Interplay between Hepatitis C Virus and Redox Cell Signaling

Abstract
:1. Introduction
2. HCV Infection Induces Oxidative Stress
3. Oxidative Stress Affects HCV Replication
4. Redox-Regulated Signaling Pathways: MAPK and PI3K/Akt Signaling Pathways Are Critical Controllers of HCV Replication
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Lavanchy, D. The global burden of hepatitis C. Liver Int 2009, 29, 74–81. [Google Scholar]
- Liang, T.J.; Heller, T. Pathogenesis of hepatitis C-associated hepatocellular carcinoma. Gastroenterology 2004, 127, S62–S71. [Google Scholar]
- Moradpour, D.; Penin, F.; Rice, C.M. Replication of hepatitis C virus. Nat. Rev. Microbiol 2007, 5, 453–463. [Google Scholar]
- Herker, E.; Ott, M. Unique ties between hepatitis C virus replication and intracellular lipids. Trends Endocrinol. Metab 2011, 22, 241–248. [Google Scholar]
- Manns, M.P.; Wedemeyer, H.; Cornberg, M. Treating viral hepatitis C: Efficacy, side effects, and complications. Gut 2006, 55, 1350–1359. [Google Scholar]
- Hadziyannis, S.J.; Sette, H., Jr; Morgan, T.R.; Balan, V.; Diago, M.; Marcellin, P.; Ramadori, G.; Bodenheimer, H., Jr; Bernstein, D.; Rizzetto, M.; et al. Peginterferon-alpha2a and ribavirin combination therapy in chronic hepatitis C: A randomized study of treatment duration and ribavirin dose. Ann. Intern. Med. 2004, 140, 346–355. [Google Scholar]
- Manns, M.P.; McHutchison, J.G.; Gordon, S.C.; Rustgi, V.K.; Shiffman, M.; Reindollar, R.; Goodman, Z.D.; Koury, K.; Ling, M.; Albrecht, J.K. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: A randomised trial. Lancet 2001, 358, 958–965. [Google Scholar]
- Welsch, C.; Jesudian, A.; Zeuzem, S.; Jacobson, I. New direct-acting antiviral agents for the treatment of hepatitis C virus infection and perspectives. Gut 2012, 61, i36–i46. [Google Scholar]
- Jacobson, I.M.; McHutchison, J.G.; Dusheiko, G.; Di Bisceglie, A.M.; Reddy, K.R.; Bzowej, N.H.; Marcellin, P.; Muir, A.J.; Ferenci, P.; Flisiak, R.; et al. Telaprevir for previously untreated chronic hepatitis C virus infection. N. Engl. J. Med 2011, 364, 2405–2416. [Google Scholar]
- Poordad, F.; McCone, J., Jr; Bacon, B.R.; Bruno, S.; Manns, M.P.; Sulkowski, M.S.; Jacobson, I.M.; Reddy, K.R.; Goodman, Z.D.; Boparai, N.; et al. Boceprevir for untreated chronic HCV genotype 1 infection. N. Engl. J. Med. 2011, 364, 1195–1206. [Google Scholar]
- Sherman, K.E.; Flamm, S.L.; Afdhal, N.H.; Nelson, D.R.; Sulkowski, M.S.; Everson, G.T.; Fried, M.W.; Adler, M.; Reesink, H.W.; Martin, M.; et al. Response-guided telaprevir combination treatment for hepatitis C virus infection. N. Engl. J. Med 2011, 365, 1014–1024. [Google Scholar]
- Sarrazin, C.; Hézode, C.; Zeuzem, S.; Pawlotsky, J.M. Antiviral strategies in hepatitis C virus infection. J. Hepatol 2012, 56, S88–S100. [Google Scholar]
- Bühler, S.; Bartenschlager, R. New targets for antiviral therapy of chronic hepatitis C. Liver Int 2012, 32, 9–16. [Google Scholar]
- Gallay, P.A. Cyclophilin inhibitors: A novel class of promising host-targeting anti-HCV agents. Immunol. Res 2012, 52, 200–210. [Google Scholar]
- Bode, J.G.; Brenndörfer, E.D.; Karthe, J.; Häussinger, D. Interplay between host cell and hepatitis C virus in regulating viral replication. Biol. Chem 2009, 390, 1013–1032. [Google Scholar]
- Menzel, N.; Fischl, W.; Hueging, K.; Bankwitz, D.; Frentzen, A.; Haid, S.; Gentzsch, J.; Kaderali, L.; Bartenschlager, R.; Pietschmann, T. MAP-kinase regulated cytosolic phospholipase A2 activity is essential for production of infectious hepatitis C virus particles. PLoS Pathog 2012, 8, e1002829. [Google Scholar]
- Mannová, P.; Beretta, L. Activation of the N-Ras-PI3K-Akt-mTOR pathway by hepatitis C virus: Control of cell survival and viral replication. J. Virol 2005, 79, 8742–8749. [Google Scholar]
- Liu, Z.; Tian, Y.; Machida, K.; Lai, M.M.; Luo, G.; Foung, S.K.; Ou, J.H. Transient activation of the PI3K-AKT pathway by HCV to enhance viral entry. J. Biol. Chem 2012, 287, 41922–41930. [Google Scholar]
