Cholesterol Dependent Uptake and Interaction of Doxorubicin in MCF-7 Breast Cancer Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
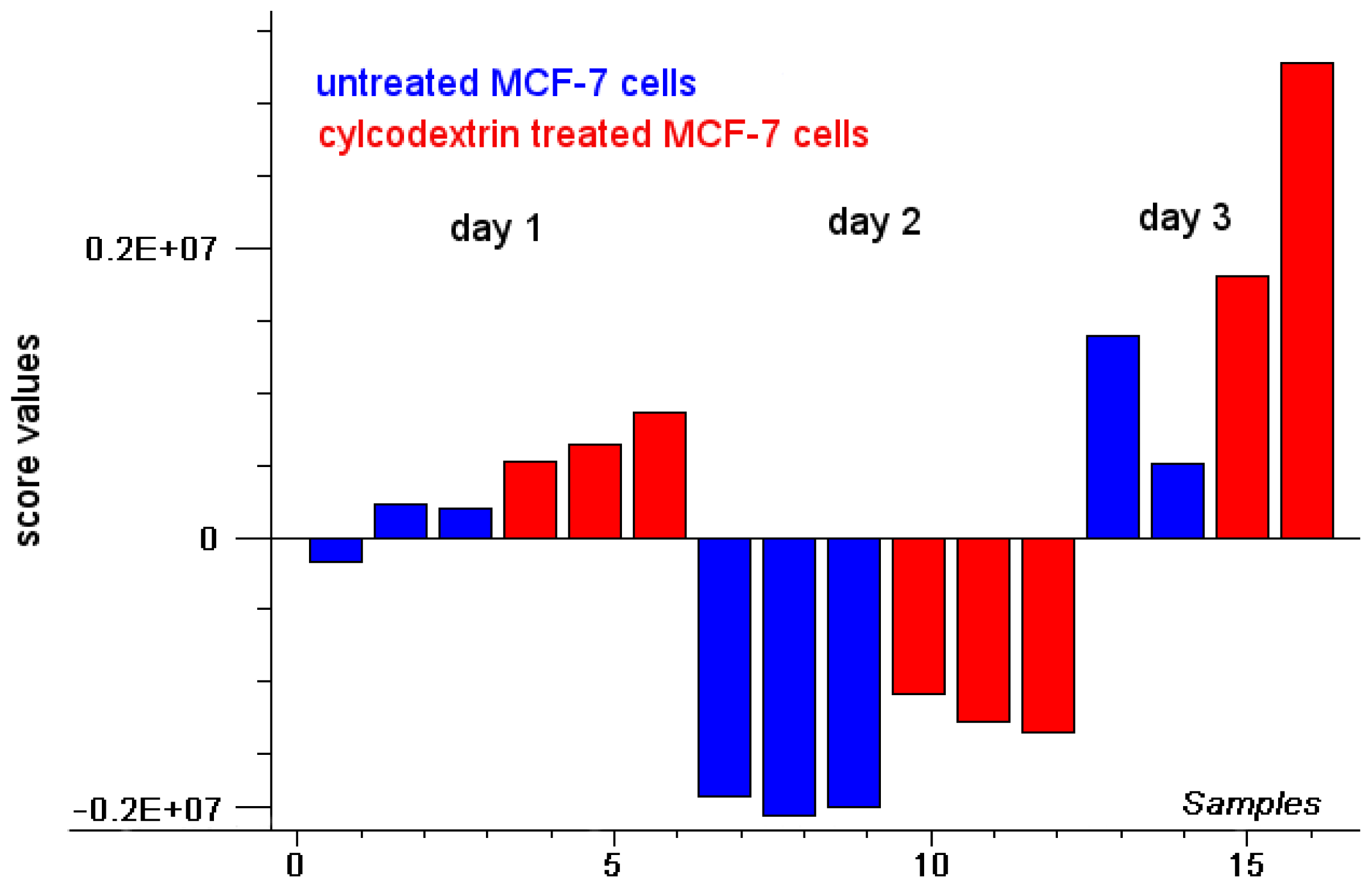
2. Results and Discussion
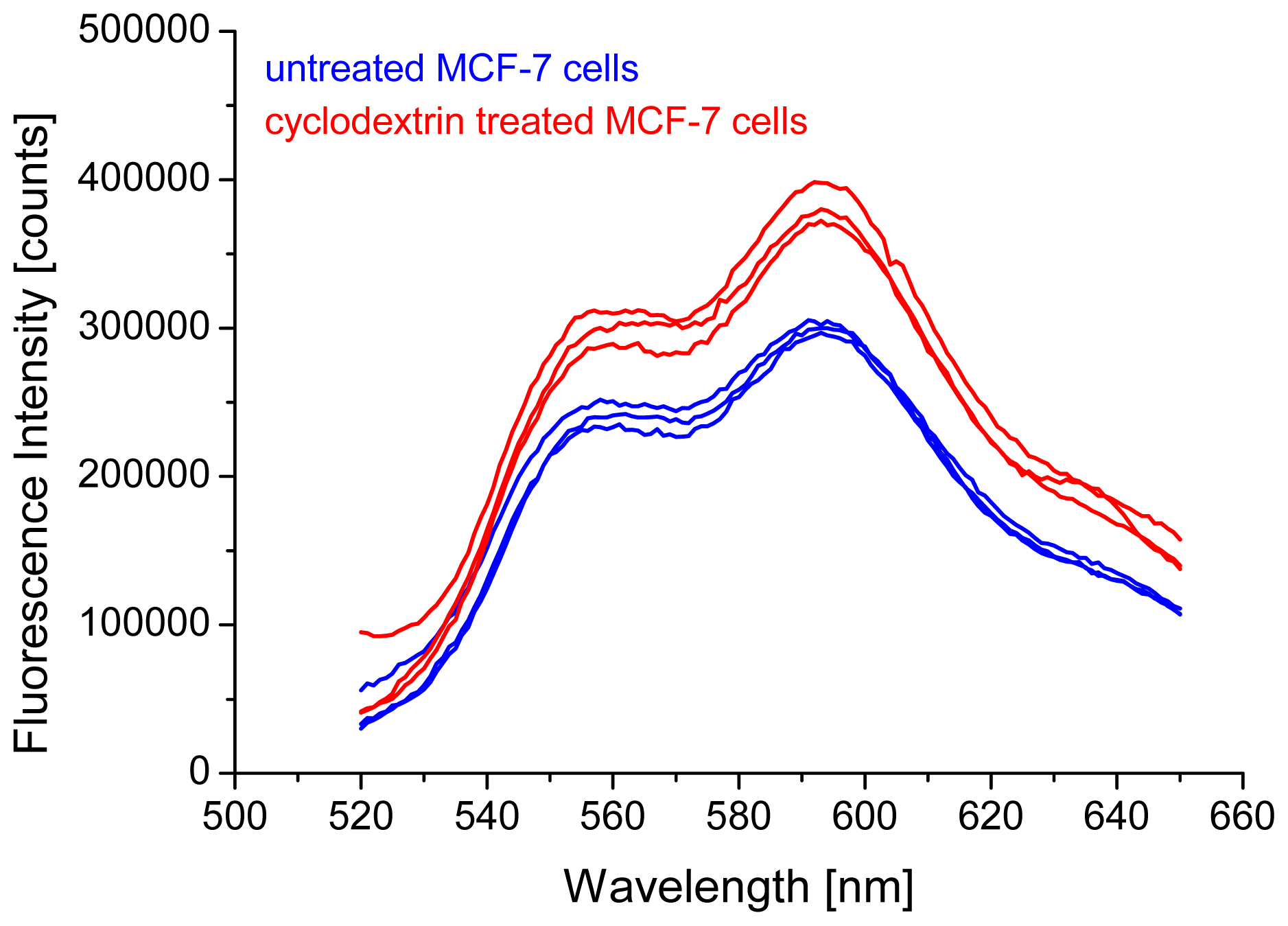
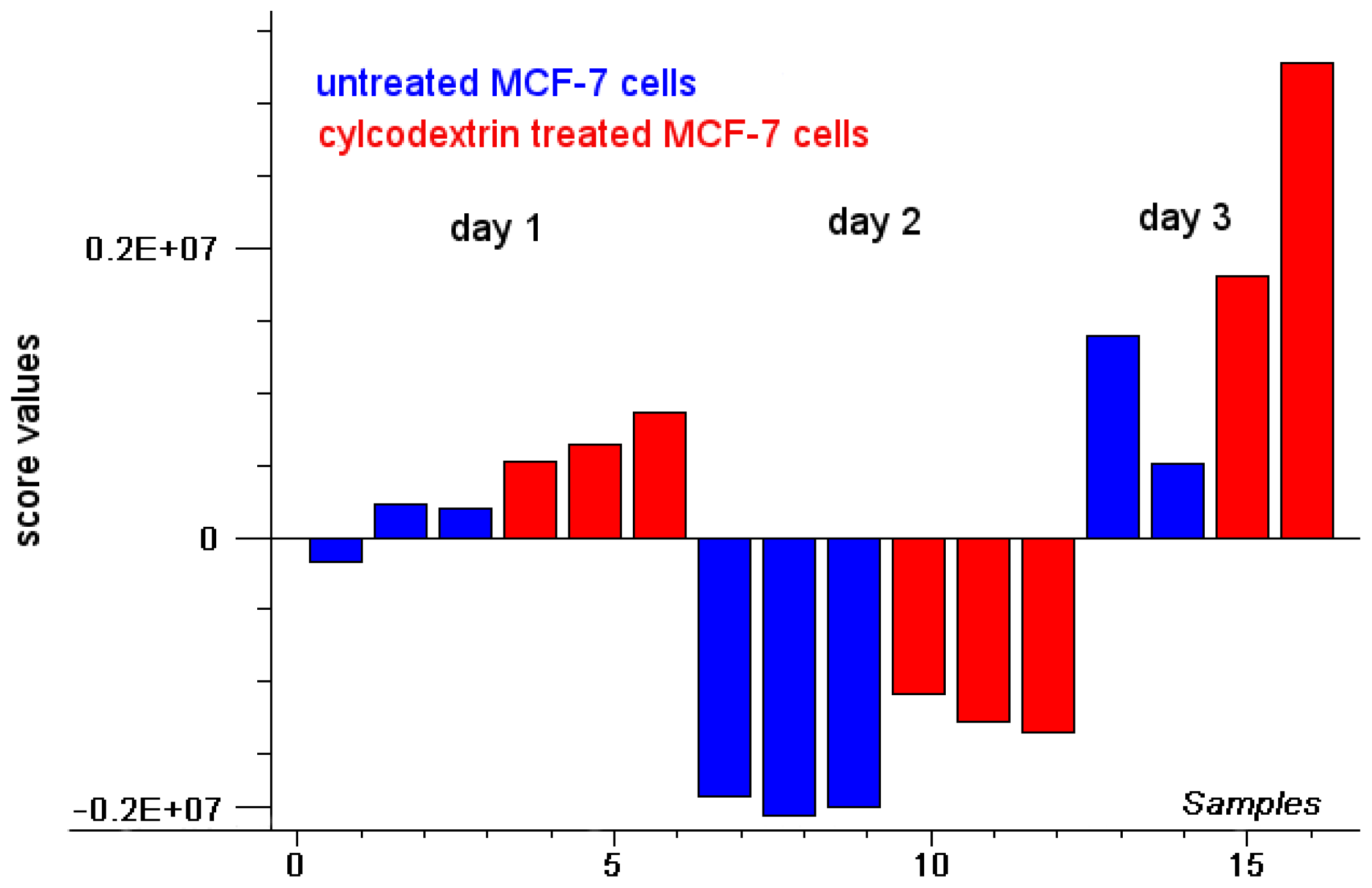
2.1. Intensity of Doxorubicin Fluorescence Increases after Cholesterol Depletion
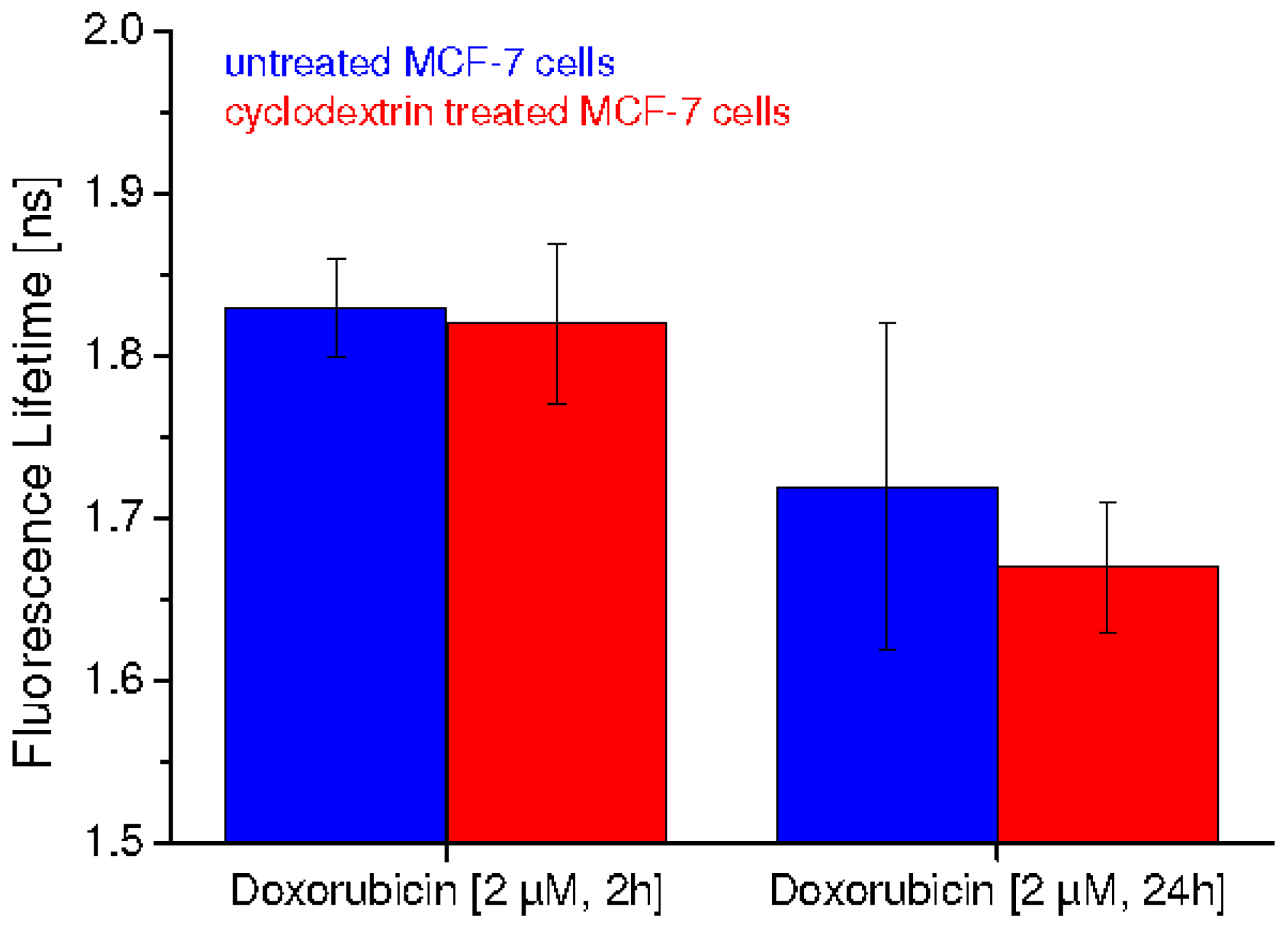
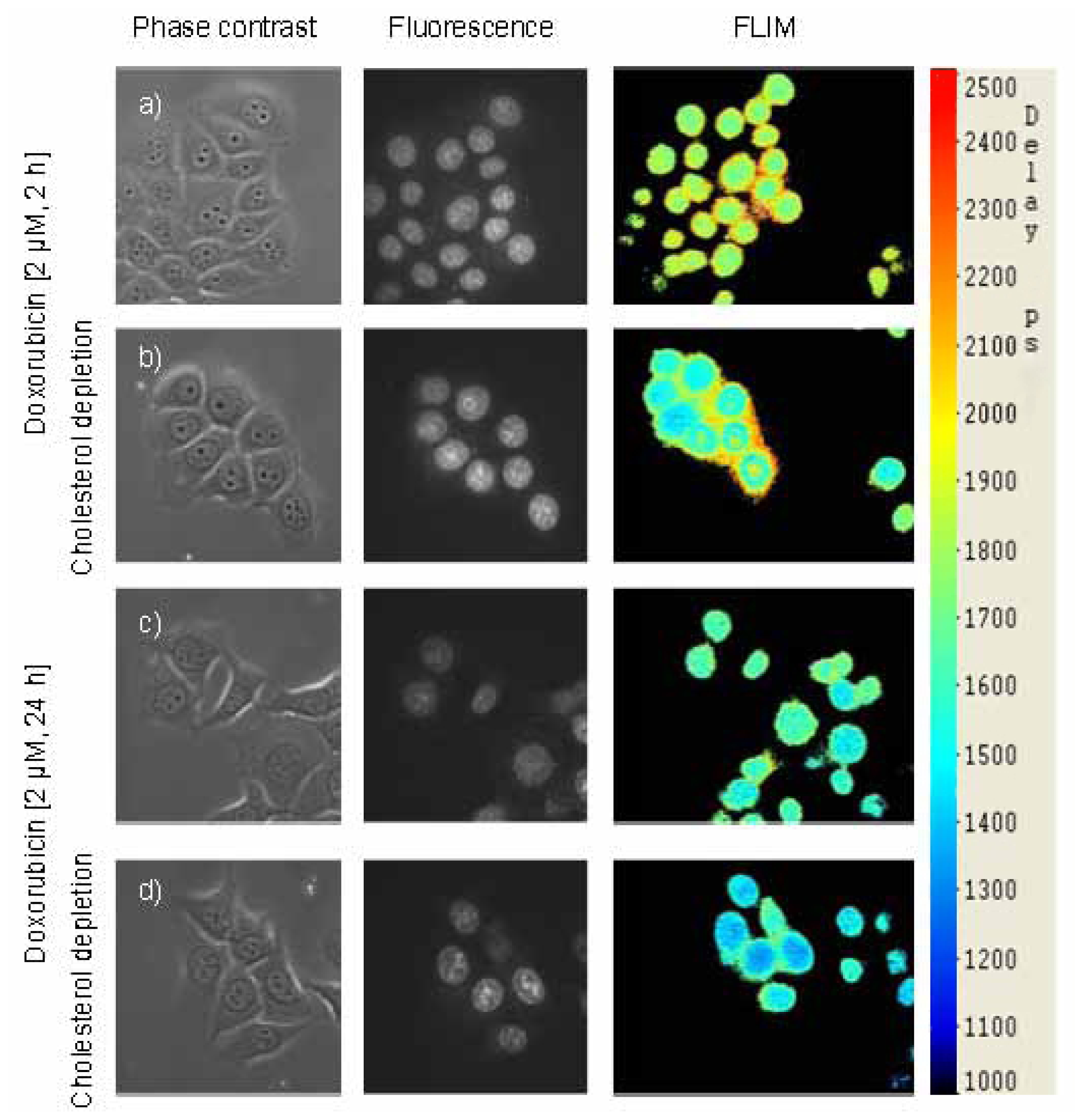
2.2. Fluorescence Lifetime Decreases as a Function of Doxorubicin Incubation Time and Cholesterol Content
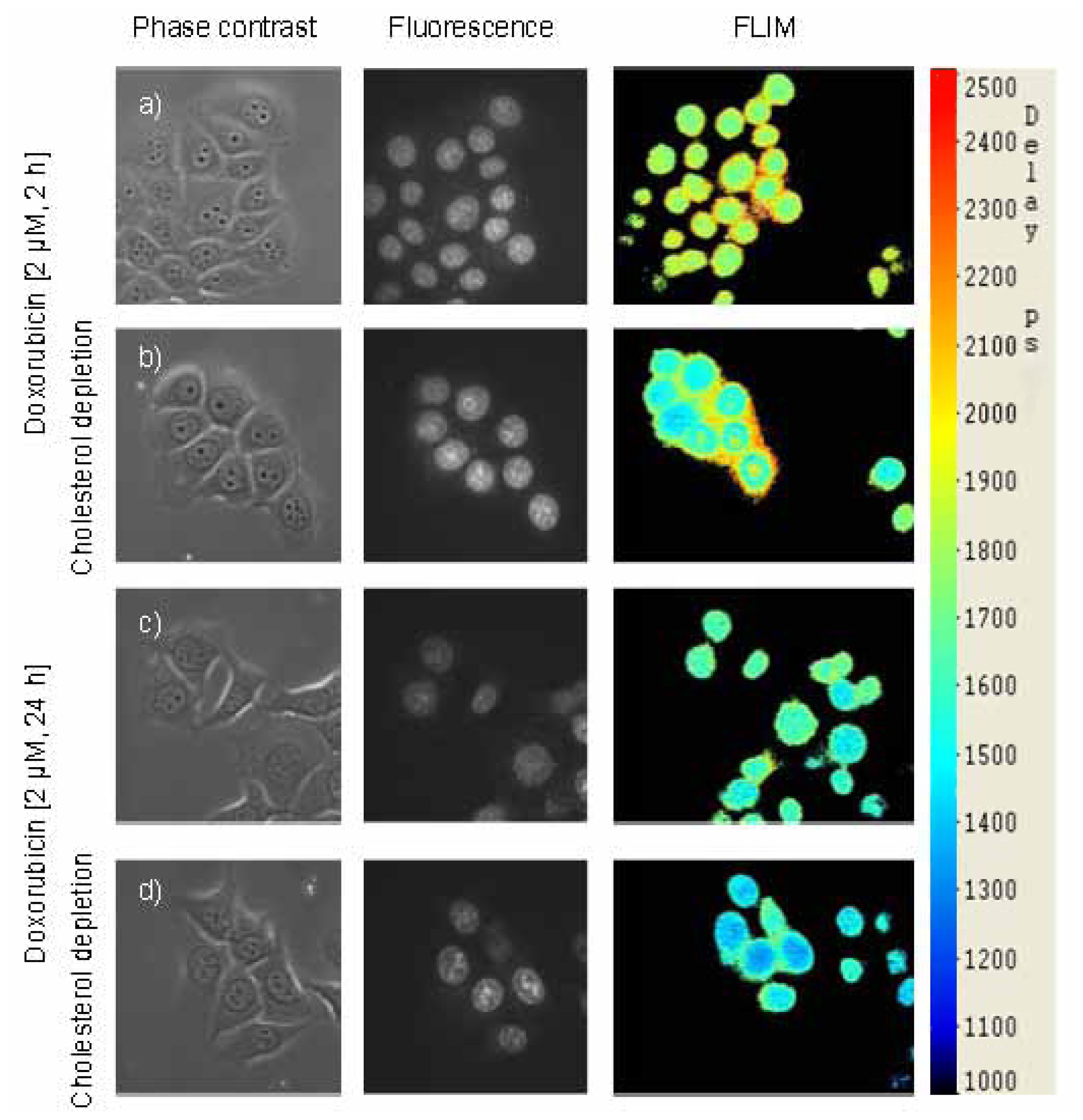
2.3. Images
3. Experimental Section
3.1. Materials
3.2. Experimental Setup
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Carter, S.K.; Blum, R.H. New chemotherapeutic agents—Bleomycin and adriamycin. CA Cancer J. Clin 1974, 24, 322–331. [Google Scholar]
- Blum, R.H.; Carter, S.K. Adriamycin. A new anticancer drug with significant clinical activity. Ann. Intern. Med 1974, 80, 249–259. [Google Scholar]
