Adsorption and Orientation of Human Islet Amyloid Polypeptide (hIAPP) Monomer at Anionic Lipid Bilayers: Implications for Membrane-Mediated Aggregation

Abstract
:1. Introduction
2. Results and Discussion
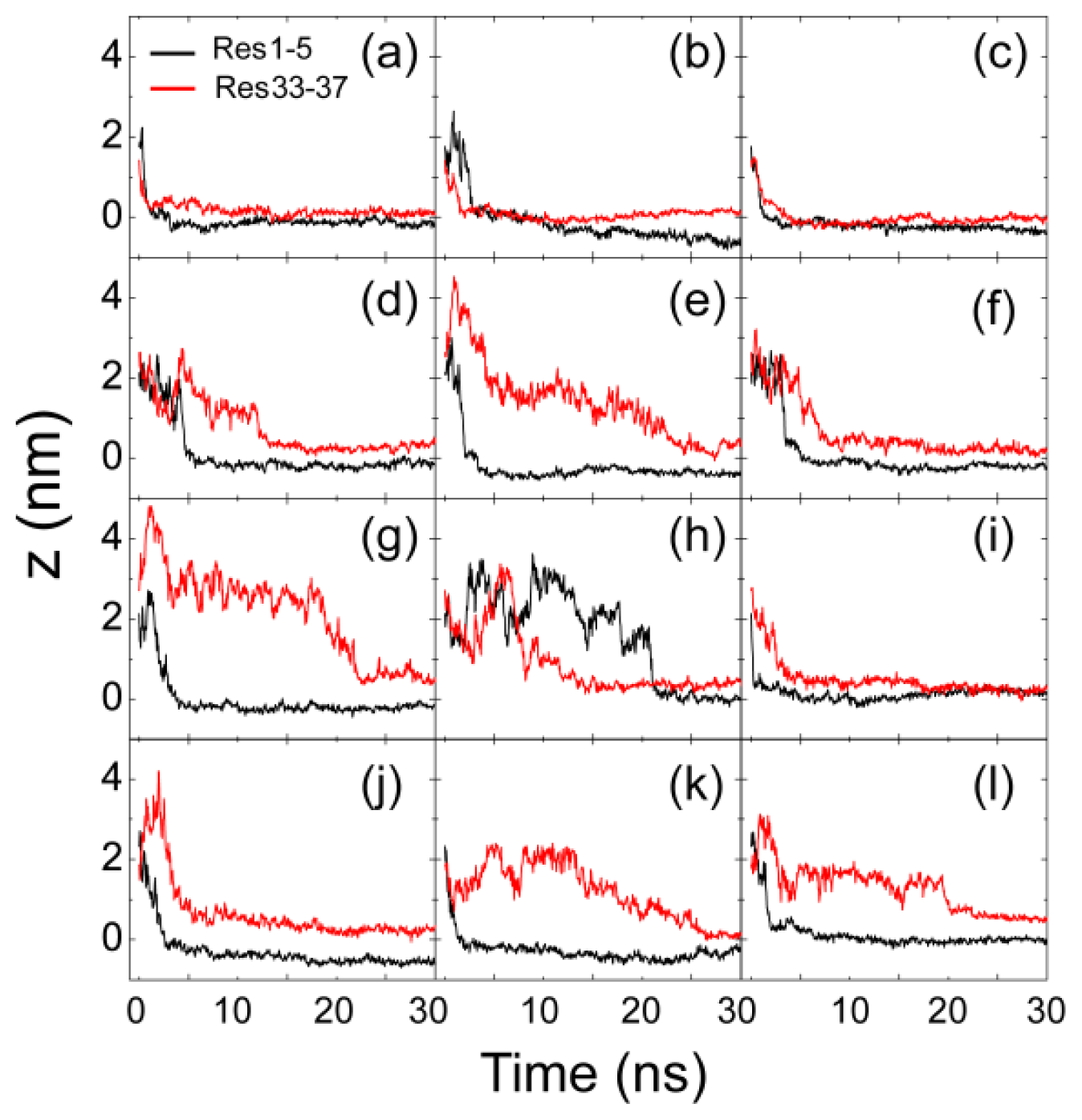
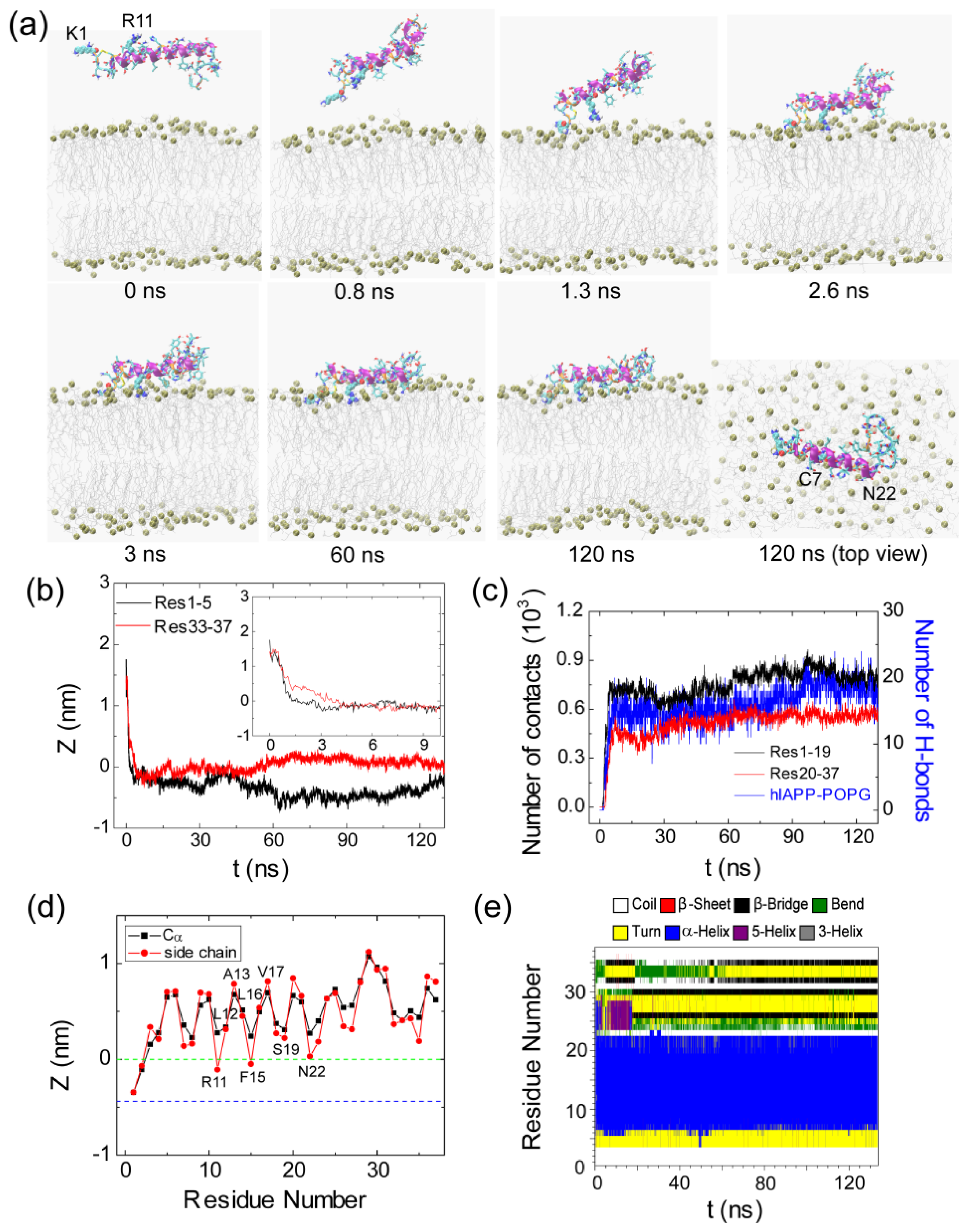
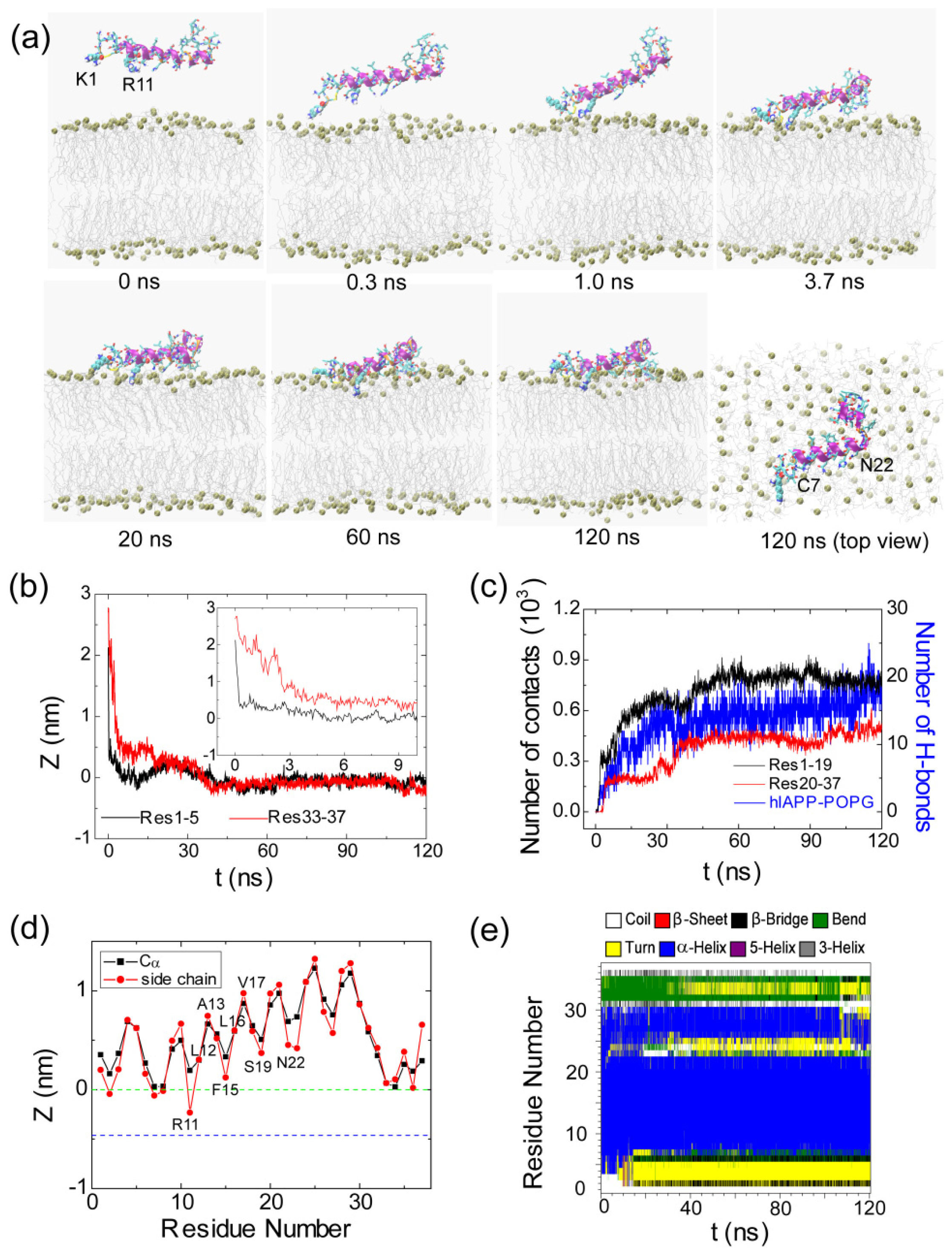
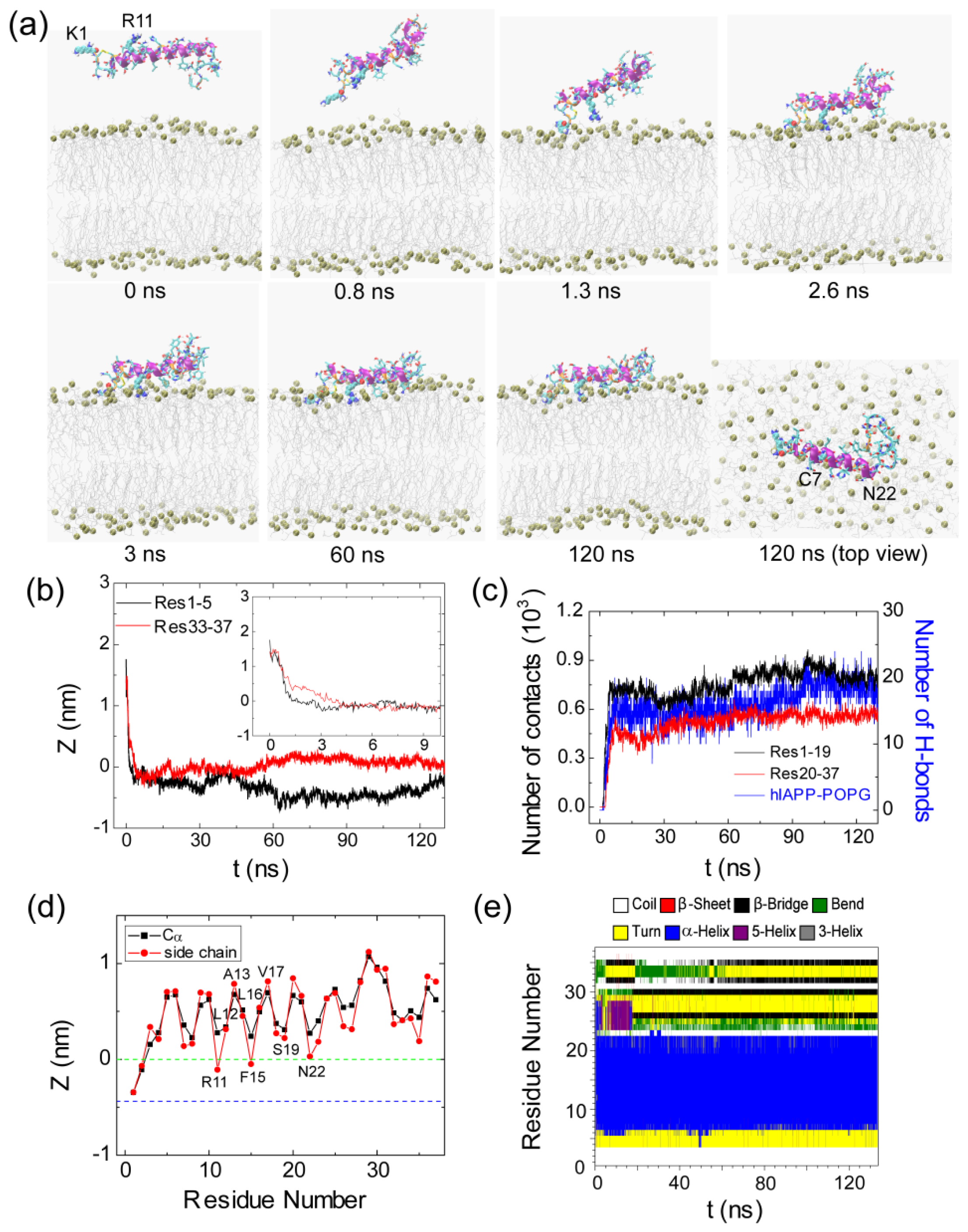
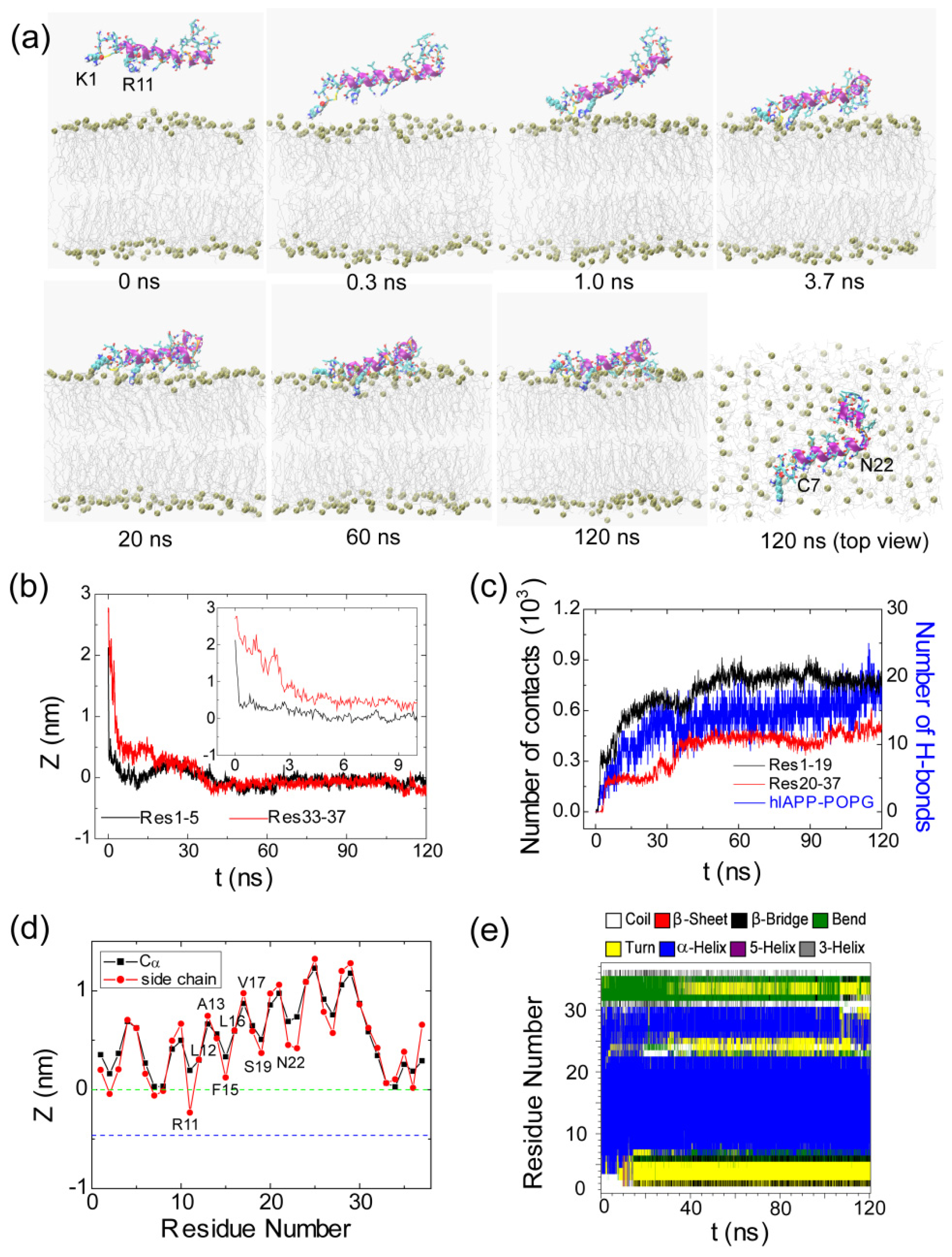
2.1. Adsorption of hIAPP Monomer from Aqueous Solution to the POPG Bilayer Is Mostly Initiated from the Positively Charged N-terminal Residues
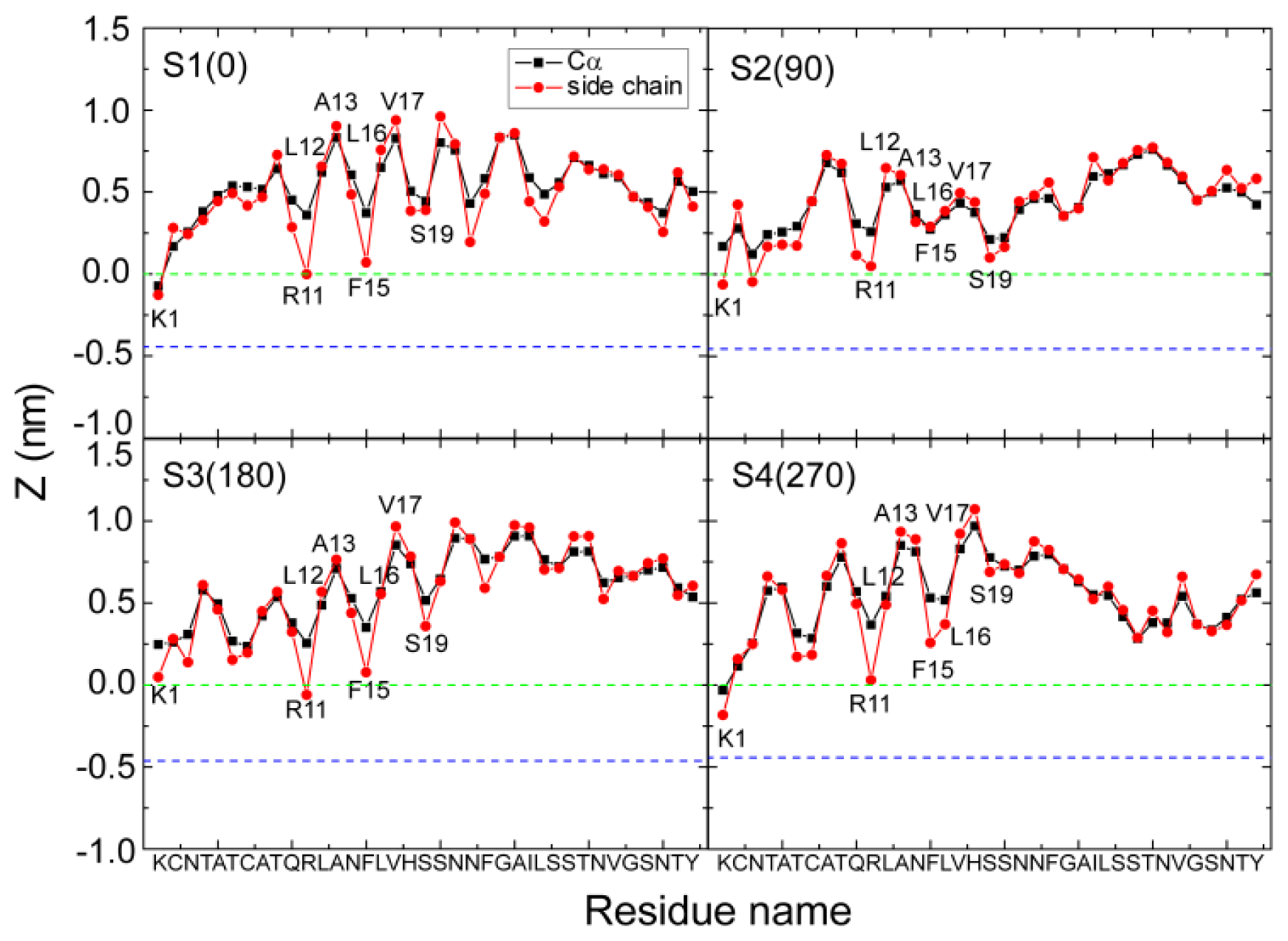
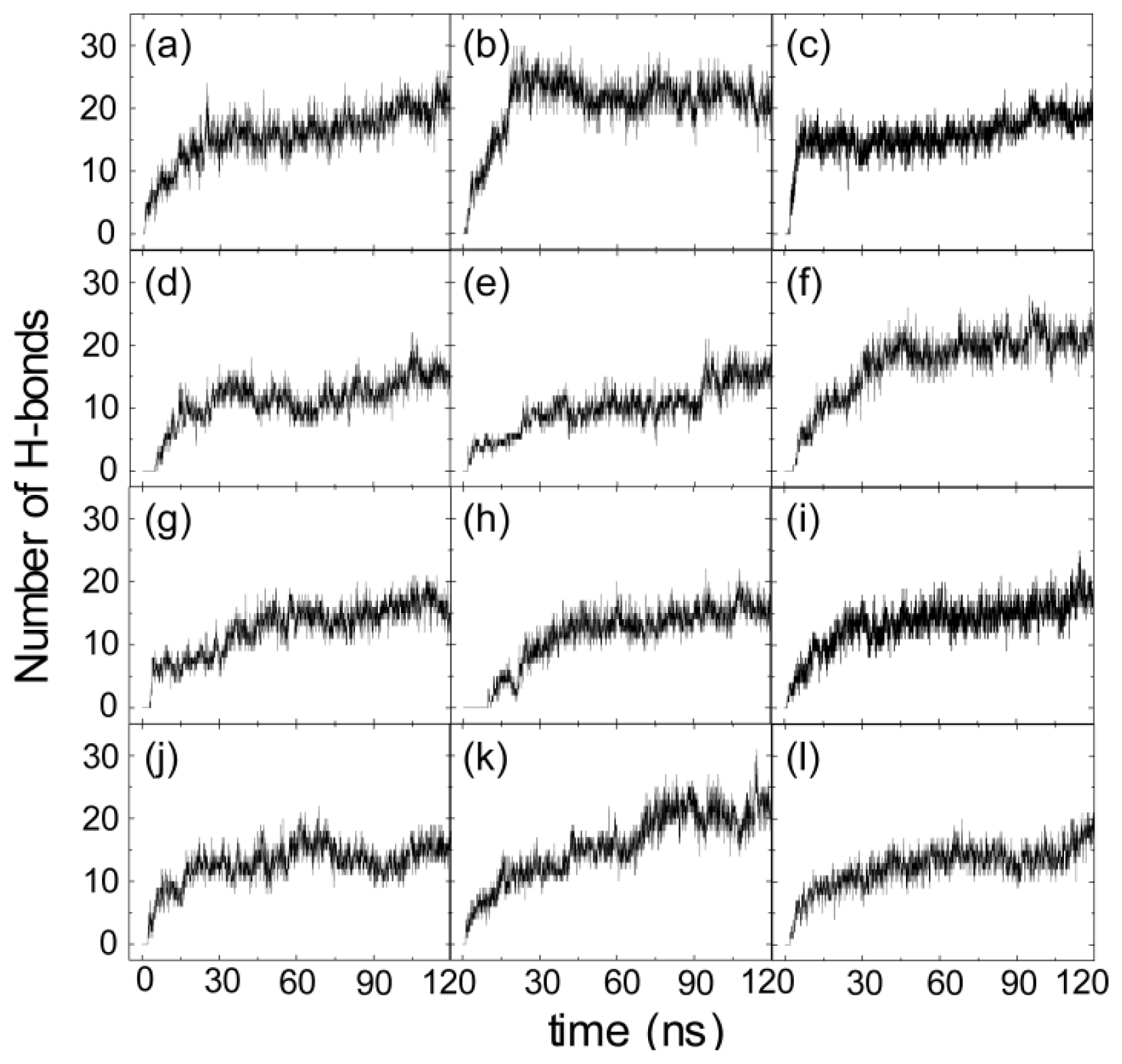
2.2. hIAPP Monomer Has a Preferred Binding Orientation to the POPG Membrane Surface
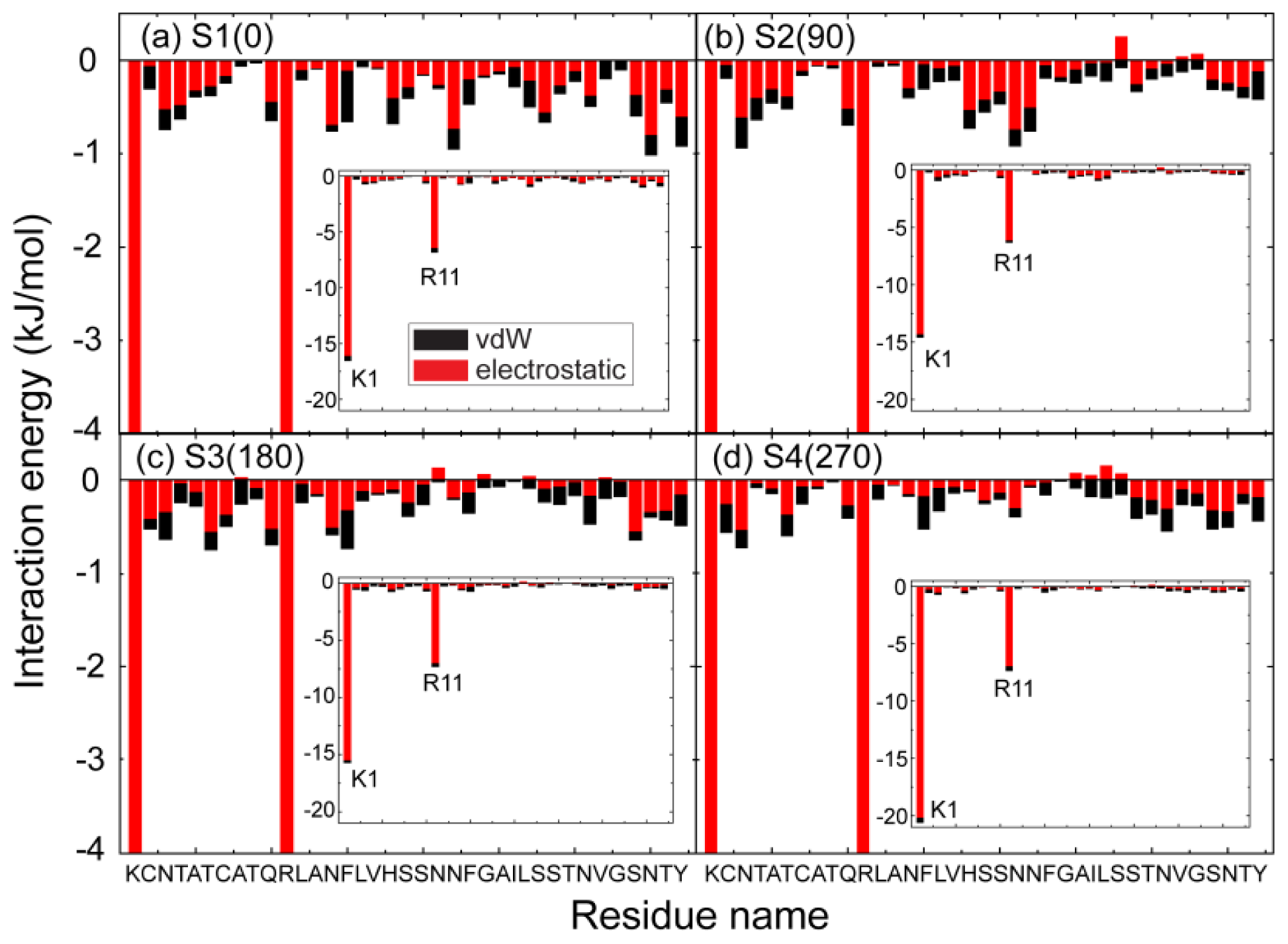
2.3. Electrostatic Interaction Plays an Important Role on the Binding Behavior of hIAPP at Bilayer Surface
2.4. hIAPP Monomer Adsorbed at Membrane Surface Displays Negligible Perturbation on Membrane Structure
2.5. Implications for Membrane-Mediated hIAPP Aggregation
3. Experimental Section
3.1. Peptide-Membrane System
3.2. MD Simulations
3.3. Analysis
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Selkoe, D.J. Folding proteins in fatal ways. Nature 2003, 426, 900–904. [Google Scholar]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem 2006, 75, 333–366. [Google Scholar]
- Ferrone, F.A. Nucleation: The connections between equilibrium and kinetic behavior. Methods Enzymol 2006, 412, 285–299. [Google Scholar]
- Westermark, P.; Andersson, A.; Westermark, G.T. Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol. Rev 2011, 91, 795–826. [Google Scholar]
- Demuro, A.; Mina, E.; Kayed, R.; Milton, S.C.; Parker, I.; Glabe, C.G. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J. Biol. Chem 2005, 280, 17294–17300. [Google Scholar]