- Cross, J.V.; Templeton, D.J. Regulation of signal transduction through protein cysteine oxidation. Antioxid. Redox Signal 2006, 8, 1819–1827. [Google Scholar]
- Filomeni, G.; Rotilio, G.; Ciriolo, M.R. Disulfide relays and phosphorylative cascades: Partners in redox-mediated signaling pathways. Cell Death Differ 2005, 12, 1555–1563. [Google Scholar]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell. Biol 2007, 39, 44–84. [Google Scholar]
- Meister, A.; Anderson, M.E. Glutathione. Ann. Rev. Biochem 1983, 52, 711–760. [Google Scholar]
- Forman, H.J.; Dickinson, D.A. Oxidative signaling and glutathione synthesis. Biofactors 2003, 17, 1–12. [Google Scholar]
- Dalle-Donne, I.; Milzani, A.; Gagliano, N.; Colombo, R.; Giustarini, D.; Rossi, R. Molecular mechanisms and potential clinical significance of S-glutathionylation. Antioxid. Redox Signal 2008, 10, 445–473. [Google Scholar]
- Pastore, A.; Piemonte, F. S-Glutathionylation signaling in cell biology: Progress and prospects. Eur. J. Pharm. Sci 2012, 46, 279–292. [Google Scholar]
- Nencioni, L.; Sgarbanti, R.; Amatore, D.; Checconi, P.; Celestino, I.; Limongi, D.; Anticoli, S.; Palamara, A.T.; Garaci, E. Intracellular redox signaling as therapeutic target for novel antiviral strategy. Curr. Pharm. Des 2011, 17, 3898–3904. [Google Scholar]
- Fraternale, A.; Paoletti, M.F.; Casabianca, A.; Oiry, J.; Clayette, P.; Vogel, J.U.; Cynatl, J., Jr.; Palamara, A.T.; Sgarbanti, R.; Garaci, E.; et al. Antiviral and immunomodulatory properties of new pro-glutathione (GSH) molecules. Curr. Med. Chem. 2006, 13, 1749–1755. [Google Scholar]
- Choi, J. Oxidative stress, endogenous antioxidants, alcohol, and hepatitis C: Pathogenic interactions and therapeutic considerations. Free Radic. Biol. Med 2012, 52, 1135–1150. [Google Scholar]
- Choi, J.; Ou, J.H. Mechanisms of liver injury. III. Oxidative stress in the pathogenesis of hepatitis C virus. Am. J. Physiol. Gastrointest. Liver Physiol 2006, 290, G847–G851. [Google Scholar]
- Sgarbanti, R.; Nencioni, L.; Amatore, D.; Coluccio, P.; Fraternale, A.; Sale, P.; Mammola, C.L.; Carpino, G.; Gaudio, E.; Magnani, M.; et al. Redox regulation of the influenza hemagglutinin maturation process: A new cell-mediated strategy for anti-influenza therapy. Antioxid. Redox Signal 2011, 15, 593–606. [Google Scholar]
- Kavouras, J.H.; Prandovszky, E.; Valyi-Nagy, K.; Kovacs, S.K.; Tiwari, V.; Kovacs, M.; Shukla, D.; Valyi-Nagy, T. Herpes simplex virus type 1 infection induces oxidative stress and the release of bioactive lipid peroxidation by-products in mouse P19N neural cell cultures. J. Neurovirol 2007, 13, 416–425. [Google Scholar]
- Peterhans, E. Oxidants and antioxidants in viral diseases: Disease mechanisms and metabolic regulation. J. Nutr 1997, 127, S962–S965. [Google Scholar]
- Seronello, S.; Sheikh, M.Y.; Choi, J. Redox regulation of hepatitis C in nonalcoholic and alcoholic liver. Free Radic. Biol. Med 2007, 43, 869–882. [Google Scholar]
- Mahmood, S.; Kawanaka, M.; Kamei, A.; Izumi, A.; Nakata, K.; Niiyama, G.; Ikeda, H.; Hanano, S.; Suehiro, M.; Togawa, K.; et al. Immunohistochemical evaluation of oxidative stress markers in chronic hepatitis C. Antioxid. Redox Signal 2004, 6, 19–24. [Google Scholar]
- Romero, M.J.; Bosch-Morell, F.; Romero, B.; Rodrigo, J.M.; Serra, M.A.; Romero, F.J. Serum malondialdehyde: Possible use for the clinical management of chronic hepatitis C patients. Free Radic. Biol. Med 1998, 25, 993–997. [Google Scholar]
- Fujita, N.; Sugimoto, R.; Ma, N.; Tanaka, H.; Iwasa, M.; Kobayashi, Y.; Kawanishi, S.; Watanabe, S.; Kaito, M.; Takei, Y. Comparison of hepatic oxidative DNA damage in patients with chronic hepatitis B and C. J. Viral Hepat 2008, 15, 498–507. [Google Scholar]
- Farias, M.S.; Budni, P.; Ribeiro, C.M.; Parisotto, E.B.; Santos, C.E.; Dias, J.F.; Dalmarco, E.M.; Fröde, T.S.; Pedrosa, R.C.; Wilhelm Filho, D. Antioxidant supplementation attenuates oxidative stress in chronic hepatitis C patients. Gastroenterol. Hepatol 2012, 35, 386–394. [Google Scholar]