- Li, Z.X.; Wang, T.T.; Wu, Y.T.; Xu, C.M.; Dong, M.Y.; Sheng, J.Z.; Huang, H.F. Adriamycin induces H2AX phosphorylation in human spermatozoa. Asian J. Androl 2008, 10, 749–757. [Google Scholar]
- Karukstis, K.K.; Thompson, E.H.; Whiles, J.A.; Rosenfeld, R.J. Deciphering the fluorescence signature of daunomycin and doxorubicin. Biophys. Chem 1998, 73, 249–263. [Google Scholar]
- Chen, N.T.; Wu, C.Y.; Chung, C.Y.; Hwu, Y.; Cheng, S.H.; Mou, C.Y.; Lo, L.W. Probing the dynamics of doxorubicin-DNA intercalation during the initial activation of apoptosis by fluorescence lifetime imaging microscopy (FLIM). PLoS One 2012, 7, e44947. [Google Scholar]
- Bakker, G.J.; Andresen, V.; Hoffman, R.M.; Friedl, P. Fluorescence lifetime microscopy of tumor cell invasion, drug delivery, and cytotoxicity. Methods Enzymol 2012, 504, 109–125. [Google Scholar]
- Dai, X.; Yue, Z.; Eccleston, M.E.; Swartling, J.; Slater, N.K.; Kaminski, C.F. Fluorescence intensity and lifetime imaging of free and micellar-encapsulated doxorubicin in living cells. Nanomedicine 2008, 4, 49–56. [Google Scholar]
