Differential Expression of Genes Associated with the Progression of Renal Disease in the Kidneys of Liver-Specific Glucokinase Gene Knockout Mice

Abstract
:1. Introduction
2. Results and Discussion
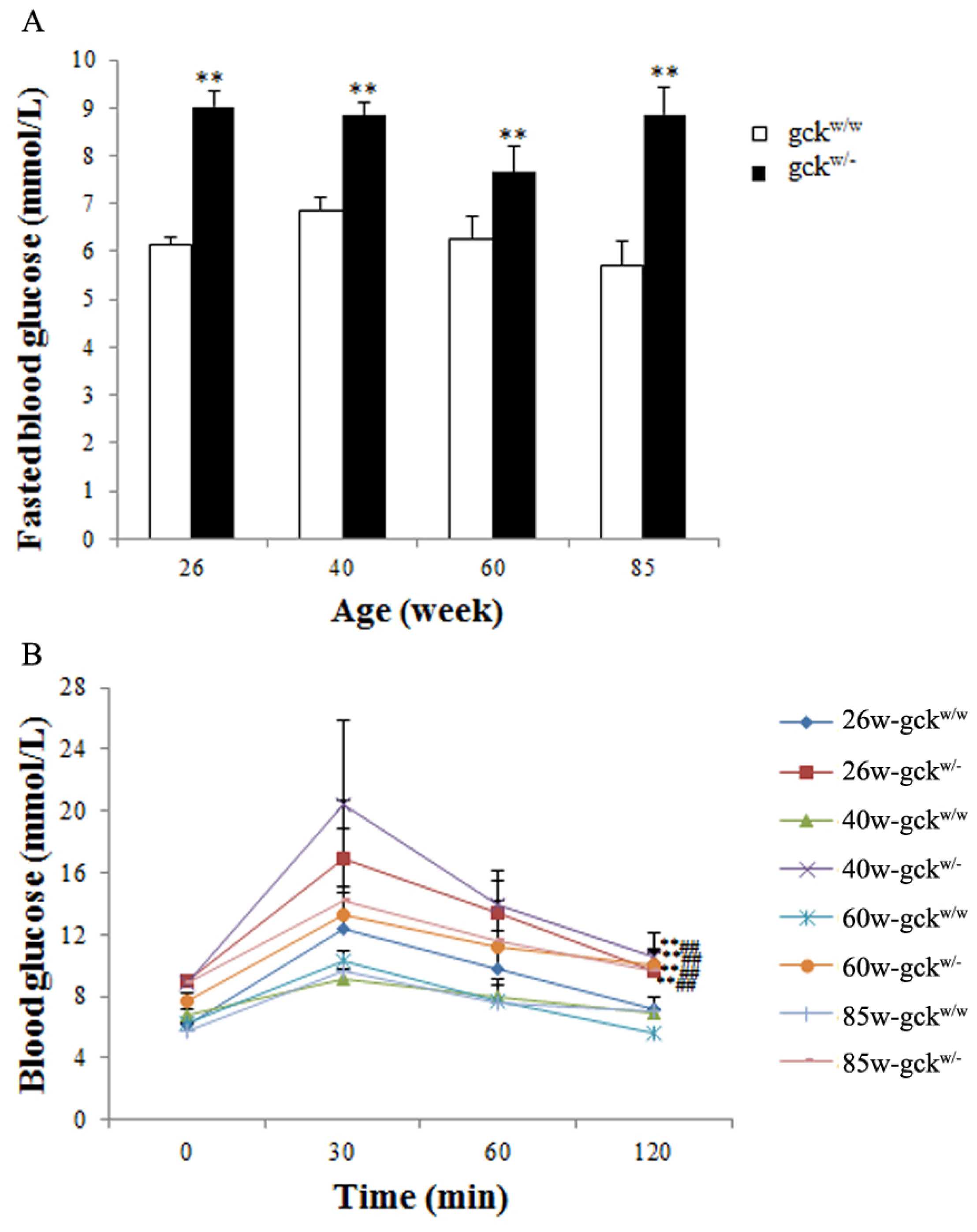
2.1. Changes in Biochemical Parameters in Gck Deficient Mice as They Age
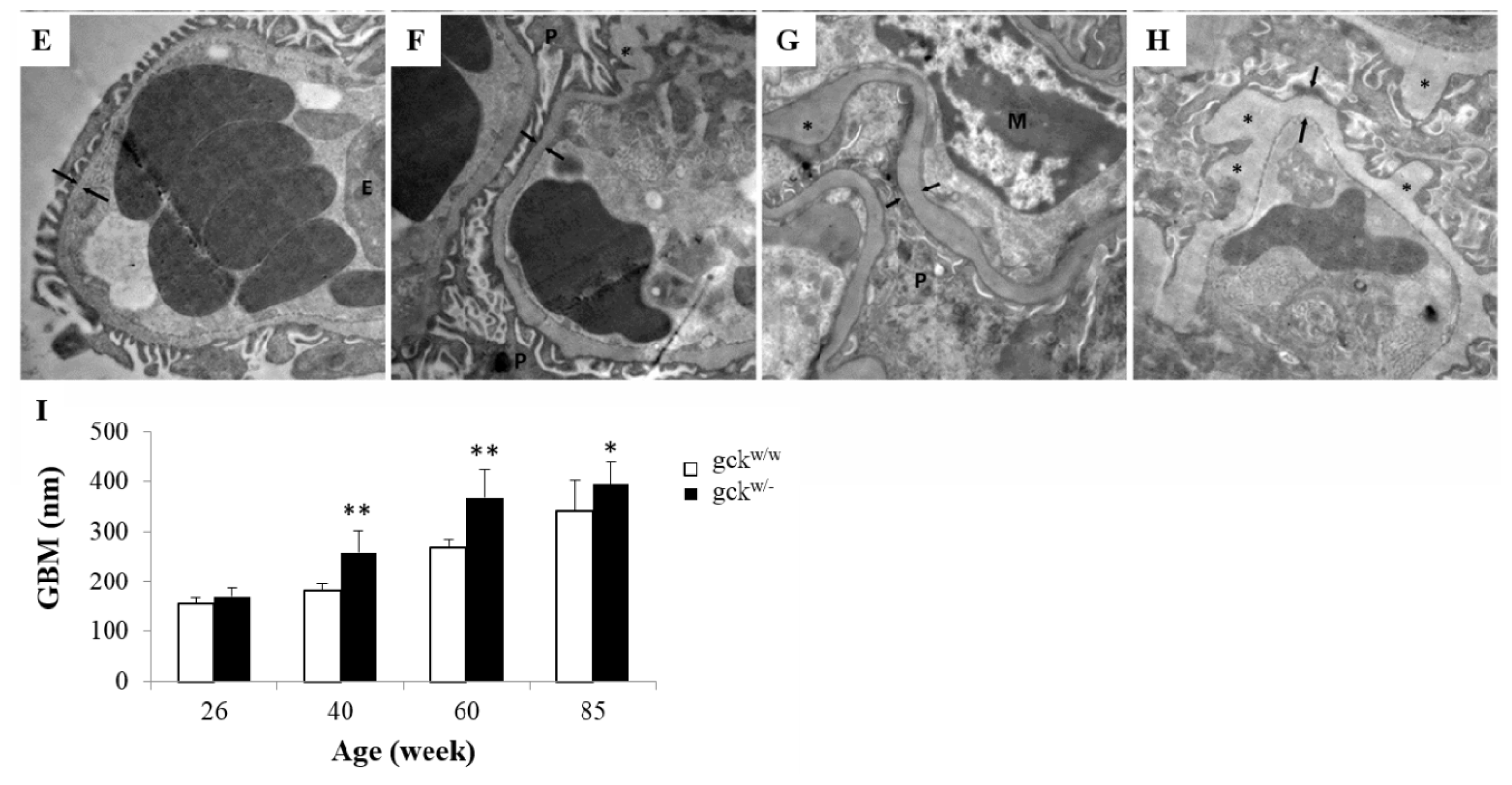
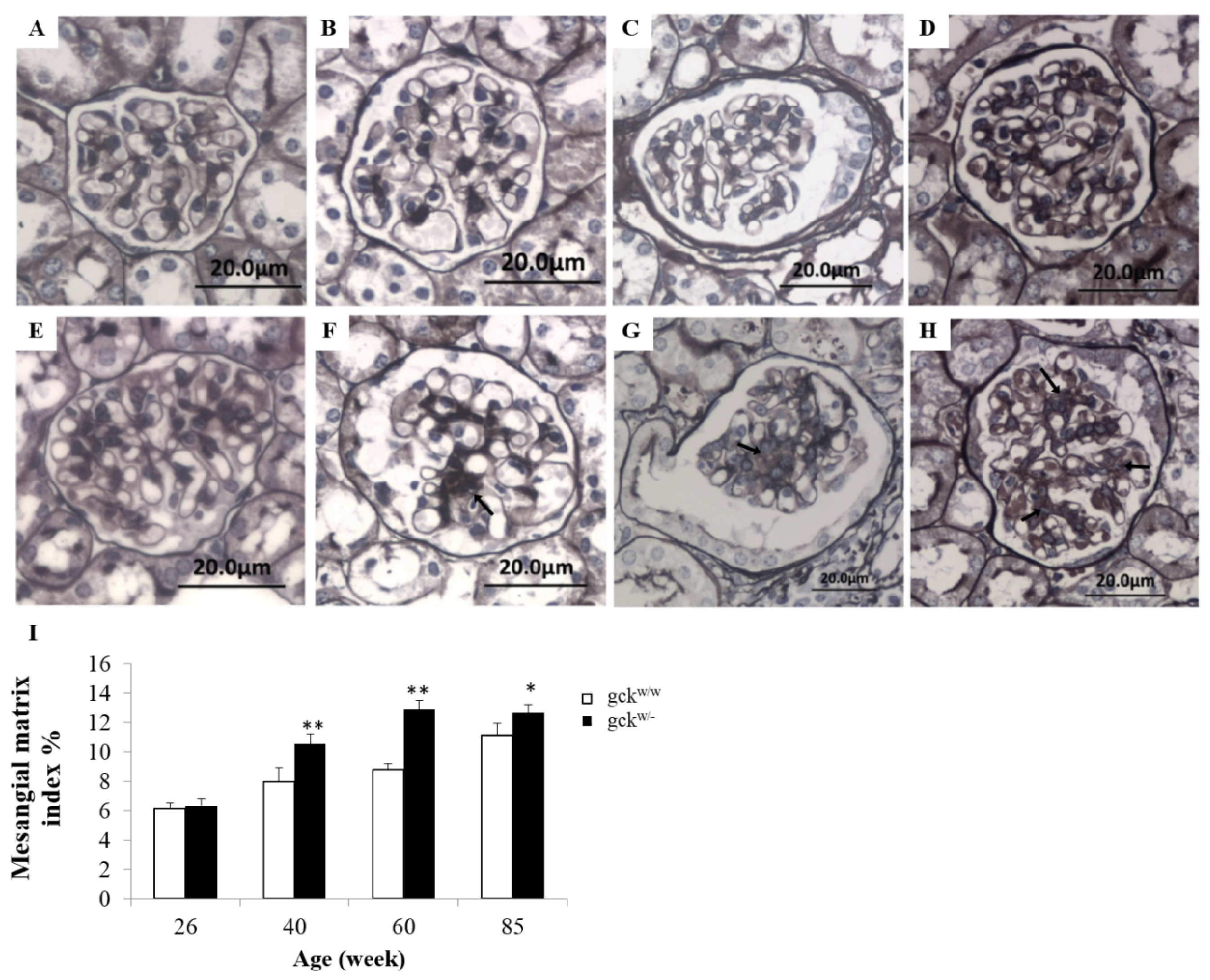
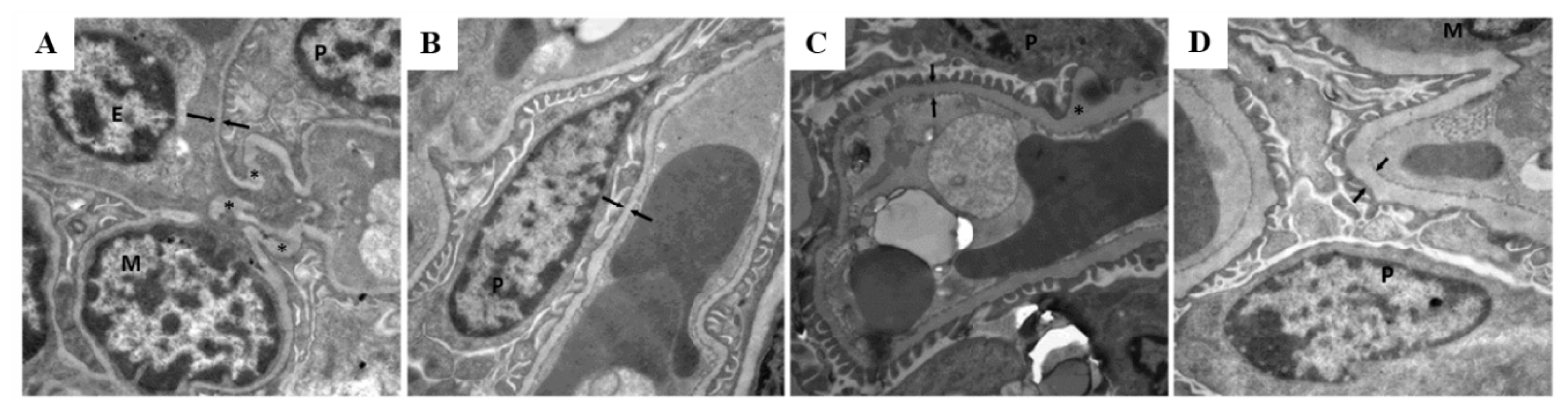
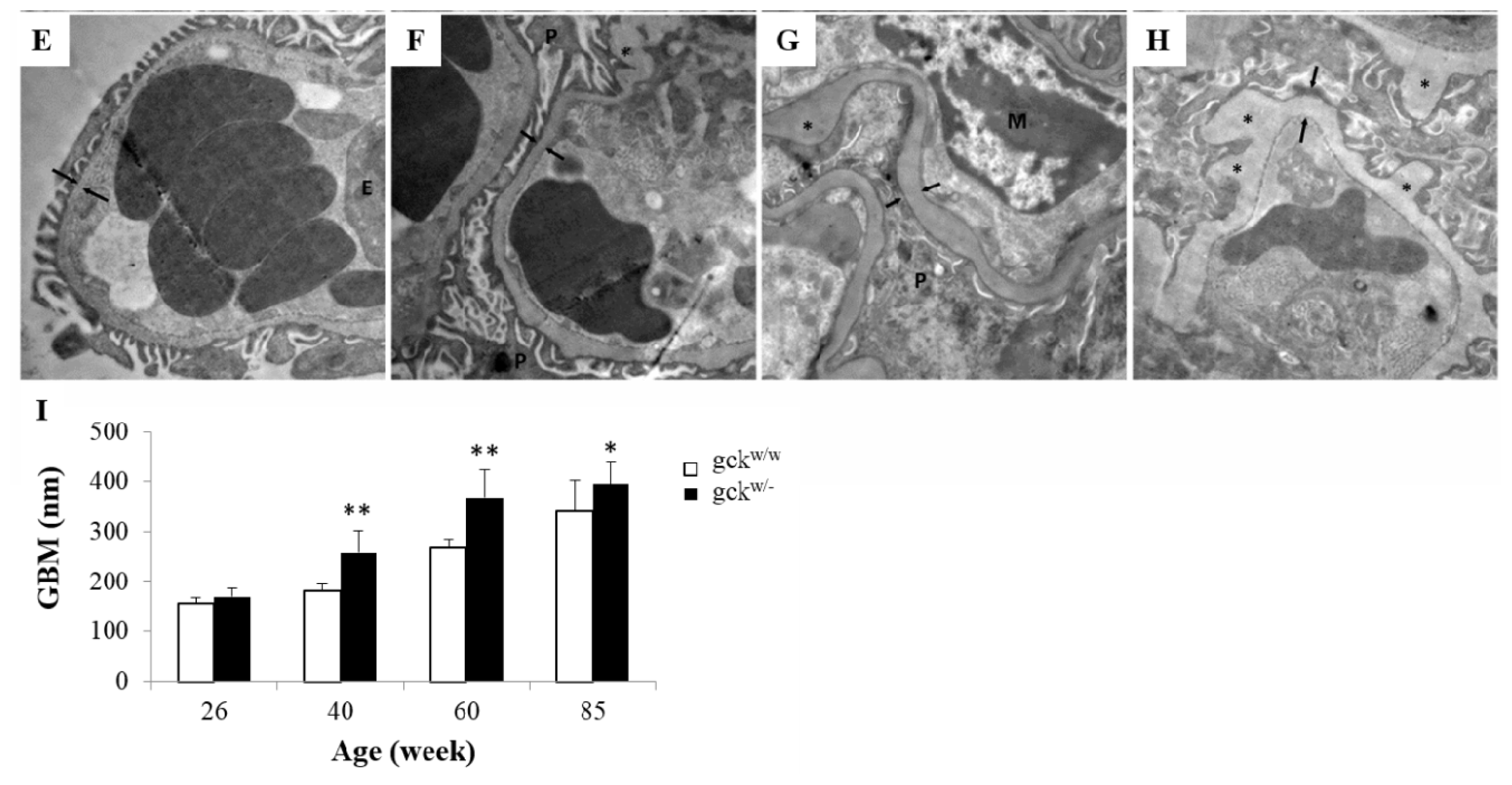
2.2. Morphological Analysis of Kidney Tissue
2.3. Identification of Differentially Expressed Genes by SSH
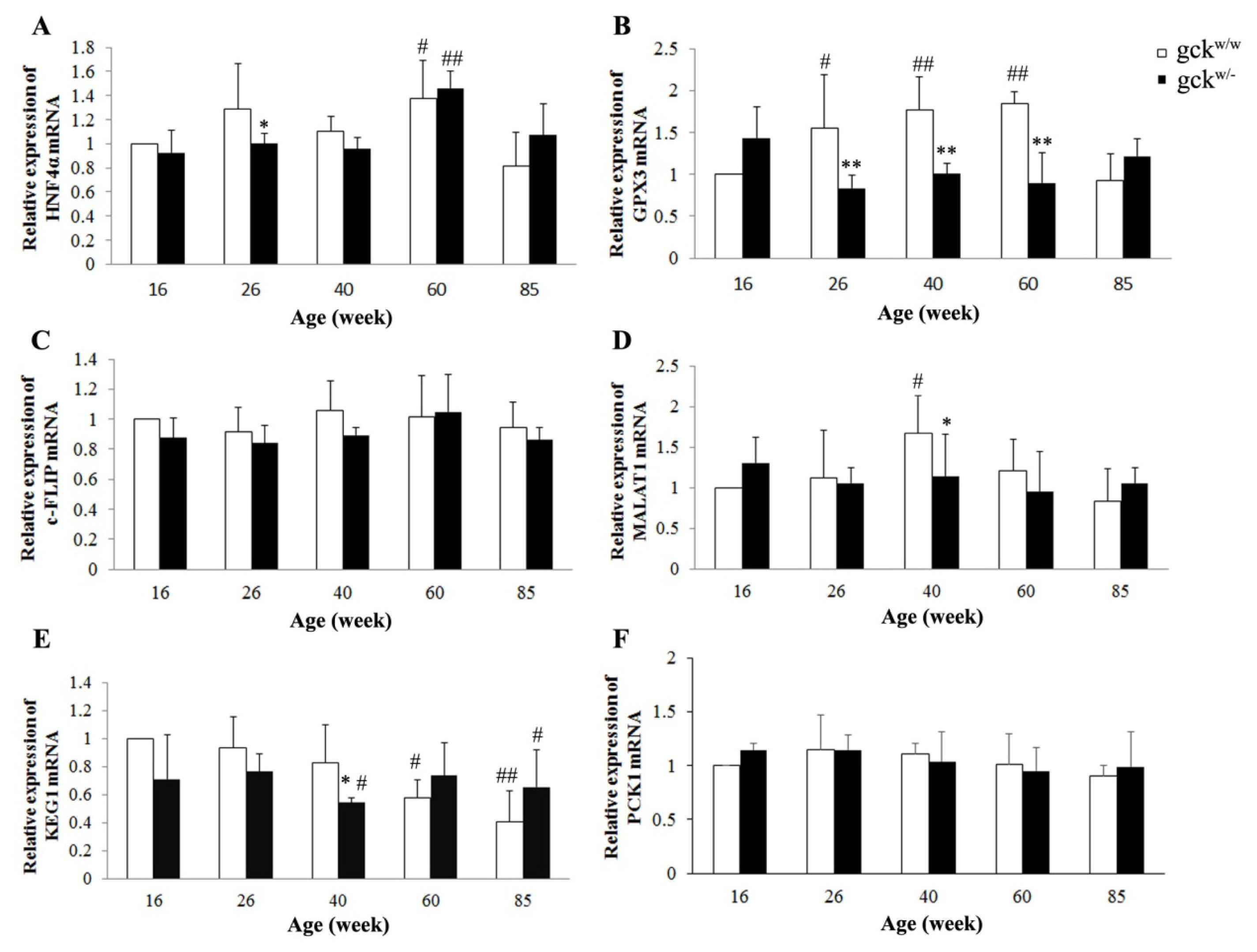
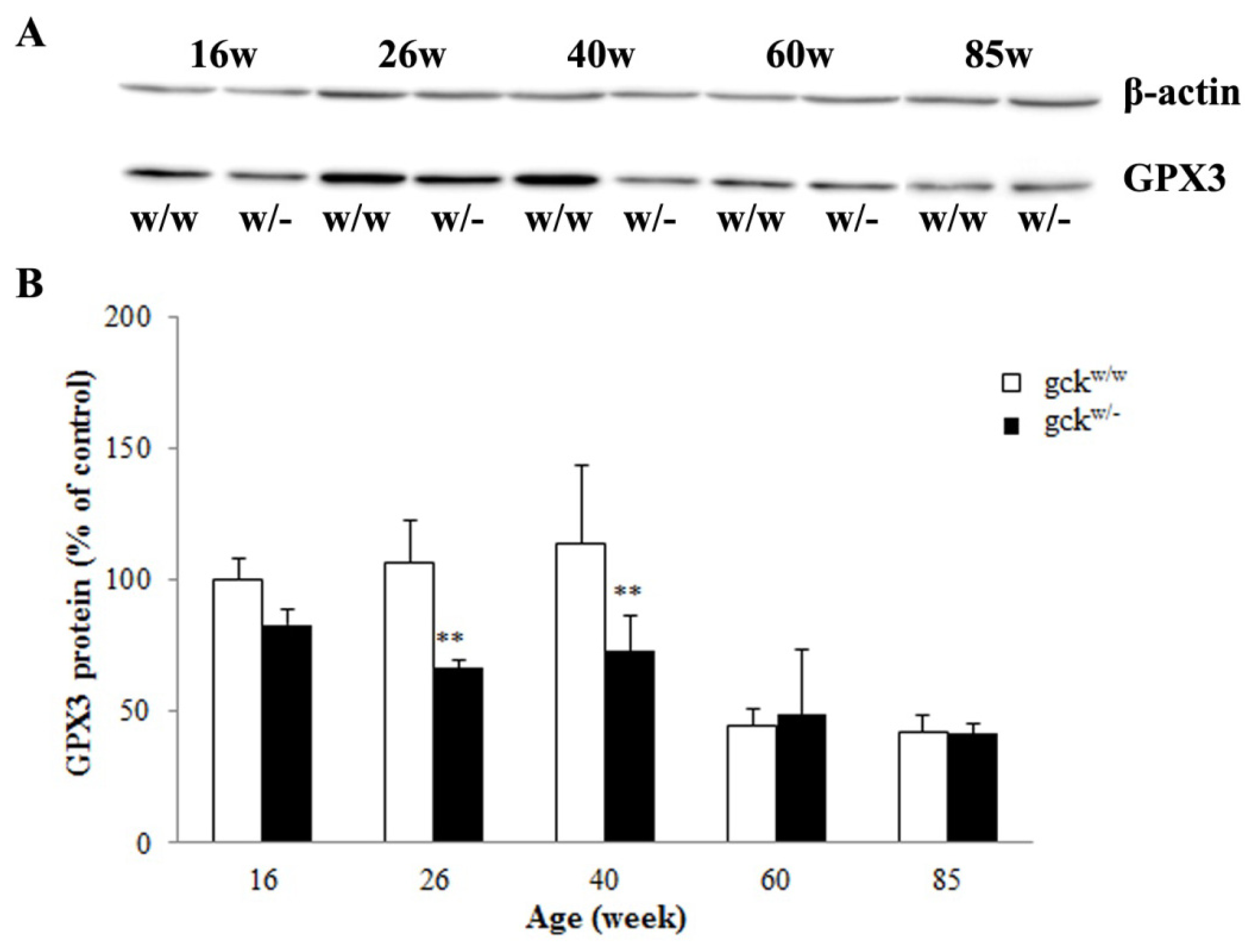
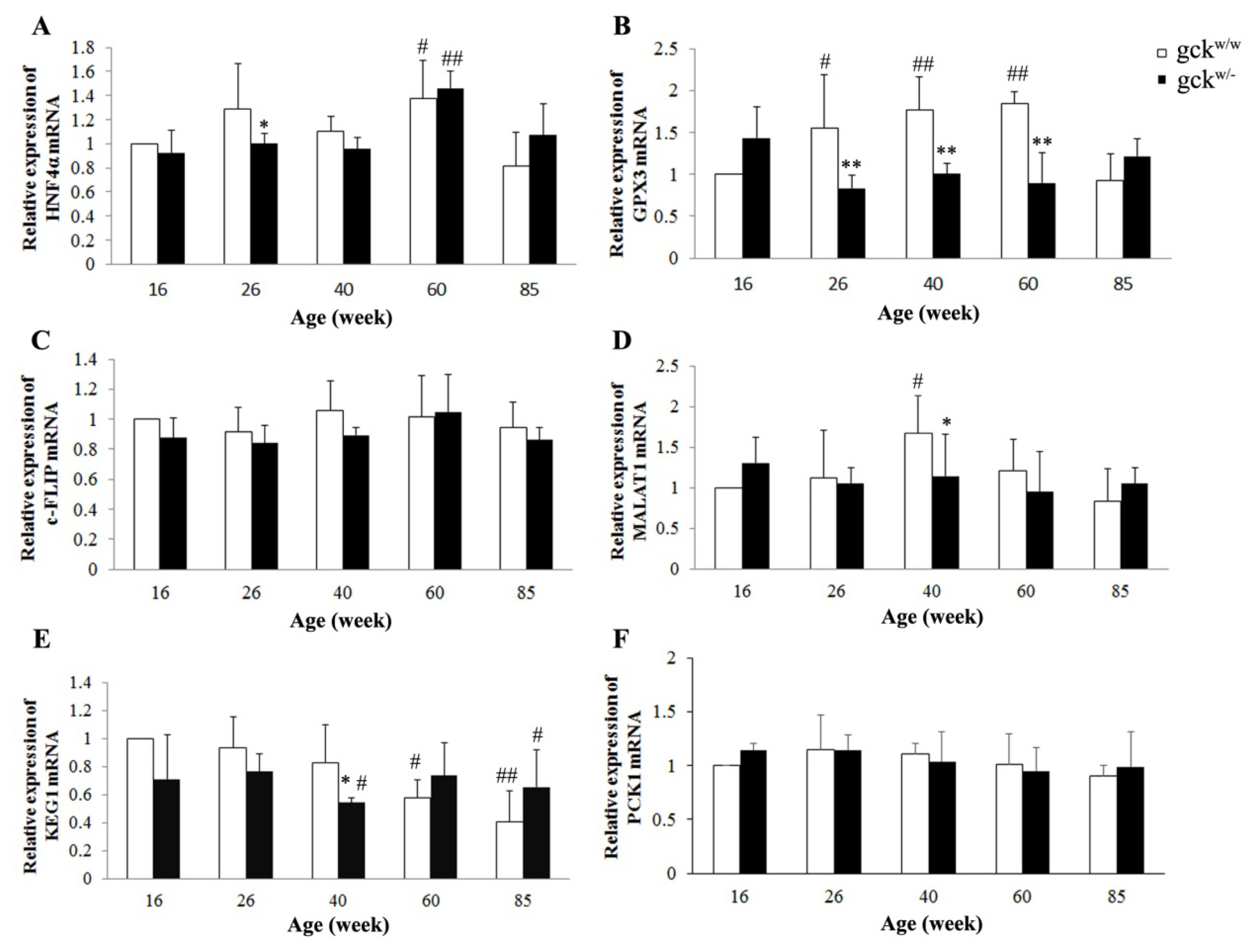
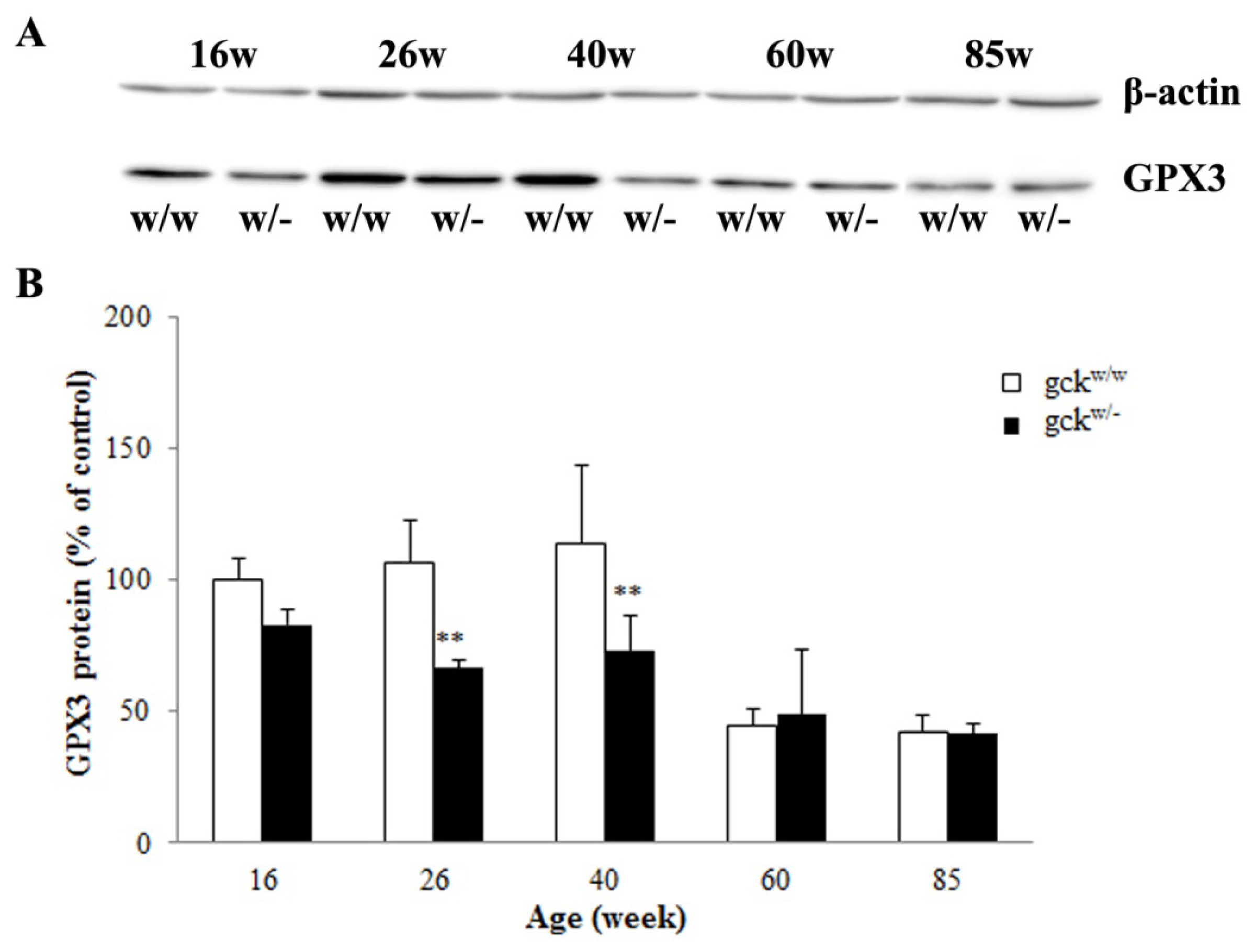
2.4. Expression Profiles of Six Candidate Genes
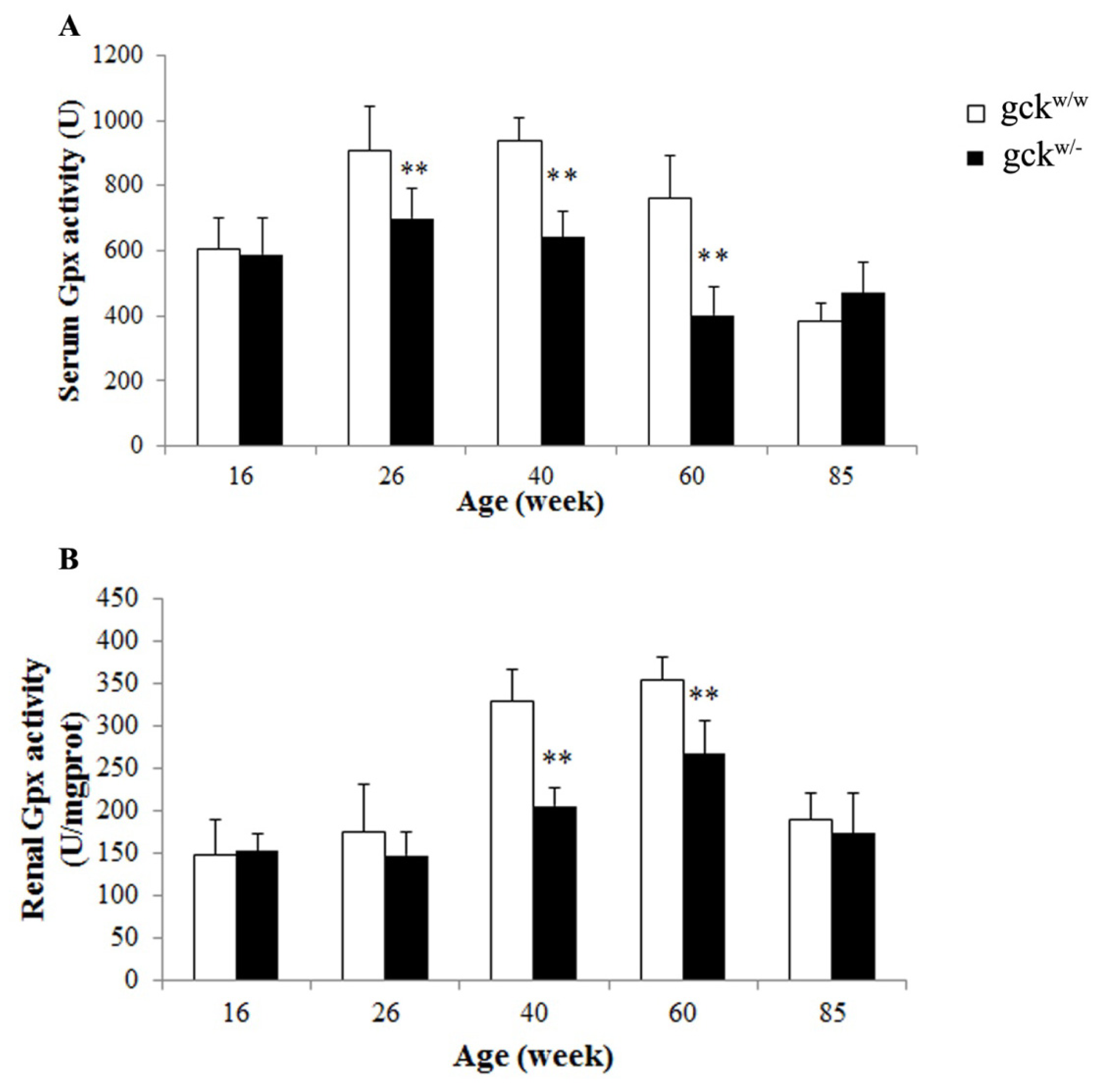
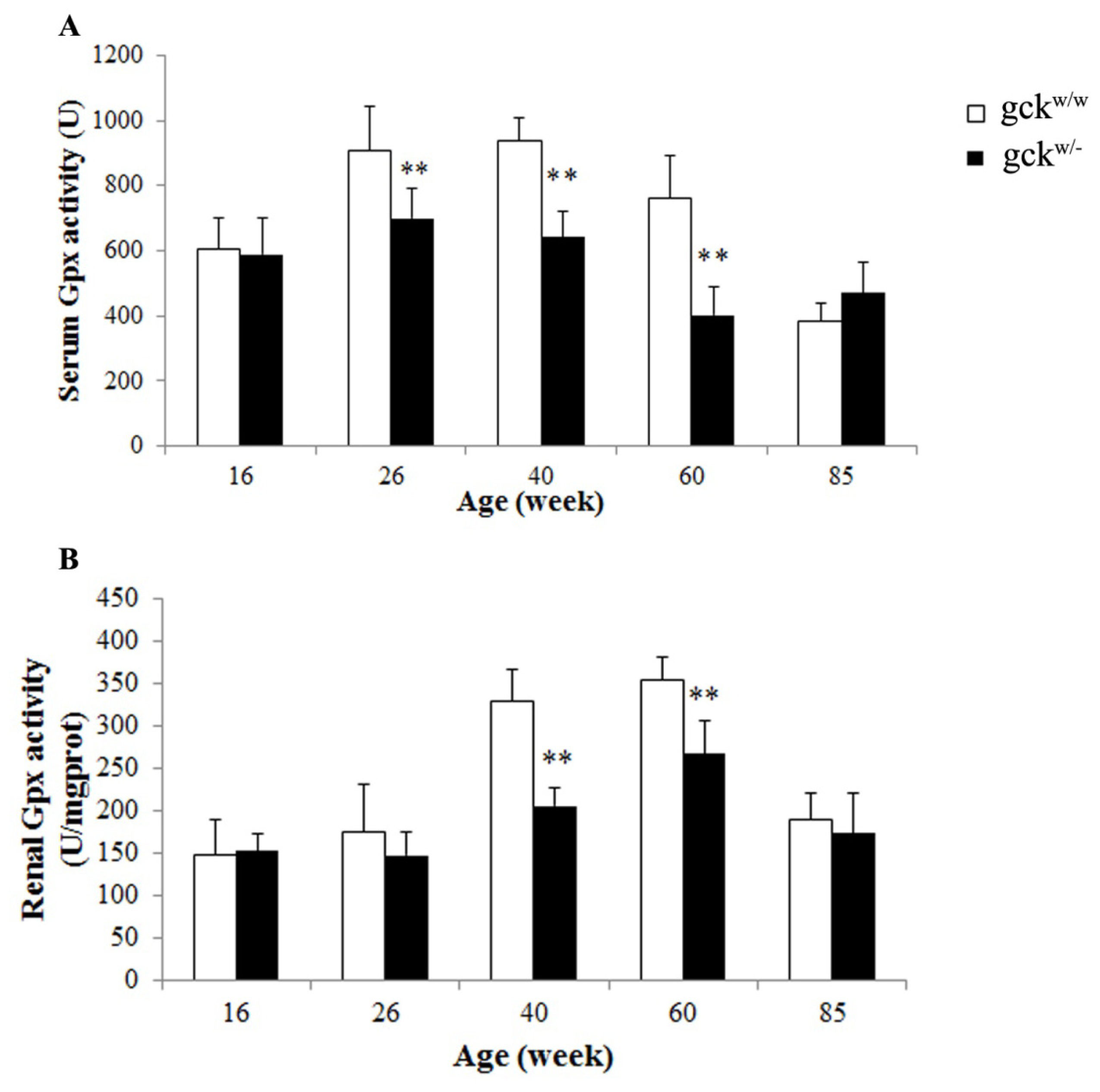
2.5. Serum and Renal Gpx Activities
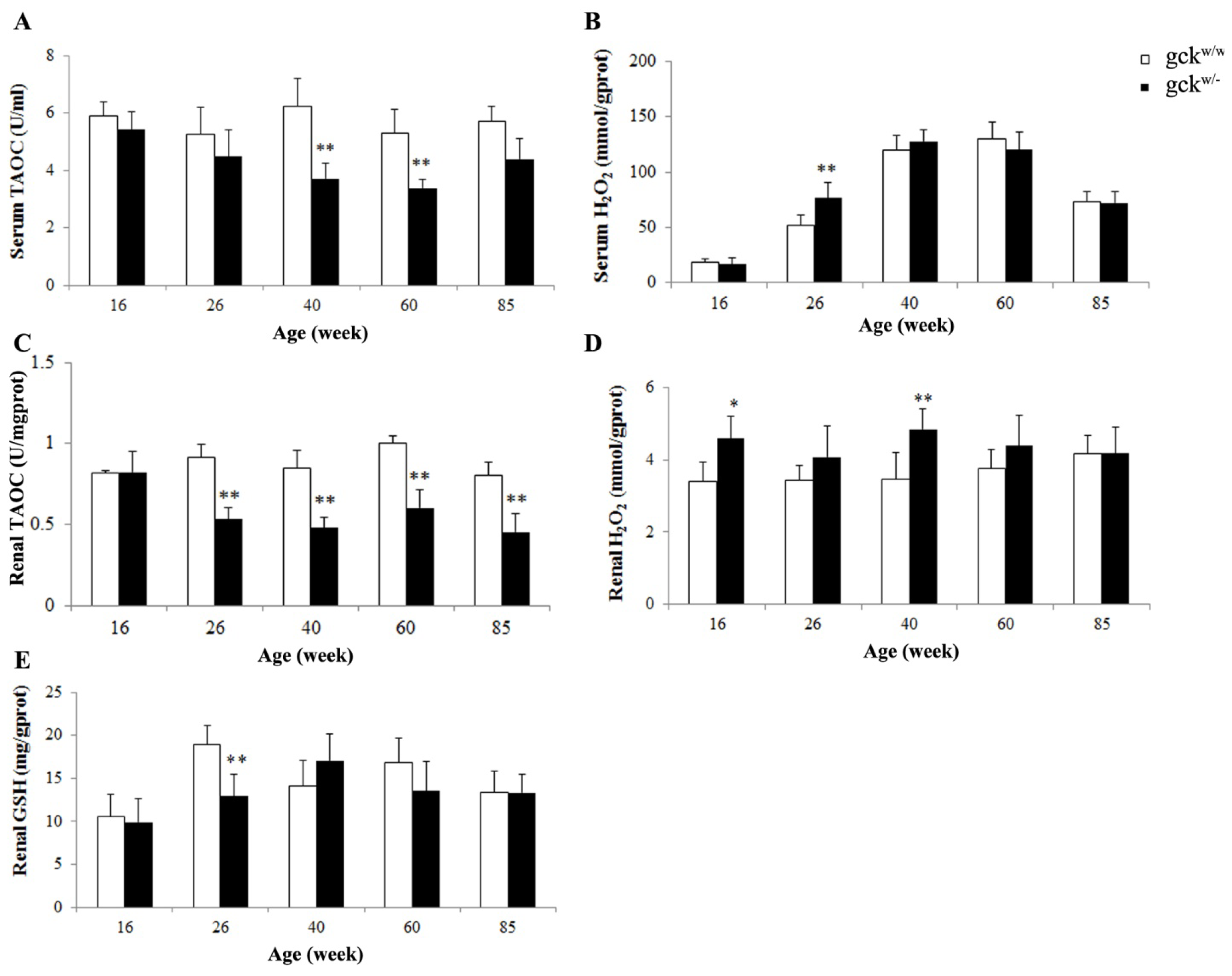
2.6. Antioxidative Capacity of Serum and Kidney in Gckw/− and Gckw/w Mice
2.7. Discussion
3. Experimental Section
3.1. Animals
3.2. Biochemical Analysis
3.3. Renal Morphology
3.4. Suppression Subtractive Hybridization (SSH)
3.5. Quantification of mRNA by Real-Time Quantitative PCR
3.6. Total Protein Extraction and Western Blot
3.7. Measurement of Serum and Renal Gpx Enzyme Activities
3.8. Measurement of TAOC, Hydrogen Peroxide and Glutathione
3.9. Statistical Analysis
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Ritz, E.; Rychlik, I.; Locatelli, F.; Halimi, S. End-stage renal failure in type 2 diabetes: A medical catastrophe of worldwide dimensions. Am. J. Kidney Dis 1999, 34, 795–808. [Google Scholar]
- Pugh, J.A.; Medina, R.; Ramirez, M. Comparison of the course to end-stage renal disease of type 1 (insulin-dependent) and type 2 (non-insulin-dependent) diabetic nephropathy. Diabetologia 1993, 36, 1094–1098. [Google Scholar]
- Schena, F.P.; Gesualdo, L. Pathogenetic mechanisms of diabetic nephropathy. J. Am. Soc. Nephrol 2005, 16, S30–S33. [Google Scholar]