- Lin, C.C.; Liu, W.H.; Wang, Z.H.; Yin, M.C. Vitamins B status and antioxidative defense in patients with chronic hepatitis B or hepatitis C virus infection. Eur. J. Nutr 2011, 50, 499–506. [Google Scholar]
- Barbaro, G.; Di Lorenzo, G.; Soldini, M.; Parrotto, S.; Bellomo, G.; Belloni, G.; Grisorio, B.; Barbarini, G. Hepatic glutathione deficiency in chronic hepatitis C: Quantitative evaluation in patients who are HIV positive and HIV negative and correlations with plasmatic and lymphocytic concentrations and with the activity of the liver disease. Am. J. Gastroenterol 1996, 91, 2569–2573. [Google Scholar]
- Loguercio, C.; Federico, A. Oxidative stress in viral and alcoholic hepatitis. Free Radic. Biol. Med 2003, 34, 1–10. [Google Scholar]
- Hosomura, N.; Kono, H.; Tsuchiya, M.; Ishii, K.; Ogiku, M.; Matsuda, M.; Fujii, H. HCV-related proteins activate Kupffer cells isolated from human liver tissues. Dig. Dis. Sci 2011, 56, 1057–1064. [Google Scholar]
- Thoren, F.; Romero, A.; Lindh, M.; Dahlgren, C.; Hellstrand, K. A hepatitis C virus-encoded, nonstructural protein (NS3) triggers dysfunction and apoptosis in lymphocytes: Role of NADPH oxidase-derived oxygen radicals. J. Leukoc. Biol 2004, 76, 1180–1186. [Google Scholar]
- Vendemiale, G.; Grattagliano, I.; Portincasa, P.; Serviddio, G.; Palasciamo, G.; Altomare, E. Oxidative stress in symptom-free HCV carriers: Relation with ALT flare-up. Eur. J. Clin. Invest 2001, 31, 54–63. [Google Scholar]
- Moriya, K.; Nakagawa, K.; Santa, T.; Shintani, Y.; Fujie, H.; Miyoshi, H.; Tsutsumi, T.; Miyazawa, T.; Ishibashi, K.; Horie, T.; et al. Oxidative stress in the absence of inflammation in a mouse model for hepatitis C virus-associated hepatocarcinogenesis. Cancer Res 2001, 61, 4365–4370. [Google Scholar]
- Korenaga, M.; Wang, T.; Li, Y.; Showalter, L.A.; Chan, T.; Sun, J.; Weinman, S.A. Hepatitis C virus core protein inhibits mitochondrial electron transport and increases reactive oxygen species (ROS) production. J. Biol. Chem 2005, 280, 37481–37488. [Google Scholar]
- Li, Y.; Boehning, D.F.; Qian, T.; Popov, V.L.; Weinman, S.A. Hepatitis C virus core protein increases mitochondrial ROS production by stimulation of Ca2+ uniporter activity. FASEB J 2007, 21, 2474–2485. [Google Scholar]
- Machida, K.; Cheng, K.T.; Lai, C.K.; Jeng, K.S.; Sung, V.M.; Lai, M.M. Hepatitis C virus triggers mitochondrial permeability transition with production of reactive oxygen species, leading to DNA damage and STAT3 activation. J. Virol 2006, 80, 7199–7207. [Google Scholar]
- Tardif, K.D.; Waris, G.; Siddiqui, A. Hepatitis C virus, ER stress, and oxidative stress. Trends Microbiol 2005, 13, 159–163. [Google Scholar]
- Robinson, L.C.; Marchant, J.S. Enhanced Ca2+ leak from ER Ca2+ stores induced by hepatitis C NS5A protein. Biochem. Biophys. Res. Commun 2008, 368, 593–599. [Google Scholar]
- Tong, W.Y.; Nagano-Fujii, M.; Hidajat, R.; Deng, L.; Takigawa, Y.; Hotta, H. Physical interaction between hepatitis C virus NS4B protein and CREB-RP/ATF6beta. Biochem. Biophys. Res. Commun 2002, 299, 366–372. [Google Scholar]
- Chan, S.W.; Egan, P.A. Hepatitis C virus envelope proteins regulate CHOP via induction of the unfolded protein response. FASEB J 2005, 19, 1510–1512. [Google Scholar]
- Santos, C.X.; Tanaka, L.Y.; Wosniak, J.; Laurindo, F.R. Mechanisms and implications of reactive oxygen species generation during the unfolded protein response: Roles of endoplasmic reticulum oxidoreductases, mitochondrial electron transport, and NADPH oxidase. Antioxid. Redox. Signal 2009, 11, 2409–2427. [Google Scholar]
- De Mochel, N.S.R.; Seronello, S.; Wang, S.H. Hepatocyte NAD(P)H oxidases as an endogenous source of reactive oxygen species during hepatitis C virus infection. Hepatology 2010, 52, 47–59. [Google Scholar]
- Boudreau, H.E.; Emerson, S.U.; Korzeniowska, A.; Jendrysik, M.A.; Leto, T.L. Hepatitis C virus (HCV) proteins induce NADPH oxidase 4 expression in a transforming growth factor beta-dependent manner: A new contributor to HCV-induced oxidative stress. J. Virol 2009, 83, 12934–12946. [Google Scholar]