- Haaland, D.M.; Jones, H.D.; van Benthem, M.H.; Sinclair, M.B.; Melgaard, D.K.; Stork, C.L.; Pedroso, M.C.; Liu, P.; Brasier, A.R.; Andrews, N.L.; et al. Hyperspectral confocal fluorescence imaging: Exploring alternative multivariate curve resolution approaches. Appl. Spectrosc 2009, 63, 271–279. [Google Scholar]
- Schneckenburger, H.; Weber, P.; Wagner, M.; Schickinger, S.; Richter, V.; Bruns, T.; Strauss, W.S.; Wittig, R. Light exposure and cell viability in fluorescence microscopy. J. Microsc 2012, 245, 311–318. [Google Scholar]
- Slingerland, M.; Guchelaar, H.J.; Gelderblom, H. Liposomal drug formulations in cancer therapy: 15 years along the road. Drug Discov. Today 2012, 17, 160–166. [Google Scholar]
- Opel, D.; Westhoff, M.A.; Bender, A.; Braun, V.; Debatin, K.M.; Fulda, S. Phosphatidylinositol 3-kinase inhibition broadly sensitizes glioblastoma cells to death receptor- and drug-induced apoptosis. Cancer Res 2008, 68, 6271–6280. [Google Scholar]
- Regev, R.; Eytan, G.D. Flip-flop of doxorubicin across erythrocyte and lipid membranes. Biochem. Pharmacol 1997, 54, 1151–1158. [Google Scholar]