- Toye, A.A.; Moir, L.; Hugill, A.; Bentley, L.; Quarterman, J.; Mijat, V.; Hough, T.; Goldsworthy, M.; Haynes, A.; Hunter, A.J.; et al. A new mouse model of type 2 diabetes, produced by N-ethyl-nitrosourea mutagenesis, is the result of a missense mutation in the glucokinase gene. Diabetes 2004, 53, 1577–1583. [Google Scholar]
- Fajans, S.S.; Bell, G.I.; Polonsky, K.S. Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. N. Engl. J. Med 2001, 345, 971–980. [Google Scholar]
- Zhang, Y.L.; Tan, X.H.; Xiao, M.F.; Li, H.; Mao, Y.Q.; Yang, X.; Tan, H.R. Establishment of liver specific glucokinase gene knockout mice: A new animal model for screening anti-diabetic drugs. Acta Pharmacol. Sin 2004, 25, 1659–1665. [Google Scholar]
- Kreunin, P.; Urquidi, V.; Lubman, D.M.; Goodison, S. Identification of metastasis-associated proteins in a human tumor metastasis model using the mass-mapping technique. Proteomics 2004, 4, 2754–2765. [Google Scholar]
- Han, Z.; Ni, J.; Smits, P.; Underhill, C.B.; Xie, B.; Chen, Y.; Liu, N.; Tylzanowski, P.; Parmelee, D.; Feng, P.; et al. Extracellular matrix protein 1 (ECM1) has angiogenic properties and is expressed by breast tumor cells. FASEB J 2001, 15, 988–994. [Google Scholar]
- Wang, H.; Maechler, P.; Antinozzi, P.A.; Hagenfeldt, K.A.; Wollheim, C.B. Hepatocyte nuclear factor 4alpha regulates the expression of pancreatic β-cell genes implicated in glucose metabolism and nutrient-induced insulin secretion. J. Biol. Chem 2000, 275, 35953–35959. [Google Scholar]
- Martinez-Jimenez, C.P.; Kyrmizi, I.; Cardot, P.; Gonzalez, F.J.; Talianidis, I. Hepatocyte nuclear factor 4alpha coordinates a transcription factor network regulating hepatic fatty acid metabolism. Mol. Cell. Biol 2010, 30, 565–577. [Google Scholar]
- Bonzo, J.A.; Ferry, C.H.; Matsubara, T.; Kim, J.H.; Gonzalez, F.J. Suppression of hepatocyte proliferation by hepatocyte nuclear factor 4alpha in adult mice. J. Biol. Chem 2012, 287, 7345–7356. [Google Scholar]