- Diamond, D.L.; Syder, A.J.; Jacobs, J.M.; Sorensen, C.M.; Walters, K.A.; Proll, S.C.; McDermott, J.E.; Gritsenko, M.A.; Zhang, Q.; Zhao, R.; et al. Temporal proteome and lipidome profiles reveal hepatitis C virus-associated reprogramming of hepatocellular metabolism and bioenergetics. PLoS Pathog 2010, 6, e1000719. [Google Scholar]
- Abdalla, M.Y.; Ahmad, I.M.; Spitz, D.R.; Schmidt, W.N.; Britigan, B.E. Hepatitis C virus-core and non structural proteins lead to different effects on cellular antioxidant defenses. J. Med. Virol 2005, 76, 489–497. [Google Scholar]
- Qadri, I.; Iwahashi, M.; Capasso, J.M.; Hopken, M.W.; Flores, S.; Schaack, J.; Simon, F.R. Induced oxidative stress and activated expression of manganese superoxide dismutase during hepatitis C virus replication: Role of JNK, p38 MAPK and AP-1. Biochem. J 2004, 378, 919–928. [Google Scholar]
- Li, K.; Prow, T.; Lemon, S.M.; Beard, M.R. Cellular response to conditional expression of hepatitis C virus core protein in Huh7 cultured human hepatoma cells. Hepatology 2002, 35, 1237–1246. [Google Scholar]
- Roe, B.; Kensicki, E.; Mohney, R.; Hall, W.W. Metabolomic profile of hepatitis C virus-infected hepatocytes. PLoS One 2011, 6, e23641. [Google Scholar]
- Burdette, D.; Olivarez, M.; Waris, G. Activation of transcription factor Nrf2 by hepatitis C virus induces the cell-survival pathway. J. Gen. Virol 2010, 91, 681–690. [Google Scholar]
- Ivanov, A.V.; Smirnova, O.A.; Ivanova, O.N.; Masalova, O.V.; Kochetkov, S.N.; Isaguliants, M.G. Hepatitis C virus proteins activate NRF2/ARE pathway by distinct ROS-dependent and independent mechanisms in HUH7 cells. PLoS One 2011, 6, e24957. [Google Scholar]
- Carvajal-Yepes, M.; Himmelsbach, K.; Schaedler, S.; Ploen, D.; Krause, J.; Ludwig, L.; Weiss, T.; Klingel, K.; Hildt, E. Hepatitis C virus impairs the induction of cytoprotective Nrf2 target genes by delocalization of small Maf proteins. J. Biol. Chem 2011, 286, 8941–8951. [Google Scholar]
- Motohashi, H.; Yamamoto, M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med 2004, 10, 549–557. [Google Scholar]
- Rushmore, T.H.; Morton, M.R.; Pickett, C.B. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. J. Biol. Chem 1991, 266, 11632–11639. [Google Scholar]
- Garaci, E.; Palamara, A.T.; Ciriolo, M.R.; D’Agostini, C.; Abdel-Latif, M.S.; Aquaro, S.; Lafavia, E.; Rotilio, G. Intracellular GSH content and HIV replication in human macrophages. J. Leukoc. Biol 1997, 62, 54–59. [Google Scholar]
- Clement, S.; Pascarella, S.; Negro, F. Hepatitis C Virus infection: Molecular pathways to steatosis, insulin resistance and oxidative stress. Viruses 2009, 1, 126–143. [Google Scholar]
- Tanikawa, K.; Torimura, T. Studies on oxidative stress in liver diseases: Important future trends in liver research. Med. Mol. Morphol 2006, 39, 22–27. [Google Scholar]
- Melhem, A.; Stern, M.; Shibolet, O.; Israeli, E.; Ackerman, Z.; Pappo, O.; Hemed, N.; Rowe, M.; Ohana, H.; Zabrecky, G.; et al. Treatment of chronic hepatitis C virus infection via antioxidants: Results of a phase I clinical trial. J. Clin. Gastroenterol 2005, 39, 737–742. [Google Scholar]
- Choi, J.; Lee, K.J.; Zheng, Y.; Yamaga, A.K.; Lai, M.M.; Ou, J.H. Reactive oxygen species suppress hepatitis C virus RNA replication in human hepatoma cells. Hepatology 2004, 39, 81–89. [Google Scholar]
- Choi, J.; Forman, H.J.; Ou, J.H.; Lai, M.M.; Seronello, S.; Nandipati, A. Redox modulation of the hepatitis C virus replication complex is calcium dependent. Free Radic. Biol. Med 2006, 41, 1488–1498. [Google Scholar]
- Seronello, S.; Ito, C.; Wakita, T.; Choi, J. Ethanol enhances hepatitis C virus replication through lipid metabolism and elevated NADH/NAD+. J. Biol. Chem 2010, 285, 845–854. [Google Scholar]