- Pacilio, C.; Florio, S.; Pagnini, U.; Crispino, A.; Claudio, P.P.; Pacilio, G.; Pagnini, G. Modification of membrane fluidity and depolarization by some anthracyclines in different cell lines. Anticancer Res 1998, 18, 4027–4034. [Google Scholar]
- Storch, C.H.; Ehehalt, R.; Haefeli, W.E.; Weiss, J. Localization of the human breast cancer resistance protein (BCRP/ABCG2) in lipid rafts/caveolae and modulation of its activity by cholesterol in vitro. J. Pharmacol. Exp. Ther 2007, 323, 257–264. [Google Scholar]
- Peetla, C.; Bhave, R.; Vijayaraghavalu, S.; Stine, A.; Kooijman, E.; Labhasetwar, V. Drug resistance in breast cancer cells: Biophysical characterization of and doxorubicin interactions with membrane lipids. Mol. Pharm 2010, 7, 2334–2348. [Google Scholar]
- Weber, P.; Wagner, M.; Schneckenburger, H. Fluorescence imaging of membrane dynamics in living cells. J. Biomed. Opt 2010, 15, 046017. [Google Scholar]
- von Arnim, C.A.; von Einem, B.; Weber, P.; Wagner, M.; Schwanzar, D.; Spoelgen, R.; Strauss, W.L.; Schneckenburger, H. Impact of cholesterol level upon APP and BACE proximity and APP cleavage. Biochem. Biophys. Res. Commun 2008, 370, 207–212. [Google Scholar]
- Christian, A.E.; Haynes, M.P.; Phillips, M.C.; Rothblat, G.H. Use of cyclodextrins for manipulating cellular cholesterol content. J. Lipid Res 1997, 38, 2264–2272. [Google Scholar]
- Eker, C.; Rydell, R.; Svanberg, K.; Andersson-Engels, S. Multivariate analysis of laryngeal fluorescence spectra recorded in vivo. Lasers Surg. Med 2001, 28, 259–266. [Google Scholar]
- Qu, J.Y.; Chang, H.; Xiong, S. Fluorescence spectral imaging for characterization of tissue based on multivariate statistical analysis. J. Opt. Soc. Am. A 2002, 19, 1823–1831. [Google Scholar]
- Xu, X.; Gerard, A.L.; Huang, B.C.; Anderson, D.C.; Payan, D.G.; Luo, Y. Detection of programmed cell death using fluorescence energy transfer. Nucleic Acids Res 1998, 26, 2034–2035. [Google Scholar]