- Beale, E.G.; Harvey, B.J.; Forest, C. PCK1 and PCK2 as candidate diabetes and obesity genes. Cell Biochem. Biophys 2007, 48, 89–95. [Google Scholar]
- Granner, D.; Pilkis, S. The genes of hepatic glucose metabolism. J. Biol. Chem 1990, 265, 10173–10176. [Google Scholar]
- Ji, P.; Diederichs, S.; Wang, W.; Boing, S.; Metzger, R.; Schneider, P.M.; Tidow, N.; Brandt, B.; Buerger, H.; Bulk, E.; et al. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 2003, 22, 8031–8041. [Google Scholar]
- Takenaka, M.; Imai, E.; Kaneko, T.; Ito, T.; Moriyama, T.; Yamauchi, A.; Hori, M.; Kawamoto, S.; Okubo, K. Isolation of genes identified in mouse renal proximal tubule by comparing different gene expression profiles. Kidney Int 1998, 53, 562–572. [Google Scholar]
- Takahashi, K.; Cohen, H.J. Selenium-dependent glutathione peroxidase protein and activity: Immunological investigations on cellular and plasma enzymes. Blood 1986, 68, 640–645. [Google Scholar]
- Piggott, L.; Omidvar, N.; Perez, S.M.; Eberl, M.; Clarkson, R.W. Suppression of apoptosis inhibitor c-FLIP selectively eliminates breast cancer stem cell activity in response to the anti-cancer agent, TRAIL. Breast Cancer Res 2011, 13, R88. [Google Scholar]
- Grupe, A.; Hultgren, B.; Ryan, A.; Ma, Y.H.; Bauer, M.; Stewart, T.A. Transgenic knockouts reveal a critical requirement for pancreatic beta cell glucokinase in maintaining glucose homeostasis. Cell 1995, 83, 69–78. [Google Scholar]
- Postic, C.; Shiota, M.; Niswender, K.D.; Jetton, T.L.; Chen, Y.; Moates, J.M.; Shelton, K.D.; Lindner, J.; Cherrington, A.D.; Magnuson, M.A. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J. Biol. Chem 1999, 274, 305–315. [Google Scholar]
- Bali, D.; Svetlanov, A.; Lee, H.W.; Fusco-DeMane, D.; Leiser, M.; Li, B.; Barzilai, N.; Surana, M.; Hou, H.; Fleischer, N.; et al. Animal model for maturity-onset diabetes of the young generated by disruption of the mouse glucokinase gene. J. Biol. Chem 1995, 270, 21464–21467. [Google Scholar]