- Engel, M.F.M.; Khemtemourian, L.; Kleijer, C.C.; Meeldijk, H.J.D.; Jacobs, J.; Verkleij, A.J.; de Kruijff, B.; Killian, J.A.; Hoeppener, J.W.M. Membrane damage by human islet amyloid polypeptide through fibril growth at the membrane. Proc. Natl. Acad. Sci. USA 2008, 105, 6033–6038. [Google Scholar]
- Jayasinghe, S.A.; Langen, R. Membrane interaction of islet amyloid polypeptide. Biochim. Biophys. Acta 2007, 1768, 2002–2009. [Google Scholar]
- Engel, M.F.M. Membrane permeabilization by islet amyloid polypeptide. Chem. Phys. Lipids 2009, 160, 1–10. [Google Scholar]
- Gurlo, T.; Ryazantsev, S.; Huang, C.J.; Yeh, M.W.; Reber, H.A.; Hines, O.J.; O’Brien, T.D.; Glabe, C.G.; Butler, P.C. Evidence for proteotoxicity in beta cells in Type 2 diabetes toxic islet amyloid polypeptide oligomers form intracellularly in the secretory pathway. Am. J. Pathol 2010, 176, 861–869. [Google Scholar]
- Brender, J.R.; Salamekh, S.; Ramamoorthy, A. Membrane disruption and early events in the aggregation of the diabetes related peptide IAPP from a molecular perspective. Acc. Chem. Res 2012, 45, 454–462. [Google Scholar]
- Höppener, J.W.M.; Ahrén, B.; Lips, C.J.M. Islet amyloid and type 2 diabetes mellitus. N. Engl. J. Med 2000, 343, 411–419. [Google Scholar]
- Butterfield, S.M.; Lashuel, H.A. Amyloidogenic protein–membrane interactions: Mechanistic insight from model systems. Angew. Chem. Int. Edit 2010, 49, 5628–5654. [Google Scholar]
- Hebda, J.A.; Miranker, A.D. The interplay of catalysis and toxicity by amyloid intermediates on lipid bilayers: Insights from type II diabetes. Annu. Rev. Biophys 2009, 38, 125–152. [Google Scholar]
