido, C.; et al. TRAIL-R4 promotes tumor growth and resistance to apoptosis in cervical carcinoma HeLa cells through AKT. PLoS One 2011, 6, e19679. [Google Scholar]
- Shirley, S.; Morizot, A.; Micheau, O. Regulating TRAIL receptor-induced cell death at the membrane: A deadly discussion. Recent Pat. Anticancer Drug Discov 2011, 6, 311–323. [Google Scholar]
- Guicciardi, M.E.; Gores, G.J. Life and death by death receptors. FASEB J 2009, 23, 1625–1637. [Google Scholar]
- Hu, W.H.; Johnson, H.; Shu, H.B. Tumor necrosis factor-related apoptosis-inducing ligand receptors signal NF-kappaB and JNK activation and apoptosis through distinct pathways. J. Biol. Chem 1999, 274, 30603–30610. [Google Scholar]
- Kim, Y.S.; Schwabe, R.F.; Qian, T.; Lemasters, J.J.; Brenner, D.A. TRAIL-mediated apoptosis requires NF-kappaB inhibition and the mitochondrial permeability transition in human hepatoma cells. Hepatology 2002, 36, 1498–1508. [Google Scholar]
- Austin, C.D.; Lawrence, D.A.; Peden, A.A.; Varfolomeev, E.E.; Totpal, K.; de Maziere, A.M.; Klumperman, J.; Arnott, D.; Pham, V.; Scheller, R.H.; et al. Death-receptor activation halts clathrin-dependent endocytosis. Proc. Natl. Acad. Sci. USA 2006, 103, 10283–10288. [Google Scholar]
- Kohlhaas, S.L.; Craxton, A.; Sun, X.M.; Pinkoski, M.J.; Cohen, G.M. Receptor-mediated endocytosis is not required for tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis. J. Biol. Chem 2007, 282, 12831–12841. [Google Scholar]
- Akazawa, Y.; Mott, J.L.; Bronk, S.F.; Werneburg, N.W.; Kahraman, A.; Guicciardi, M.E.; Meng, X.W.; Kohno, S.; Shah, V.H.; Kaufmann, S.H.; et al. Death receptor 5 internalization is required for lysosomal permeabilization by TRAIL in malignant liver cell lines. Gastroenterology 2009, 136, 2365–2376. [Google Scholar]
- Takahashi, T.; Tanaka, M.; Inazawa, J.; Abe, T.; Suda, T.; Nagata, S. Human Fas ligand: Gene structure, chromosomal location and species specificity. Int. Immunol 1994, 6, 1567–1574. [Google Scholar]
- Janssen, O.; Qian, J.; Linkermann, A.; Kabelitz, D. CD95 ligand—Death factor and costimulatory molecule? Cell Death Differ 2003, 10, 1215–1225. [Google Scholar]
- Blott, E.J.; Bossi, G.; Clark, R.; Zvelebil, M.; Griffiths, G.M. Fas ligand is targeted to secretory lysosomes via a proline-rich domain in its cytoplasmic tail. J. Cell Sci 2001, 114, 2405–2416. [Google Scholar]
- Tanaka, M.; Itai, T.; Adachi, M.; Nagata, S. Downregulation of Fas ligand by shedding. Nat. Med 1998, 4, 31–36. [Google Scholar]
- Mitsiades, N.; Yu, W.H.; Poulaki, V.; Tsokos, M.; Stamenkovic, I. Matrix metalloproteinase-7-mediated cleavage of Fas ligand protects tumor cells from chemotherapeutic drug cytotoxicity. Cancer Res 2001, 61, 577–581. [Google Scholar]
- Kayagaki, N.; Kawasaki, A.; Ebata, T.; Ohmoto, H.; Ikeda, S.; Inoue, S.; Yoshino, K.; Okumura, K.; Yagita, H. Metalloproteinase-mediated release of human Fas ligand. J. Exp. Med 1995, 182, 1777–1783. [Google Scholar]