- Bennett, K.; James, C.; Mutair, A.; Al-Shaikh, H.; Sinani, A.; Hussain, K. Four novel cases of permanent neonatal diabetes mellitus caused by homozygous mutations in the glucokinase gene. Pediatr. Diabetes 2011, 12, 192–196. [Google Scholar]
- Martin, D.; Bellanne-Chantelot, C.; Deschamps, I.; Froguel, P.; Robert, J.J.; Velho, G. Long-term follow-up of oral glucose tolerance test-derived glucose tolerance and insulin secretion and insulin sensitivity indexes in subjects with glucokinase mutations (MODY2). Diabetes Care 2008, 31, 1321–1323. [Google Scholar]
- Gardner, D.S.; Tai, E.S. Clinical features and treatment of maturity onset diabetes of the young (MODY). Diabetes Metab. Syndr. Obes 2012, 5, 101–108. [Google Scholar]
- Inoue, M.; Sakuraba, Y.; Motegi, H.; Kubota, N.; Toki, H.; Matsui, J.; Toyoda, Y.; Miwa, I.; Terauchi, Y.; Kadowaki, T.; et al. A series of maturity onset diabetes of the young, type 2 (MODY2) mouse models generated by a large-scale ENU mutagenesis program. Hum. Mol. Genet 2004, 13, 1147–1157. [Google Scholar]
- Sikaris, K. Biochemistry on the human scale. Clin. Biochem. Rev 2010, 31, 121–128. [Google Scholar]
- Killard, A.J.; Smyth, M.R. Creatinine biosensors: Principles and designs. Trends Biotechnol 2000, 18, 433–437. [Google Scholar]
- Olson, G.E.; Whitin, J.C.; Hill, K.E.; Winfrey, V.P.; Motley, A.K.; Austin, L.M.; Deal, J.; Cohen, H.J.; Burk, R.F. Extracellular glutathione peroxidase (Gpx3) binds specifically to basement membranes of mouse renal cortex tubule cells. Am. J. Physiol. Renal Physiol 2010, 298, F1244–F1253. [Google Scholar]
- Avissar, N.; Ornt, D.B.; Yagil, Y.; Horowitz, S.; Watkins, R.H.; Kerl, E.A.; Takahashi, K.; Palmer, I.S.; Cohen, H.J. Human kidney proximal tubules are the main source of plasma glutathione peroxidase. Am. J. Physiol 1994, 266, C367–C375. [Google Scholar]
- Brigelius-Flohe, R. Glutathione peroxidases and redox-regulated transcription factors. Biol. Chem 2006, 387, 1329–1335. [Google Scholar]