- Angres, B.; Steuer, H.; Weber, P.; Wagner, M.; Schneckenburger, H. A membrane-bound FRET-based caspase sensor for detection of apoptosis using fluorescence lifetime and total internal reflection microscopy. Cytometry A 2009, 75, 420–427. [Google Scholar]
- Mulvey, C.S.; Sherwood, C.A.; Bigio, I.J. Wavelength-dependent backscattering measurements for quantitative real-time monitoring of apoptosis in living cells. J. Biomed. Opt 2009, 14, 064013. [Google Scholar]
- Kunz-Schughart, L.A.; Freyer, J.P.; Hofstaedter, F.; Ebner, R. The use of 3-D cultures for high-throughput screening: The multicellular spheroid model. J. Biomol. Screen 2004, 9, 273–285. [Google Scholar]
- Pawley, J. Handbook of Biological Confocal Microscopy; Plenum Press: New York NY, USA, 1990. [Google Scholar]
- Webb, R.H. Confocal optical microscopy. Rep. Prog. Phys 1996, 59, 427–471. [Google Scholar]
- Neil, M.A.; Juskaitis, R.; Wilson, T. Method of obtaining optical sectioning by using structured light in a conventional microscope. Opt. Lett 1997, 22, 1905–1907. [Google Scholar]