- Cooper, G.J.S.; Willis, A.C.; Clark, A.; Turner, R.C.; Sim, R.B.; Reid, K.B.M. Purification and characterization of a peptide from amyloid-rich pancreases of type-2 diabetic-patients. Proc. Natl. Acad. Sci. USA 1987, 84, 8628–8632. [Google Scholar]
- Miranker, A.D.; Williamson, J.A. Direct detection of transient alpha-helical states in islet amyloid polypeptide. Protein Sci 2007, 16, 110–117. [Google Scholar]
- Yonemoto, I.T.; Kroon, G.J.A.; Dyson, H.J.; Balch, W.E.; Kelly, J.W. Amylin proprotein processing generates progressively more amyloidogenic peptides that initially sample the helical state. Biochemistry 2008, 47, 9900–9910. [Google Scholar]
- Vaiana, S.M.; Best, R.B.; Yau, W.M.; Eaton, W.A.; Hofrichter, J. Evidence for a partially structured state of the amylin monomer. Biophys. J 2009, 97, 2948–2957. [Google Scholar]
- Jayasinghe, S.A.; Langen, R. Lipid membranes modulate the structure of islet amyloid polypeptide. Biochemistry 2005, 44, 12113–12119. [Google Scholar]
- Brender, J.R.; Lee, E.L.; Cavitt, M.A.; Gafni, A.; Steel, D.G.; Ramamoorthy, A. Amyloid fiber formation and membrane disruption are separate processes localized in two distinct regions of IAPP, the type-2-diabetes-related peptide. J. Am. Chem. Soc 2008, 130, 6424–6429. [Google Scholar]
- Williamson, J.A.; Loria, J.P.; Miranker, A.D. Helix stabilization precedes aqueous and bilayer-catalyzed fiber formation in islet amyloid polypeptide. J. Mol. Biol 2009, 393, 383–396. [Google Scholar]
- Apostolidou, M.; Jayasinghe, S.A.; Langen, R. Structure of alpha-helical membrane-bound human islet amyloid polypeptide and its implications for membrane-mediated misfolding. J. Biol. Chem 2008, 283, 17205–17210. [Google Scholar]
- Patil, S.M.; Xu, S.; Sheftic, S.R.; Alexandrescu, A.T. Dynamic alpha-helix structure of micelle-bound human amylin. J. Biol. Chem 2009, 284, 11982–11991. [Google Scholar]