- Cheng, J.; Liu, C.; Koopman, W.J.; Mountz, J.D. Characterization of human Fas gene. Exon/intron organization and promoter region. J. Immunol 1995, 154, 1239–1245. [Google Scholar]
- Wajant, H.; Pfizenmaier, K.; Scheurich, P. Non-apoptotic Fas signaling. Cytokine Growth Factor Rev 2003, 14, 53–66. [Google Scholar]
- Starling, G.C.; Bajorath, J.; Emswiler, J.; Ledbetter, J.A.; Aruffo, A.; Kiener, P.A. Identification of amino acid residues important for ligand binding to Fas. J. Exp. Med 1997, 185, 1487–1492. [Google Scholar]
- Edmond, V.; Ghali, B.; Penna, A.; Taupin, J.L.; Daburon, S.; Moreau, J.F.; Legembre, P. Precise mapping of the CD95 pre-ligand assembly domain. PLoS One 2012, 7, e46236. [Google Scholar]
- Cohen, P.L.; Eisenberg, R.A. Lpr and gld: Single gene models of systemic autoimmunity and lymphoproliferative disease. Annu. Rev. Immunol 1991, 9, 243–269. [Google Scholar]
- Bouillet, P.; O’Reilly, L.A. CD95, BIM and T cell homeostasis. Nat. Rev. Immunol 2009, 9, 514–519. [Google Scholar]
- Ferguson, T.A.; Griffith, T.S. A vision of cell death: Fas ligand and immune privilege 10 years later. Immunol. Rev 2006, 213, 228–238. [Google Scholar]
- Cullen, S.P.; Henry, C.M.; Kearney, C.J.; Logue, S.E.; Feoktistova, M.; Tynan, G.A.; Lavelle, E.C.; Leverkus, M.; Martin, S.J. Fas/CD95-induced chemokines can serve as “find-me” signals for apoptotic cells. Mol. Cell 2013, 49, 1034–1048. [Google Scholar]
- Strasser, A.; Jost, P.J.; Nagata, S. The many roles of FAS receptor signaling in the immune system. Immunity 2009, 30, 180–192. [Google Scholar]
- Kleber, S.; Sancho-Martinez, I.; Wiestler, B.; Beisel, A.; Gieffers, C.; Hill, O.; Thiemann, M.; Mueller, W.; Sykora, J.; Kuhn, A.; et al. Yes and PI3K bind CD95 to signal invasion of glioblastoma. Cancer Cell 2008, 13, 235–248. [Google Scholar]
- Steller, E.J.; Borel Rinkes, I.H.; Kranenburg, O. How CD95 stimulates invasion. Cell Cycle 2011, 10, 3857–3862. [Google Scholar]
- Steller, E.J.; Ritsma, L.; Raats, D.A.; Hoogwater, F.J.; Emmink, B.L.; Govaert, K.M.; Laoukili, J.; Rinkes, I.H.; van Rheenen, J.; Kranenburg, O. The death receptor CD95 activates the cofilin pathway to stimulate tumour cell invasion. EMBO Rep 2011, 12, 931–937. [Google Scholar]
- Chen, L.; Park, S.M.; Tumanov, A.V.; Hau, A.; Sawada, K.; Feig, C.; Turner, J.R.; Fu, Y.X.; Romero, I.L.; Lengyel, E.; et al. CD95 promotes tumour growth. Nature 2010, 465, 492–496. [Google Scholar]
- Ehrenschwender, M.; Wajant, H. The role of FasL and Fas in health and disease. Adv. Exp. Med. Biol 2009, 647, 64–93. [Google Scholar]
- Schneider, P.; Holler, N.; Bodmer, J.L.; Hahne, M.; Frei, K.; Fontana, A.; Tschopp, J. Conversion of membrane-bound Fas(CD95) ligand to its soluble form is associated with downregulation of its proapoptotic activity and loss of liver toxicity. J. Exp. Med 1998, 187, 1205–1213. [Google Scholar]