- Gustafsson, M.G.; Shao, L.; Carlton, P.M.; Wang, C.J.; Golubovskaya, I.N.; Cande, W.Z.; Agard, D.A.; Sedat, J.W. Three-dimensional resolution doubling in wide-field fluorescence microscopy by structured illumination. Biophys. J 2008, 94, 4957–4970. [Google Scholar]
- Huisken, J.; Swoger, J.; Del Bene, F.; Wittbrodt, J.; Stelzer, E.H. Optical sectioning deep inside live embryos by selective plane illumination microscopy. Science 2004, 305, 1007–1009. [Google Scholar]
- Santi, P.A. Light sheet fluorescence microscopy: A review. J. Histochem. Cytochem 2011, 59, 129–138. [Google Scholar]
- Bruns, T.; Schickinger, S.; Wittig, R.; Schneckenburger, H. Preparation strategy and illumination of three-dimensional cell cultures in light sheet-based fluorescence microscopy. J. Biomed. Opt 2012, 17, 101518. [Google Scholar]
- Coates, C.G.; Denvir, D.J.; McHale, N.G.; Thornbury, K.D.; Hollywood, M.A. Optimizing low-light microscopy with back-illuminated electron multiplying charge-coupled device: Enhanced sensitivity, speed, and resolution. J. Biomed. Opt 2004, 9, 1244–1252. [Google Scholar]