- Nanga, R.P.R.; Brender, J.R.; Vivekanandan, S.; Ramamoorthy, A. Structure and membrane orientation of IAPP in its natively amidated form at physiological pH in a membrane environment. Biochim. Biophys. Acta 2011, 1808, 2337–2342. [Google Scholar]
- Knight, J.D.; Hebda, J.A.; Miranker, A.D. Conserved and cooperative assembly of membrane-bound alpha-helical states of islet amyloid polypeptide. Biochemistry 2006, 45, 9496–9508. [Google Scholar]
- Laghaei, R.; Mousseau, N.; Wei, G. Effect of the disulfide bond on the monomeric structure of human amylin studied by combined Hamiltonian and temperature replica exchange molecular dynamics simulations. J. Phys. Chem. B 2010, 114, 7071–7077. [Google Scholar]
- Andrews, M.N.; Winter, R. Comparing the structural properties of human and rat islet amyloid polypeptide by MD computer simulations. Biophys. Chem 2011, 156, 43–50. [Google Scholar]
- Dupuis, N.F.; Wu, C.; Shea, J.E.; Bowers, M.T. The amyloid formation mechanism in human IAPP: Dimers have beta-strand monomer-monomer interfaces. J. Am. Chem. Soc 2011, 133, 7240–7243. [Google Scholar]
- Laghaei, R.; Mousseau, N.; Wei, G.H. Structure and thermodynamics of amylin dimer studied by hamiltonian-temperature replica exchange molecular dynamics simulations. J. Phys. Chem. B 2011, 115, 3146–3154. [Google Scholar]
- Jiang, P.; Xu, W.; Mu, Y. Amyloidogenesis abolished by proline substitutions but enhanced by lipid binding. PLoS Comput. Biol 2009, 5, e1000357. [Google Scholar]
- Xu, W.; Ping, J.; Li, W.; Mu, Y. Assembly dynamics of two-beta sheets revealed by molecular dynamics simulations. J. Chem. Phys 2009, 130, 164709. [Google Scholar]