- Lee, K.H.; Feig, C.; Tchikov, V.; Schickel, R.; Hallas, C.; Schutze, S.; Peter, M.E.; Chan, A.C. The role of receptor internalization in CD95 signaling. EMBO J 2006, 25, 1009–1023. [Google Scholar]
- Chinnaiyan, A.M.; O’Rourke, K.; Tewari, M.; Dixit, V.M. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell 1995, 81, 505–512. [Google Scholar]
- Muzio, M.; Chinnaiyan, A.M.; Kischkel, F.C.; O’Rourke, K.; Shevchenko, A.; Ni, J.; Scaffidi, C.; Bretz, J.D.; Zhang, M.; Gentz, R.; et al. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death-inducing signaling complex. Cell 1996, 85, 817–827. [Google Scholar]
- Donepudi, M.; Mac Sweeney, A.; Briand, C.; Grutter, M.G. Insights into the regulatory mechanism for caspase-8 activation. Mol. Cell 2003, 11, 543–549. [Google Scholar]
- Cardone, M.H.; Salvesen, G.S.; Widmann, C.; Johnson, G.; Frisch, S.M. The regulation of anoikis: MEKK-1 activation requires cleavage by caspases. Cell 1997, 90, 315–323. [Google Scholar]
- Parlato, S.; Giammarioli, A.M.; Logozzi, M.; Lozupone, F.; Matarrese, P.; Luciani, F.; Falchi, M.; Malorni, W.; Fais, S. CD95 (APO-1/Fas) linkage to the actin cytoskeleton through ezrin in human T lymphocytes: A novel regulatory mechanism of the CD95 apoptotic pathway. EMBO J 2000, 19, 5123–5134. [Google Scholar]
- Algeciras-Schimnich, A.; Shen, L.; Barnhart, B.C.; Murmann, A.E.; Burkhardt, J.K.; Peter, M.E. Molecular ordering of the initial signaling events of CD95. Mol. Cell Biol 2002, 22, 207–220. [Google Scholar]
- Feig, C.; Tchikov, V.; Schutze, S.; Peter, M.E. Palmitoylation of CD95 facilitates formation of SDS-stable receptor aggregates that initiate apoptosis signaling. EMBO J 2007, 26, 221–231. [Google Scholar]
- Chakrabandhu, K.; Huault, S.; Garmy, N.; Fantini, J.; Stebe, E.; Mailfert, S.; Marguet, D.; Hueber, A.O. The extracellular glycosphingolipid-binding motif of Fas defines its internalization route, mode and outcome of signals upon activation by ligand. Cell Death Differ 2008, 15, 1824–1837. [Google Scholar]
- Chakrabandhu, K.; Herincs, Z.; Huault, S.; Dost, B.; Peng, L.; Conchonaud, F.; Marguet, D.; He, H.T.; Hueber, A.O. Palmitoylation is required for efficient Fas cell death signaling. EMBO J 2007, 26, 209–220. [Google Scholar]
- Anathy, V.; Aesif, S.W.; Guala, A.S.; Havermans, M.; Reynaert, N.L.; Ho, Y.S.; Budd, R.C.; Janssen-Heininger, Y.M. Redox amplification of apoptosis by caspase-dependent cleavage of glutaredoxin 1 and S-glutathionylation of Fas. J. Cell Biol 2009, 184, 241–252. [Google Scholar]
- Leon-Bollotte, L.; Subramaniam, S.; Cauvard, O.; Plenchette-Colas, S.; Paul, C.; Godard, C.; Martinez-Ruiz, A.; Legembre, P.; Jeannin, J.F.; Bettaieb, A. S-nitrosylation of the death receptor fas promotes fas ligand-mediated apoptosis in cancer cells. Gastroenterology 2011, 140, 2009–2018. [Google Scholar]