- Brigelius-Flohe, R.; Kipp, A. Glutathione peroxidases in different stages of carcinogenesis. Biochim. Biophys. Acta 2009, 1790, 1555–1568. [Google Scholar]
- Hayes, J.D.; McLellan, L.I. Glutathione and glutathione-dependent enzymes represent a co-ordinately regulated defence against oxidative stress. Free Radic. Res 1999, 31, 273–300. [Google Scholar]
- Herbette, S.; Roeckel-Drevet, P.; Drevet, J.R. Seleno-independent glutathione peroxidases. More than simple antioxidant scavengers. FEBS J 2007, 274, 2163–2180. [Google Scholar]
- Wingler, K.; Brigelius-Flohe, R. Gastrointestinal glutathione peroxidase. BioFactors 1999, 10, 245–249. [Google Scholar]
- Brigelius-Flohe, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2012. [Google Scholar] [CrossRef]
- De Haan, J.B.; Bladier, C.; Griffiths, P.; Kelner, M.; O’Shea, R.D.; Cheung, N.S.; Bronson, R.T.; Silvestro, M.J.; Wild, S.; Zheng, S.S.; et al. Mice with a homozygous null mutation for the most abundant glutathione peroxidase, Gpx1, show increased susceptibility to the oxidative stress-inducing agents paraquat and hydrogen peroxide. J. Biol. Chem 1998, 273, 22528–22536. [Google Scholar]
- Wang, R.; Gao, H.; Xu, W.; Li, H.; Mao, Y.; Wang, Y.; Guo, T.; Wang, X.; Song, R.; Li, Z.; et al. Differential expression of genes and changes in glucose metabolism in the liver of liver-specific glucokinase gene knockout mice. Gene 2013, 516, 248–254. [Google Scholar]
- Okada, S.; Shikata, K.; Matsuda, M.; Ogawa, D.; Usui, H.; Kido, Y.; Nagase, R.; Wada, J.; Shikata, Y.; Makino, H. Intercellular adhesion molecule-1-deficient mice are resistant against renal injury after induction of diabetes. Diabetes 2003, 52, 2586–2593. [Google Scholar]
- Sato, S.; Sasaki, Y.; Adachi, A.; Ghazizadeh, M. Validation of glomerular basement membrane thickness changes with aging in minimal change disease. Pathobiology 2010, 77, 315–319. [Google Scholar]