- Mo, Y.; Lu, Y.; Wei, G.; Derreumaux, P. Structural diversity of the soluble trimers of the human amylin(20–29) peptide revealed by molecular dynamics simulations. J. Chem. Phys 2009, 130, 125101–125106. [Google Scholar]
- Sciacca, M.F.M.; Pappalardo, M.; Attanasio, F.; Milardi, D.; La Rosa, C.; Grasso, D.M. Are fibril growth and membrane damage linked processes? An experimental and computational study of IAPP(12–18) and IAPP(21–27) peptides. New J. Chem 2010, 34, 200–207. [Google Scholar]
- Duan, M.; Fan, J.; Huo, S. Conformations of islet amyloid polypeptide monomers in a membrane environment: Implications for fibril formation. PLoS One 2012, 7, e47150. [Google Scholar]
- Zhao, J.; Luo, Y.; Jang, H.; Yu, X.; Wei, G.; Nussinov, R.; Zheng, J. Probing ion channel activity of human islet amyloid polypeptide (amylin). Biochim. Biophys. Acta 2012, 1818, 3121–3130. [Google Scholar]
- Zhang, Y.; Luo, Y.; Deng, Y.; Mu, Y.; Wei, G. Lipid Interaction and membrane perturbation of human islet amyloid polypeptide monomer and dimer by molecular dynamics simulations. PLoS One 2012, 7, e38191. [Google Scholar]
- Engel, M.F.; Yigittop, H.; Elgersma, R.C.; Rijkers, D.T.; Liskamp, R.M.; de Kruijff, B.; Hoppener, J.W.; Antoinette Killian, J. Islet amyloid polypeptide inserts into phospholipid monolayers as monomer. J. Mol. Biol 2006, 356, 783–789. [Google Scholar]
- Knight, J.D.; Miranker, A.D. Phospholipid catalysis of diabetic amyloid assembly. J. Mol. Biol 2004, 341, 1175–1187. [Google Scholar]
- Lopes, D.H.J.; Meister, A.; Gohlke, A.; Hauser, A.; Blume, A.; Winter, R. Mechanism of islet amyloid polypeptide fibrillation at lipid interfaces studied by infrared reflection absorption spectroscopy. Biophys. J 2007, 93, 3132–3141. [Google Scholar]