- Kuroki, M.; Ariumi, Y.; Ikeda, M.; Dansako, H.; Wakita, T.; Kato, N. Arsenic trioxide inhibits hepatitis C virus RNA replication through modulation of the glutathione redox system and oxidative stress. J. Virol. 2009, 83, 2338–2348. [Google Scholar]
- Yano, M.; Ikeda, M.; Abe, K.; Dansako, H.; Ohkoshi, S.; Aoyagi, Y.; Kato, N. Comprehensive analysis of the effects of ordinary nutrients on hepatitis C virus RNA replication in cell culture. Antimicrob. Agents Chemother 2007, 51, 2016–2027. [Google Scholar]
- Nakamura, M.; Saito, H.; Ikeda, M.; Hokari, R.; Kato, N.; Hibi, T.; Miura, S. An antioxidant resveratrol significantly enhanced replication of hepatitis C virus. World J. Gastroenterol 2010, 16, 184–192. [Google Scholar]
- McCartney, E.M.; Beard, M.R. Impact of alcohol on hepatitis C virus replication and interferon signaling. World J. Gastroenterol 2010, 16, 1337–1343. [Google Scholar]
- Waris, G.; Turkson, J.; Hassanein, T.; Siddiqui, A. Hepatitis C virus (HCV) constitutively activates STAT-3 via oxidative stress: Role of STAT-3 in HCV replication. J. Virol 2005, 79, 1569–1580. [Google Scholar]
- Rivas-Estilla, A.M.; Bryan-Marrugo, O.L.; Trujillo-Murillo, K.; Pérez-Ibave, D.; Charles-Niño, C.; Pedroza-Roldan, C.; Ríos-Ibarra, C.; Ramírez-Valles, E.; Ortiz-López, R.; Islas-Carbajal, M.C.; et al. Cu/Zn superoxide dismutase (SOD1) induction is implicated in the antioxidative and antiviral activity of acetylsalicylic acid in HCV-expressing cells. Am. J. Physiol. Gastrointest. Liver Physiol 2012, 302, G1264–G1273. [Google Scholar]
- Wagoner, J.; Negash, A.; Kane, O.J.; Martinez, L.E.; Nahmias, Y.; Bourne, N.; Owen, D.M.; Grove, J.; Brimacombe, C.; McKeating, J.A.; et al. Multiple effects of silymarin on the hepatitis C virus life cycle. Hepatology 2010, 51, 1912–1921. [Google Scholar]
- Wagoner, J.; Morishima, C.; Graf, T.N.; Oberlies, N.H.; Teissier, E.; Pécheur, E.I.; Tavis, J.E.; Polyak, S.J. Differential in vitro effects of intravenous versus oral formulations of silibinin on the HCV life cycle and inflammation. PLoS One 2011, 6, e16464. [Google Scholar]
- Goldwasser, J.; Cohen, P.Y.; Lin, W.; Kitsberg, D.; Balaguer, P.; Polyak, S.J.; Chung, R.T.; Yarmush, M.L.; Nahmias, Y. Naringenin inhibits the assembly and long-term production of infectious hepatitis C virus particles through a PPAR-mediated mechanism. J. Hepatol 2011, 55, 963–971. [Google Scholar]
- Nahmias, Y.; Goldwasser, J.; Casali, M.; van Poll, D.; Wakita, T.; Chung, R.T.; Yarmush, M.L. Apolipoprotein B-dependent hepatitis C virus secretion is inhibited by the grapefruit flavonoid naringenin. Hepatology 2008, 47, 1437–1445. [Google Scholar]
- Khachatoorian, R.; Arumugaswami, V.; Raychaudhuri, S.; Yeh, G.K.; Maloney, E.M.; Wang, J.; Dasgupta, A.; French, S.W. Divergent antiviral effects of bioflavonoids on the hepatitis C virus life cycle. Virology 2012, 433, 346–355. [Google Scholar]
- Bachmetov, L.; Gal-Tanamy, M.; Shapira, A.; Vorobeychik, M.; Giterman-Galam, T.; Sathiyamoorthy, P.; Golan-Goldhirsh, A.; Benhar, I.; Tur-Kaspa, R.; Zemel, R. Suppression of hepatitis C virus by the flavonoid quercetin is mediated by inhibition of NS3 protease activity. J. Viral. Hepat 2012, 19, e81–e88. [Google Scholar]
- Kim, K.; Kim, K.H.; Kim, H.Y.; Cho, H.K.; Sakamoto, N.; Cheong, J. Curcumin inhibits hepatitis C virus replication via suppressing the Akt-SREBP-1 pathway. FEBS Lett 2010, 584, 707–712. [Google Scholar]
- Guedj, J.; Dahari, H.; Pohl, R.T.; Ferenci, P.; Perelson, A.S. Understanding silibinin’s modes of action against HCV using viral kinetic modeling. J. Hepatol 2012, 56, 1019–1024. [Google Scholar]
- Falasca, K.; Ucciferri, C.; Mancino, P.; Vitacolonna, E.; De Tullio, D.; Pizzigallo, E.; Conti, P.; Vecchiet, J. Treatment with silybin-vitamin E-phospholipid complex in patients with hepatitis C infection. J. Med. Virol 2008, 80, 1900–1906. [Google Scholar]
- Powis, G. Free radical formation by antitumor quinones. Free Radic. Biol. Med 1989, 6, 63–101. [Google Scholar]
- Monks, T.J.; Hanzlik, R.P.; Cohen, G.M.; Ross, D.; Graham, D.G. Quinone chemistry and toxicity. Toxicol. Appl. Pharmacol 1992, 112, 2–16. [Google Scholar]