- Dawson, H.D.; Beshah, E.; Nishi, S.; Solano-Aguilar, G.; Morimoto, M.; Zhao, A.; Madden, K.B.; Ledbetter, T.K.; Dubey, J.P.; Shea-Donohue, T.; et al. Localized multigene expression patterns support an evolving Th1/Th2-like paradigm in response to infections with Toxoplasma gondii and Ascaris suum. Infect. Immun 2005, 73, 1116–1128. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar]
- Cao, H.; Hininger-Favier, I.; Kelly, M.A.; Benaraba, R.; Dawson, H.D.; Coves, S.; Roussel, A.M.; Anderson, R.A. Green tea polyphenol extract regulates the expression of genes involved in glucose uptake and insulin signaling in rats fed a high fructose diet. J. Agric. Food Chem 2007, 55, 6372–6378. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 26 weeks | 40 weeks | 60 weeks | 85 weeks | ||
|---|---|---|---|---|---|
| Body weight (g) | gckw/w | 36.76 ± 2.64 | 47.65 ± 1.83 | 45.11 ± 4.13 | 39.36 ± 5.37 |
| gckw/− | 39.61 ± 4.47 | 47.55 ± 1.93 | 47.42 ± 2.63 | 45.67 ± 4.16 | |
| Kidney/body weight (%) | gckw/w | 1.05 ± 0.12 | 1.29 ± 0.35 | 1.07 ± 0.19 | 1.18 ± 0.19 |
| gckw/− | 1.08 ± 0.33 | 1.22 ± 0.32 | 1.00 ± 0.08 | 1.22 ± 0.19 | |
| Serum | |||||
| Triglyceride (mg/dL) | gckw/w | 137.75 ± 6.45 | 107.33 ± 16.46 | 120.16 ± 37.62 | 87.67 ± 15.89 |
| gckw/− | 123 ± 14.31 | 106.41 ± 21.64 | 131.62 ± 26.66 | 91.00 ± 27.06 | |
| Total cholesterol (mg/dL) | gckw/w | 128.67 ± 20.26 | 131.11 ± 16.66 | 147.72 ± 21.44 | 147.33 ± 17.39 |
| gckw/− | 124.40 ± 14.45 | 137.27 ± 25.05 | 125.69 ± 13.22 | 124.25 ± 34.24 | |
| Urea nitrogen (mmol/L) | gckw/w | 7.37 ± 0.19 | 7.39 ± 0.09 | 7.16 ± 0.24 | 6.90 ± 0.07 |
| gckw/− | 7.29 ± 0.08 | 7.37 ± 0.11 | 7.16 ± 0.11 | 6.82 ± 0.15 | |
| Creatinine (mg/dL) | gckw/w | 0.73 ± 0.07 | 0.65 ± 0.11 | 0.81 ± 0.04 | 0.74 ± 0.20 |
| gckw/− | 0.84 ± 0.10 | 1.04 ± 0.20 * | 0.89 ± 0.19 | 0.57 ± 0.15 | |
| Urine | |||||
| Volume (mL/24h) | gckw/w | 0.65 ± 0.13 | 2.90 ± 0.37 | 1.28 ± 0.51 | 1.73 ± 0.31 |
| gckw/− | 0.77 ± 0.21 | 3.17 ± 0.67 | 2.07 ± 0.49 * | 2.07 ± 0.21 | |
| Protein (μg/mL) | gckw/w | 49.32 ± 9.67 | 27.52 ± 4.57 | 20.68 ± 7.41 | 18.17 ± 2.72 |
| gckw/− | 32.42 ± 14.22 | 152.18 ± 39.54 ** | 163.63 ± 50.28 ** | 100.00 ± 18.58 ** |