- Yu, X.; Zheng, J. Cholesterol promotes the interaction of Alzheimer β-amyloid monomer with lipid bilayer. J. Mol. Biol 2012, 421, 561–571. [Google Scholar]
- Tofoleanu, F.; Buchete, N.-V. Molecular interactions of alzheimer’s Aβ protofilaments with lipid membranes. J. Mol. Biol 2012, 421, 572–586. [Google Scholar]
- Vermeer, L.S.; de Groot, B.L.; Reat, V.; Milon, A.; Czaplicki, J. Acyl chain order parameter profiles in phospholipid bilayers: Computation from molecular dynamics simulations and comparison with H-2 NMR experiments. Eur. Biophys. J. Biophy 2007, 36, 919–931. [Google Scholar]
- Janson, J.; Ashley, R.H.; Harrison, D.; McIntyre, S.; Butler, P.C. The mechanism of islet amyloid polypeptide toxicity is membrane disruption by intermediate-sized toxic amyloid particles. Diabetes 1999, 48, 491–498. [Google Scholar]
- Jha, S.; Sellin, D.; Seidel, R.; Winter, R. Amyloidogenic propensities and conformational properties of ProIAPP and IAPP in the presence of lipid bilayer membranes. J. Mol. Biol 2009, 389, 907–920. [Google Scholar]
- Abedini, A.; Raleigh, D.P. A role for helical intermediates in amyloid formation by natively unfolded polypeptides? Phys. Biol. 2009, 6. [Google Scholar] [CrossRef]
- Manna, M.; Mukhopadhyay, C. Molecular dynamics simulations of the interactions of kinin peptides with an anionic POPG bilayer. Langmuir 2011, 27, 3713–3722. [Google Scholar]
- Chang, Z.W.; Luo, Y.; Zhang, Y.; Wei, G.H. Interactions of A beta 25–35 beta-Barrel-like oligomers with anionic lipid bilayer and resulting membrane leakage: An all-atom molecular dynamics study. J. Phys. Chem. B 2011, 115, 1165–1174. [Google Scholar]
- Elmore, D.E. Molecular dynamics simulation of a phosphatidylglycerol membrane. Febs. Lett 2006, 580, 144–148. [Google Scholar]