- Wang, A.L.; Wang, J.P.; Wang, H.; Chen, Y.H.; Zhao, L.; Wang, L.S.; Wei, W.; Xu, D.X. A dual effect of N-acetylcysteine on acute ethanol-induced liver damage in mice. Hepatol. Res 2006, 34, 199–206. [Google Scholar]
- Palamara, A.T.; Nencioni, L.; Aquilano, K; De Chiara, G.; Hernandez, L.; Cozzolino, F.; Ciriolo, M.R.; Garaci, E. Inhibition of influenza A virus replication by resveratrol. J. Infect. Dis. 2005, 191, 1719–1729. [Google Scholar]
- Burkitt, M.J.; Duncan, J. Effects of trans-resveratrol on copper-dependent hydroxyl-radical formation and DNA damage: Evidence for hydroxyl-radical scavenging and a novel, glutathione-sparing mechanism of action. Archiv. Biochem. Biophys 2000, 381, 253–263. [Google Scholar]
- Chang, Y.C.; Chuang, L.M. The role of oxidative stress in the pathogenesis of type 2 diabetes: From molecular mechanism to clinical implication. Am. J. Transl. Res 2010, 2, 316–331. [Google Scholar]
- Torres, M.; Forman, H.J. Redox signaling and the MAP kinase pathways. Biofactors 2003, 17, 287–296. [Google Scholar]
- McCubrey, J.A.; Lahair, M.M.; Franklin, R.A. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxid. Redox. Signal 2006, 8, 1775–1789. [Google Scholar]
- Leslie, N.R. The redox regulation of PI 3-kinase-dependent signaling. Antioxid. Redox. Signal 2006, 8, 1765–1774. [Google Scholar]
- Nencioni, L.; de Chiara, G.; Sgarbanti, R.; Amatore, D.; Aquilano, K.; Marcocci, M.E.; Serafino, A.; Torcia, M.; Cozzolino, F.; Ciriolo, M.R.; et al. Bcl-2 expression and p38MAPK activity in cells infected with influenza A virus: Impact on virally induced apoptosis and viral replication. J. Biol. Chem 2009, 284, 16004–16015. [Google Scholar]
- Nencioni, L.; Sgarbanti, R.; De Chiara, G.; Garaci, E.; Palamara, A.T. Influenza virus and redox mediated cell signaling: A complex network of virus/host interaction. New Microbiol 2007, 30, 367–375. [Google Scholar]
- Shin, Y.K.; Liu, Q.; Tikoo, S.K.; Babiuk, L.A.; Zhou, Y. Effect of the phosphatidylinositol 3-kinase/Akt pathway on influenza A virus propagation. J. Gen. Virol 2007, 88, 942–950. [Google Scholar]
- Muthumani, K.; Wadsworth, S.A.; Dayes, N.S.; Hwang, D.S.; Choo, A.Y.; Abeysinghe, H.R.; Siekierka, J.J.; Weiner, D.B. Suppression of HIV-1 viral replication and cellular pathogenesis by a novel p38/JNK kinase inhibitor. AIDS 2004, 18, 739–748. [Google Scholar]
- Yu, Y.; Alwine, J.C. Human cytomegalovirus major immediate-early proteins and simian virus 40 large T antigen can inhibit apoptosis through activation of the phosphatidylinositide 3′-OH kinase pathway and the cellular kinase Akt. J. Virol 2002, 76, 3731–3738. [Google Scholar]
- Rahaus, M.; Desloges, N.; Wolff, M.H. Varicella-zoster virus requires a functional PI3K/Akt/GSK-3alpha/beta signaling cascade for efficient replication. Cell Signal 2007, 19, 312–320. [Google Scholar]
- Lahair, M.M.; Howe, C.J.; Rodriguez-Mora, O.; McCubrey, J.A.; Franklin, R.A. Molecular pathways leading to oxidative stress-induced phosphorylation of Akt. Antioxid. Redox Signal 2006, 8, 1749–1756. [Google Scholar]
- Waris, G.; Siddiqui, A. Hepatitis C virus stimulates the expression of cyclooxygenase-2 via oxidative stress: Role of prostaglandin E2 in RNA replication. J. Virol. 2005, 79, 9725–9734. [Google Scholar]
- Blanc, A.; Pandey, N.R.; Srivastava, A.K. Distinct roles of Ca2+, calmodulin, and protein kinase C in H2O2-induced activation of ERK1/2, p38 MAPK, and protein kinase B signaling in vascular smooth muscle cells. Antioxid. Redox Signal 2004, 6, 353–366. [Google Scholar]
- Dossumbekova, A.; Berdyshev, E.V.; Gorshkova, I.; Shao, Z.; Li, C.; Long, P.; Joshi, A.; Natarajan, V.; Vanden Hoek, T.L. Akt activates NOS3 and separately restores barrier integrity in H2O2-stressed human cardiac microvascular endothelium. Am. J. Physiol. Heart Circ. Physiol 2008, 295, H2417–H2426. [Google Scholar]