| GenBank Accession | Blast result | Number of clones | Function |
|---|---|---|---|
| NM_007899.2 | Ecm1, extracellular matrix protein 1 | 30 | Signal transduction |
| NM_008261.2 | Hnf4α, hepatic nuclear factor 4, alpha | 27 | Transcription regulation |
| NM_011044.2 | Pck1, phosphoenolpyruvate carboxykinase 1, cytosolic | 26 | Glucose metabolism |
| NR_002847.2 | Malat1, metastasis associated lung adenocarcinoma transcript 1 | 17 | Unknown, long non-coding RNA |
| NM_029550.4 | Keg1, kidney expressed gene 1 | 13 | Transferase |
| NM_016805.2 | Hnrnpu, heterogeneous nuclear ribonucleoprotein U | 12 | Ribosomal protein |
| NM_025337.3 | Akr7a5, aldo-keto reductase family 7, member A5 | 9 | Oxidoreductase |
| NM_001081158.2 | 1300001I01Rik, RIKEN cDNA 1300001I01 gene | 7 | Unknown |
| NM_007390.3 | Chrna7, cholinergic receptor, nicotinic, alpha polypeptide 7 | 5 | Transporter |
| NM_001130526.1 | Lzts2, leucine zipper, putative tumor suppressor 2 | 2 | Cell cycle |
| NM_018860.4 | Rpl41, ribosomal protein L41 | 2 | Ribosomal protein |
| NM_021607.3 | Ncstn, nicastrin | 1 | Signal transduction |
| NM_133218.2 | Zfp704, zinc finger protein 704 | 1 | Metal ion binding |
| GenBank accession | Blast result | Number of clones | Function |
|---|---|---|---|
| NM_008161.3 | Gpx3, glutathione peroxidase 3 | 17 | Oxidoreductase |
| NM_145758.1 | 0610010K14Rik, RIKEN cDNA 0610010K14 gene | 15 | Unknown |
| NM_207653.3 | c-Flip, cellular FLICE-like inhibitory protein | 12 | Inhibitor of apoptosis |
| NM_001159571.1 | Ephb4, Eph receptor B4 | 9 | Developmental protein |
| NM_025974.2 | Rpl14, ribosomal protein L14 | 5 | Ribosomal protein |
| NM_025701.4 | Trappc5, trafficking protein particle complex 5 | 3 | Transporter |
| NM_019883.3 | Uba52, ubiquitin A-52 residue ribosomal protein fusion product 1 | 3 | Ribosomal protein |
| NM_010106.2 | Eef1a1, eukaryotic translation elongation factor 1 alpha 1 | 2 | Protein biosynthesis |