- Shatnyeva, O.M.; Kubarenko, A.V.; Weber, C.E.; Pappa, A.; Schwartz-Albiez, R.; Weber, A.N.; Krammer, P.H.; Lavrik, I.N. Modulation of the CD95-induced apoptosis: The role of CD95 N-glycosylation. PLoS One 2011, 6, e19927. [Google Scholar]
- Zuccato, E.; Blott, E.J.; Holt, O.; Sigismund, S.; Shaw, M.; Bossi, G.; Griffiths, G.M. Sorting of Fas ligand to secretory lysosomes is regulated by mono-ubiquitylation and phosphorylation. J. Cell Sci 2007, 120, 191–199. [Google Scholar]
- Migone, T.S.; Zhang, J.; Luo, X.; Zhuang, L.; Chen, C.; Hu, B.; Hong, J.S.; Perry, J.W.; Chen, S.F.; Zhou, J.X.; et al. TL1A is a TNF-like ligand for DR3 and TR6/DcR3 and functions as a T cell costimulator. Immunity 2002, 16, 479–492. [Google Scholar]
- Marsters, S.A.; Sheridan, J.P.; Donahue, C.J.; Pitti, R.M.; Gray, C.L.; Goddard, A.D.; Bauer, K.D.; Ashkenazi, A. Apo-3, a new member of the tumor necrosis factor receptor family, contains a death domain and activates apoptosis and NF-kappa B. Curr. Biol 1996, 6, 1669–1676. [Google Scholar]
- Bodmer, J.L.; Burns, K.; Schneider, P.; Hofmann, K.; Steiner, V.; Thome, M.; Bornand, T.; Hahne, M.; Schroter, M.; Becker, K.; et al. TRAMP, a novel apoptosis-mediating receptor with sequence homology to tumor necrosis factor receptor 1 and Fas(Apo-1/CD95). Immunity 1997, 6, 79–88. [Google Scholar]
- Screaton, G.R.; Xu, X.N.; Olsen, A.L.; Cowper, A.E.; Tan, R.; McMichael, A.J.; Bell, J.I. LARD: A new lymphoid-specific death domain containing receptor regulated by alternative pre-mRNA splicing. Proc. Natl. Acad. Sci. USA 1997, 94, 4615–4619. [Google Scholar]
- Kitson, J.; Raven, T.; Jiang, Y.P.; Goeddel, D.V.; Giles, K.M.; Pun, K.T.; Grinham, C.J.; Brown, R.; Farrow, S.N. A death-domain-containing receptor that mediates apoptosis. Nature 1996, 384, 372–375. [Google Scholar]
- Borysenko, C.W.; Furey, W.F.; Blair, H.C. Comparative modeling of TNFRSF25 (DR3) predicts receptor destabilization by a mutation linked to rheumatoid arthritis. Biochem. Biophys. Res. Commun 2005, 328, 794–799. [Google Scholar]
- Marsters, S.A.; Sheridan, J.P.; Pitti, R.M.; Brush, J.; Goddard, A.; Ashkenazi, A. Identification of a ligand for the death-domain-containing receptor Apo3. Curr. Biol 1998, 8, 525–528. [Google Scholar]
- Kaptein, A.; Jansen, M.; Dilaver, G.; Kitson, J.; Dash, L.; Wang, E.; Owen, M.J.; Bodmer, J.L.; Tschopp, J.; Farrow, S.N. Studies on the interaction between TWEAK and the death receptor WSL-1/TRAMP (DR3). FEBS Lett 2000, 485, 135–141. [Google Scholar]
- Wiley, S.R.; Cassiano, L.; Lofton, T.; Davis-Smith, T.; Winkles, J.A.; Lindner, V.; Liu, H.; Daniel, T.O.; Smith, C.A.; Fanslow, W.C. A novel TNF receptor family member binds TWEAK and is implicated in angiogenesis. Immunity 2001, 15, 837–846. [Google Scholar]