- Mehdi, M.Z.; Pandey, N.R.; Pandey, S.K.; Srivastava, A.K. H2O2-induced phosphorylation of ERK1/2 and PKB requires tyrosine kinase activity of insulin receptor and c-Src. Antioxid. Redox Signal 2005, 7, 1014–1020. [Google Scholar]
- Conde de la Rosa, L.; Schoemaker, M.H.; Vrenken, T.E.; Buist-Homan, M.; Havinga, R.; Jansen, P.L.; Moshage, H. Superoxide anions and hydrogen peroxide induce hepatocyte death by different mechanisms: Involvement of JNK and ERK MAP kinases. J. Hepatol 2006, 44, 918–929. [Google Scholar]
- Aikawa, R.; Komuro, I.; Yamazaki, T.; Zou, Y.; Kudoh, S.; Tanaka, M.; Shiojima, I.; Hiroi, Y.; Yazaki, Y. Oxidative stress activates extracellular signal-regulated kinases through Src and Ras in cultured cardiac myocytes of neonatal rats. J. Clin. Invest 1997, 100, 1813–1821. [Google Scholar]
- Lee, S.R.; Kwon, K.S.; Kim, S.R.; Rhee, S.G. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J. Biol. Chem 1998, 273, 15366–15372. [Google Scholar]
- Liu, H.; Nishitoh, H.; Ichijo, H.; Kyriakis, J.M. Activation of apoptosis signal-regulating kinase 1 (ASK1) by tumor necrosis factor receptor-associated factor 2 requires prior dissociation of the ASK1 inhibitor thioredoxin. Mol. Cell Biol 2000, 20, 2198–2208. [Google Scholar]
- Saitoh, M.; Nishitoh, H.; Fujii, M.; Takeda, K.; Tobiume, K.; Sawada, Y.; Kawabata, M.; Miyazono, K.; Ichijo, H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J 1998, 17, 2596–2606. [Google Scholar]
- Matsukawa, J.; Matsuzawa, A.; Takeda, K.; Ichijo, H. The ASK1-MAP kinase cascades in mammalian stress response. J. Biochem 2004, 136, 261–265. [Google Scholar]
- Adler, V.; Yin, Z.; Fuchs, S.Y.; Benezra, M.; Rosario, L.; Tew, K.D.; Pincus, M.R.; Sardana, M.; Henderson, C.J.; Wolf, C.R.; et al. Regulation of JNK signaling by GSTp. EMBO J 1999, 18, 1321–1334. [Google Scholar]
- Lee, S.R.; Yang, K.S.; Kwon, J.; Lee, C.; Jeong, W.; Rhee, S.G. Reversible inactivation of the tumor suppressor PTEN by H2O2. J. Biol. Chem 2002, 277, 20336–20342. [Google Scholar]
- Leslie, N.R.; Bennett, D.; Lindsay, Y.E.; Stewart, H.; Gray, A.; Downes, C.P. Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J 2003, 22, 5501–5510. [Google Scholar]
- Mazzocca, A.; Sciammetta, S.C.; Carloni, V.; Cosmi, L.; Annunziato, F.; Harada, T.; Abrignani, S.; Pinzani, M. Binding of hepatitis C virus envelope protein E2 to CD81 up-regulates matrix metalloproteinase-2 in human hepatic stellate cells. J. Biol. Chem 2005, 280, 11329–11339. [Google Scholar]
- Ming-Ju, H.; Yih-Shou, H.; Tzy-Yen, C.; Hui-Ling, C. Hepatitis C virus E2 protein induce reactive oxygen species (ROS)-related fibrogenesis in the HSC-T6 hepatic stellate cell line. J. Cell Biochem 2011, 112, 233–243. [Google Scholar]
- Lin, W.; Tsai, W.L.; Shao, R.X.; Wu, G.; Peng, L.F.; Barlow, L.L.; Chung, W.J.; Zhang, L.; Zhao, H.; Jang, J.Y.; et al. Hepatitis C virus regulates transforming growth factor beta1 production through the generation of reactive oxygen species in a nuclear factor kappaB-dependent manner. Gastroenterology 2010, 138, 2509–2518. [Google Scholar]
- Street, A.; Macdonald, A.; McCormick, C.; Harris, M. Hepatitis C virus NS5A-mediated activation of phosphoinositide 3-kinase results in stabilization of cellular beta-catenin and stimulation of beta-catenin-responsive transcription. J. Virol 2005, 79, 5006–5016. [Google Scholar]
- Street, A.; Macdonald, A.; Crowder, K.; Harris, M. The Hepatitis C virus NS5A protein activates a phosphoinositide 3-kinase-dependent survival signaling cascade. J. Biol. Chem 2004, 279, 12232–12241. [Google Scholar]
- Frelin, L.; Brenndörfer, E.D.; Ahlén, G.; Weiland, M.; Hultgren, C.; Alheim, M.; Glaumann, H.; Rozell, B.; Milich, D.R.; Bode, J.G.; et al. The hepatitis C virus and immune evasion: Non-structural 3/4A transgenic mice are resistant to lethal tumour necrosis factor alpha mediated liver disease. Gut 2006, 55, 1475–1483. [Google Scholar]
- Brazzoli, M.; Bianchi, A.; Filippini, S.; Weiner, A.; Zhu, Q.; Pizza, M.; Crotta, S. CD81 is a central regulator of cellular events required for hepatitis C virus infection of human hepatocytes. J. Virol 2008, 82, 8316–8329. [Google Scholar]