- Soliman, W.; Bhattacharjee, S.; Kaur, K. Interaction of an antimicrobial peptide with a model lipid bilayer using molecular dynamics simulation. Langmuir 2009, 25, 6591–6595. [Google Scholar]
- Lindahl, E.; Hess, B.; van der Spoel, D. GROMACS 3.0: A package for molecular simulation and trajectory analysis. J. Mol. Model 2001, 7, 306–317. [Google Scholar]
- Berger, O.; Edholm, O.; Jahnig, F. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys. J 1997, 72, 2002–2013. [Google Scholar]
- Van Gunsteren, W.F.; Berendsen, H.J.C. Groningen Molecular Simulation (GROMOS) Library Manual; Biomos: Groningen, The Netherlands, 1987; pp. 1–221. [Google Scholar]
- Berendsen, H.J.C.; Postma, J.P.M.; Gunsteren, W.F.V.; Hermans, J. Interaction Models for Water in Relation to Protein Hydration; D. Reidel Publishing Co.: Dordrecht, The Netherlands, 1981; pp. 331–342. [Google Scholar]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem 1997, 18, 1463–1472. [Google Scholar]
- Miyamoto, S.; Kollman, P.A. Settle—An analytical version of the shake and rattle algorithm for rigid water models. J. Comput. Chem 1992, 13, 952–962. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Particle mesh ewald—An N.Log(N) method for ewald sums in large systems. J. Chem. Phys 1993, 98, 10089–10092. [Google Scholar]
- Patra, M.; Karttunen, M.; Hyvonen, M.T.; Falck, E.; Lindqvist, P.; Vattulainen, I. Molecular dynamics simulations of lipid bilayers: Major artifacts due to truncating electrostatic interactions. Biophys. J 2003, 84, 3636–3645. [Google Scholar]
- Wiedmann, T.; Salmon, A.; Wong, V. Phase-behavior of mixtures of dppc and popg. Biochim. Biophys. Acta 1993, 1167, 114–120. [Google Scholar]