| NM_033080.2 | Nudt19, nudix (nucleoside diphosphate linked moiety X)-type motif 19 | 2 | Hydrolase |
| NM_148932.2 | Pom121, nuclear pore membrane protein 121 | 1 | Transporter |
| NM_001168623.1 | Znrf1, zinc and ring finger 1 | 1 | Metal ion binding |
| NM_001159483.1 | Rpl19, ribosomal protein L19 | 1 | Ribosomal protein |
| Gene | Sense primer | Antisense primer |
|---|---|---|
| HNF4α | GGTCCATGGTGTTTAAGGACGTG | GTCATCAATCTGCAGCTCTTGGAA |
| GPX3 | AACGTAGCCAGCTACTGAGGTCTGA | CTGTTTGCCAAATTGGTTGGAAG |
| c-FLIP | TGCACAGCAGACGTATCTCACTTG | TGTTCCACGCATACACTTTGTCC |
| MALAT1 | GAAGACAGGAGCGGCAGACA | GCTTCACCACCACATCCGTATG |
| KEG1 | ACTAACTTGGGCAAGGTCAAGCA | GCAGCATGTGTAACATCCAGTGAG |
| PCK1 | GAACTGAGACTCGCCCTATGTG | GTTGCAGGCCCAGTTGTTGA |
| β-actin | CATCCGTAAAGACCTCTATGCCAAC | ATGGAGCCACCGATCCACA |
| PCR conditions 50 °C × 5 s, 95 °C × 10 min, 40 cycles of 92 °C × 15 s and 60 °C × 60 s. | ||
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Xu, W.; Li, H.; Wang, R.; Lei, Z.; Mao, Y.; Wang, X.; Zhang, Y.; Guo, T.; Song, R.; Zhang, X.; et al. Differential Expression of Genes Associated with the Progression of Renal Disease in the Kidneys of Liver-Specific Glucokinase Gene Knockout Mice. Int. J. Mol. Sci. 2013, 14, 6467-6486. https://doi.org/10.3390/ijms14036467
Xu W, Li H, Wang R, Lei Z, Mao Y, Wang X, Zhang Y, Guo T, Song R, Zhang X, et al. Differential Expression of Genes Associated with the Progression of Renal Disease in the Kidneys of Liver-Specific Glucokinase Gene Knockout Mice. International Journal of Molecular Sciences. 2013; 14(3):6467-6486. https://doi.org/10.3390/ijms14036467
Chicago/Turabian StyleXu, Wei, Hui Li, Rong Wang, Zhen Lei, Yiqing Mao, Xi Wang, Yizhuang Zhang, Tingting Guo, Rongjing Song, Xiaojing Zhang, and et al. 2013. "Differential Expression of Genes Associated with the Progression of Renal Disease in the Kidneys of Liver-Specific Glucokinase Gene Knockout Mice" International Journal of Molecular Sciences 14, no. 3: 6467-6486. https://doi.org/10.3390/ijms14036467