- Pei, R.; Chen, H.; Lu, L.; Zhu, W.; Beckebaum, S.; Cicinnati, V.; Lu, M.; Chen, X. Hepatitis C virus infection induces the expression of amphiregulin, a factor related to the activation of cellular survival pathways and required for efficient viral assembly. J. Gen. Virol 2011, 92, 2237–2248. [Google Scholar]
- Murata, T.; Hijikata, M.; Shimotohno, K. Enhancement of internal ribosome entry site-mediated translation and replication of hepatitis C virus by PD98059. Virology 2005, 340, 105–115. [Google Scholar]
- Huang, Y.; Chen, X.C.; Konduri, M.; Fomina, N.; Lu, J.; Jin, L.; Kolykhalov, A.; Tan, S.L. Mechanistic link between the anti-HCV effect of interferon gamma and control of viral replication by a Ras-MAPK signaling cascade. Hepatology 2006, 43, 81–90. [Google Scholar]
- Yano, M.; Ikeda, M.; Abe, K.; Kawai, Y.; Kuroki, M.; Mori, K.; Dansako, H.; Ariumi, Y.; Ohkoshi, S.; Aoyagi, Y.; et al. Oxidative stress induces anti-hepatitis C virus status via the activation of extracellular signal-regulated kinase. Hepatology 2009, 50, 678–688. [Google Scholar]
- Brenndörfer, E.D.; Karthe, J.; Frelin, L.; Cebula, P.; Erhardt, A.; Schulteam Esch, J.; Hengel, H.; Bartenschlager, R.; Sällberg, M.; Häussinger, D.; et al. Nonstructural 3/4A protease of hepatitis C virus activates epithelial growth factor-induced signal transduction by cleavage of the T-cell protein tyrosine phosphatase. Hepatology 2009, 49, 1810–1820. [Google Scholar]
- Mankouri, J.; Tedbury, P.R.; Gretton, S.; Hughes, M.E.; Griffin, S.D.; Dallas, M.L.; Green, K.A.; Hardie, D.G.; Peers, C.; Harris, M. Enhanced hepatitis C virus genome replication and lipid accumulation mediated by inhibition of AMP-activated protein kinase. Proc. Natl. Acad. Sci. USA 2010, 107, 11549–11554. [Google Scholar]
- Fukuhara, T.; Matsuura, Y. Role of miR-122 and lipid metabolism in HCV infection. J. Gastroenterol 2012, 48, 169–176. [Google Scholar]
- Syed, G.H.; Amako, Y.; Siddiqui, A. Hepatitis C virus hijacks host lipid metabolism. Trends Endocrinol. Metab 2010, 21, 33–40. [Google Scholar]
- Aizaki, H.; Lee, K.J.; Sung, V.M.; Ishiko, H.; Lai, M.M. Characterization of the hepatitis C virus RNA replication complex associated with lipid rafts. Virology 2004, 324, 450–461. [Google Scholar]
- Miyanari, Y.; Atsuzawa, K.; Usuda, N.; Watashi, K.; Hishiki, T.; Zayas, M.; Bartenschlager, R.; Wakita, T.; Hijikata, M.; Shimotohno, K. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell. Biol 2007, 9, 1089–1097. [Google Scholar]
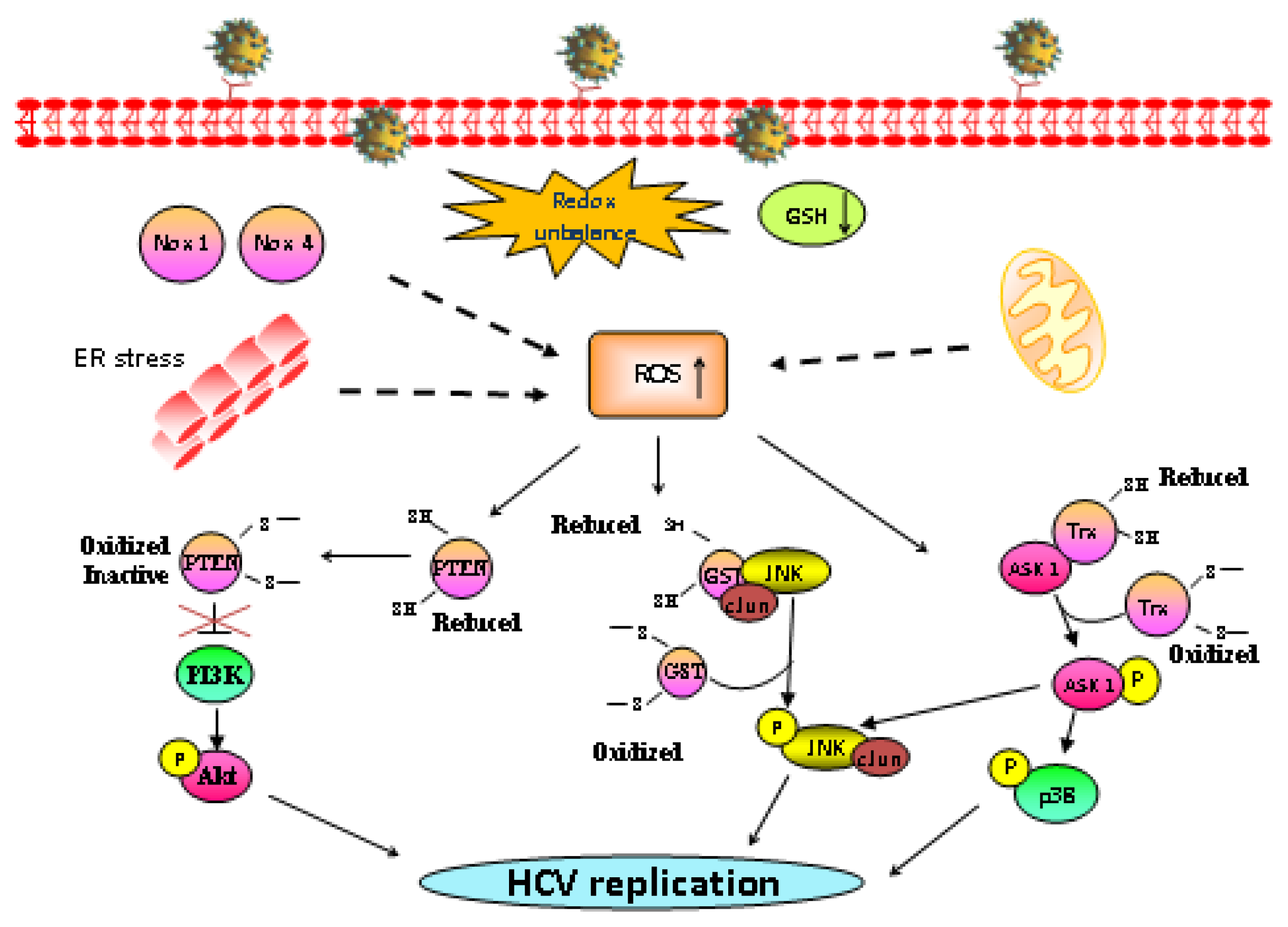

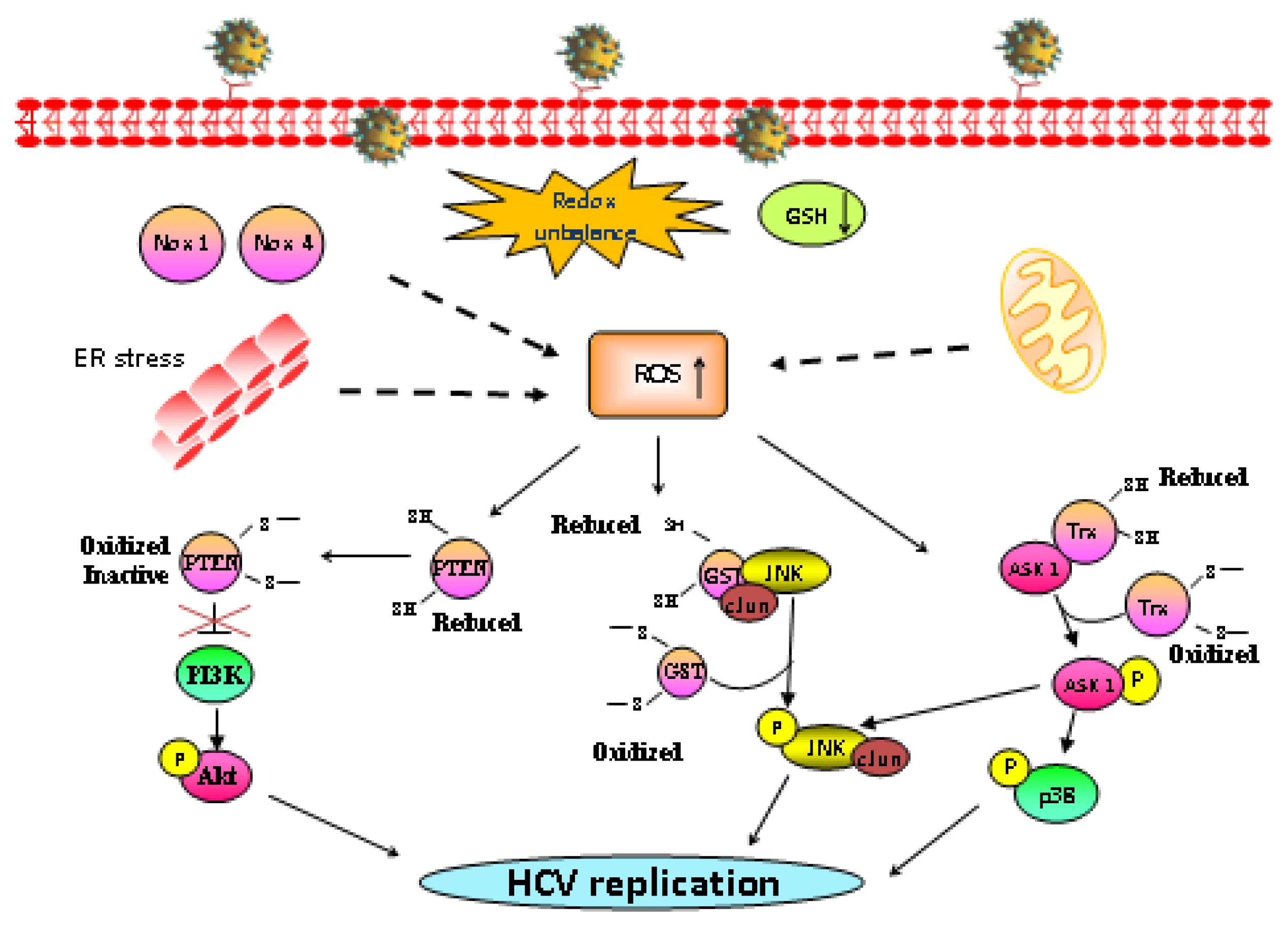{kind=link}
| Source of ROS/RNS | HCV proteins | References |
|---|---|---|
| Transcriptional up-regulation of iNOS | core, NS3 | [47] |
| Activation of Nox2 in PBMCs | NS3 | [41] |
| GSH depletion | Core | [56] |
| Increased production of mitochondrial ROS by the electron transport chain | Core | [45] |
| Activation of Nox2 in Kupffer cells | HCV | [29,40] |
| Endoplasmic reticulum stress (ER stress) | NS4B, NS5A, E1, E2 | [49–51] |
| Increase of proinflammatory cytokines | HCV, core | [54] |
| Activation of Nox1 and 4 proteins in hepatocytes | HCV | [53,54] |
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ruggieri, A.; Anticoli, S.; Nencioni, L.; Sgarbanti, R.; Garaci, E.; Palamara, A.T. Interplay between Hepatitis C Virus and Redox Cell Signaling. Int. J. Mol. Sci. 2013, 14, 4705-4721. https://doi.org/10.3390/ijms14034705
Ruggieri A, Anticoli S, Nencioni L, Sgarbanti R, Garaci E, Palamara AT. Interplay between Hepatitis C Virus and Redox Cell Signaling. International Journal of Molecular Sciences. 2013; 14(3):4705-4721. https://doi.org/10.3390/ijms14034705
Chicago/Turabian StyleRuggieri, Anna, Simona Anticoli, Lucia Nencioni, Rossella Sgarbanti, Enrico Garaci, and Anna Teresa Palamara. 2013. "Interplay between Hepatitis C Virus and Redox Cell Signaling" International Journal of Molecular Sciences 14, no. 3: 4705-4721. https://doi.org/10.3390/ijms14034705