- Gout, S.; Morin, C.; Houle, F.; Huot, J. Death receptor-3, a new E-Selectin counter-receptor that confers migration and survival advantages to colon carcinoma cells by triggering p38 and ERK MAPK activation. Cancer Res 2006, 66, 9117–9124. [Google Scholar]
- Porquet, N.; Poirier, A.; Houle, F.; Pin, A.L.; Gout, S.; Tremblay, P.L.; Paquet, E.R.; Klinck, R.; Auger, F.A.; Huot, J. Survival advantages conferred to colon cancer cells by E-selectin-induced activation of the PI3K-NFkappaB survival axis downstream of Death receptor-3. BMC Cancer 2011, 11, 285. [Google Scholar]
- McLaren, J.E.; Calder, C.J.; McSharry, B.P.; Sexton, K.; Salter, R.C.; Singh, N.N.; Wilkinson, G.W.; Wang, E.C.; Ramji, D.P. The TNF-like protein 1A-death receptor 3 pathway promotes macrophage foam cell formation in vitro. J. Immunol 2010, 184, 5827–5834. [Google Scholar]
- Tan, K.B.; Harrop, J.; Reddy, M.; Young, P.; Terrett, J.; Emery, J.; Moore, G.; Truneh, A. Characterization of a novel TNF-like ligand and recently described TNF ligand and TNF receptor superfamily genes and their constitutive and inducible expression in hematopoietic and non-hematopoietic cells. Gene 1997, 204, 35–46. [Google Scholar]
- Buchan, S.L.; Taraban, V.Y.; Slebioda, T.J.; James, S.; Cunningham, A.F.; Al-Shamkhani, A. Death receptor 3 is essential for generating optimal protective CD4(+) T-cell immunity against Salmonella. Eur. J. Immunol 2012, 42, 580–588. [Google Scholar]
- Meylan, F.; Richard, A.C.; Siegel, R.M. TL1A and DR3, a TNF family ligand-receptor pair that promotes lymphocyte costimulation, mucosal hyperplasia, and autoimmune inflammation. Immunol. Rev 2011, 244, 188–196. [Google Scholar]
- Wang, E.C.; Thern, A.; Denzel, A.; Kitson, J.; Farrow, S.N.; Owen, M.J. DR3 regulates negative selection during thymocyte development. Mol. Cell Biol 2001, 21, 3451–3461. [Google Scholar]
- Meylan, F.; Davidson, T.S.; Kahle, E.; Kinder, M.; Acharya, K.; Jankovic, D.; Bundoc, V.; Hodges, M.; Shevach, E.M.; Keane-Myers, A.; et al. The TNF-family receptor DR3 is essential for diverse T cell-mediated inflammatory diseases. Immunity 2008, 29, 79–89. [Google Scholar]
- Pappu, B.P.; Borodovsky, A.; Zheng, T.S.; Yang, X.; Wu, P.; Dong, X.; Weng, S.; Browning, B.; Scott, M.L.; Ma, L.; et al. TL1A-DR3 interaction regulates Th17 cell function and Th17-mediated autoimmune disease. J. Exp. Med 2008, 205, 1049–1062. [Google Scholar]
- Zhang, J.; Wang, X.; Fahmi, H.; Wojcik, S.; Fikes, J.; Yu, Y.; Wu, J.; Luo, H. Role of TL1A in the pathogenesis of rheumatoid arthritis. J. Immunol 2009, 183, 5350–5357. [Google Scholar]
- Bamias, G.; Mishina, M.; Nyce, M.; Ross, W.G.; Kollias, G.; Rivera-Nieves, J.; Pizarro, T.T.; Cominelli, F. Role of TL1A and its receptor DR3 in two models of chronic murine ileitis. Proc. Natl. Acad. Sci. USA 2006, 103, 8441–8446. [Google Scholar]