- Tolokh, I.S.; Vivcharuk, V.; Tomberli, B.; Gray, C.G. Binding free energy and counterion release for adsorption of the antimicrobial peptide lactoferricin B on a POPG membrane. Phys. Rev. E 2009, 80, 031911. [Google Scholar]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys 1984, 81, 3684–3690. [Google Scholar]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph 1996, 14, 33–38. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Simulation box size a × b × c (nm3) | # of water molecules | Initial distance Res1–5 (nm) | Initial distance Res33–37 (nm) | Simulation time (ns)a | Initial state |
|---|---|---|---|---|---|---|
| S1(0) | 5 × 7 × 11 | 7,102 | 1.8 | 1.4 | 120 (3) | Figure 1a |
| S2(90) | 5 × 7 × 12 | 8,325 | 2.1 | 2.6 | 120 (3) | Figure 1b |
| S3(180) | 5 × 7 × 12 | 8,327 | 2.1 | 2.7 | 120 (3) | Figure 1c |
| S4(270) | 5 × 7 × 12 | 8,311 | 2.3 | 1.9 | 120 (3) | Figure 1d |
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jia, Y.; Qian, Z.; Zhang, Y.; Wei, G. Adsorption and Orientation of Human Islet Amyloid Polypeptide (hIAPP) Monomer at Anionic Lipid Bilayers: Implications for Membrane-Mediated Aggregation. Int. J. Mol. Sci. 2013, 14, 6241-6258. https://doi.org/10.3390/ijms14036241
Jia Y, Qian Z, Zhang Y, Wei G. Adsorption and Orientation of Human Islet Amyloid Polypeptide (hIAPP) Monomer at Anionic Lipid Bilayers: Implications for Membrane-Mediated Aggregation. International Journal of Molecular Sciences. 2013; 14(3):6241-6258. https://doi.org/10.3390/ijms14036241
Chicago/Turabian StyleJia, Yan, Zhenyu Qian, Yun Zhang, and Guanghong Wei. 2013. "Adsorption and Orientation of Human Islet Amyloid Polypeptide (hIAPP) Monomer at Anionic Lipid Bilayers: Implications for Membrane-Mediated Aggregation" International Journal of Molecular Sciences 14, no. 3: 6241-6258. https://doi.org/10.3390/ijms14036241