- Schneckenburger, H.; Wagner, M.; Kretzschmar, M.; Strauss, W.S.; Sailer, R. Laser-assisted fluorescence microscopy for measuring cell membrane dynamics. Photochem. Photobiol. Sci 2004, 3, 817–822. [Google Scholar]
- Wilcoxon, F. Individual comparisons by ranking methods. Biometrics Bull 1945, 1, 80–83. [Google Scholar]


© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Weber, P.; Wagner, M.; Schneckenburger, H. Cholesterol Dependent Uptake and Interaction of Doxorubicin in MCF-7 Breast Cancer Cells. Int. J. Mol. Sci. 2013, 14, 8358-8366. https://doi.org/10.3390/ijms14048358
Weber P, Wagner M, Schneckenburger H. Cholesterol Dependent Uptake and Interaction of Doxorubicin in MCF-7 Breast Cancer Cells. International Journal of Molecular Sciences. 2013; 14(4):8358-8366. https://doi.org/10.3390/ijms14048358
Chicago/Turabian StyleWeber, Petra, Michael Wagner, and Herbert Schneckenburger. 2013. "Cholesterol Dependent Uptake and Interaction of Doxorubicin in MCF-7 Breast Cancer Cells" International Journal of Molecular Sciences 14, no. 4: 8358-8366. https://doi.org/10.3390/ijms14048358