- Bamias, G.; Martin, C., III; Marini, M.; Hoang, S.; Mishina, M.; Ross, W.G.; Sachedina, M.A.; Friel, C.M.; Mize, J.; Bickston, S.J.; et al. Expression, localization, and functional activity of TL1A, a novel Th1-polarizing cytokine in inflammatory bowel disease. J. Immunol. 2003, 171, 4868–4874. [Google Scholar]
- Meylan, F.; Song, Y.J.; Fuss, I.; Villarreal, S.; Kahle, E.; Malm, I.J.; Acharya, K.; Ramos, H.L.; Lo, L.; Mentink-Kane, M.M.; et al. The TNF-family cytokine TL1A drives IL-13-dependent small intestinal inflammation. Mucosal Immunol 2011, 4, 172–185. [Google Scholar]
- Takedatsu, H.; Michelsen, K.S.; Wei, B.; Landers, C.J.; Thomas, L.S.; Dhall, D.; Braun, J.; Targan, S.R. TL1A (TNFSF15) regulates the development of chronic colitis by modulating both T-helper 1 and T-helper 17 activation. Gastroenterology 2008, 135, 552–567. [Google Scholar]
- Murtaza, I.; Adhami, V.M.; Hafeez, B.B.; Saleem, M.; Mukhtar, H. Fisetin, a natural flavonoid, targets chemoresistant human pancreatic cancer AsPC-1 cells through DR3-mediated inhibition of NF-kappaB. Int. J. Cancer 2009, 125, 2465–2473. [Google Scholar]
- Ge, Z.; Sanders, A.J.; Ye, L.; Mansel, R.E.; Jiang, W.G. Expression of death receptor-3 in human breast cancer and its functional effects on breast cancer cells in vitro. Oncol. Rep 2013, 29, 1356–1364. [Google Scholar]
- Chinnaiyan, A.M.; O’Rourke, K.; Yu, G.L.; Lyons, R.H.; Garg, M.; Duan, D.R.; Xing, L.; Gentz, R.; Ni, J.; Dixit, V.M. Signal transduction by DR3, a death domain-containing receptor related to TNFR-1 and CD95. Science 1996, 274, 990–992. [Google Scholar]
- Wen, L.; Zhuang, L.; Luo, X.; Wei, P. TL1A-induced NF-kappaB activation and c-IAP2 production prevent DR3-mediated apoptosis in TF-1 cells. J. Biol. Chem 2003, 278, 39251–39258. [Google Scholar]
- Pobezinskaya, Y.L.; Choksi, S.; Morgan, M.J.; Cao, X.; Liu, Z.G. The adaptor protein TRADD is essential for TNF-like ligand 1A/death receptor 3 signaling. J. Immunol 2011, 186, 5212–5216. [Google Scholar]
- Kozik, P.; Francis, R.W.; Seaman, M.N.; Robinson, M.S. A screen for endocytic motifs. Traffic 2010, 11, 843–855. [Google Scholar]

© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Schneider-Brachert, W.; Heigl, U.; Ehrenschwender, M. Membrane Trafficking of Death Receptors: Implications on Signalling. Int. J. Mol. Sci. 2013, 14, 14475-14503. https://doi.org/10.3390/ijms140714475
Schneider-Brachert W, Heigl U, Ehrenschwender M. Membrane Trafficking of Death Receptors: Implications on Signalling. International Journal of Molecular Sciences. 2013; 14(7):14475-14503. https://doi.org/10.3390/ijms140714475
Chicago/Turabian StyleSchneider-Brachert, Wulf, Ulrike Heigl, and Martin Ehrenschwender. 2013. "Membrane Trafficking of Death Receptors: Implications on Signalling" International Journal of Molecular Sciences 14, no. 7: 14475-14503. https://doi.org/10.3390/ijms140714475