Atypical Protein Phosphatases: Emerging Players in Cellular Signaling

Abstract
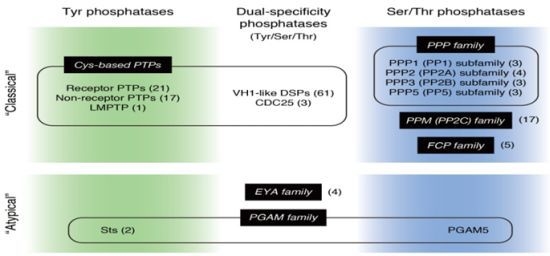
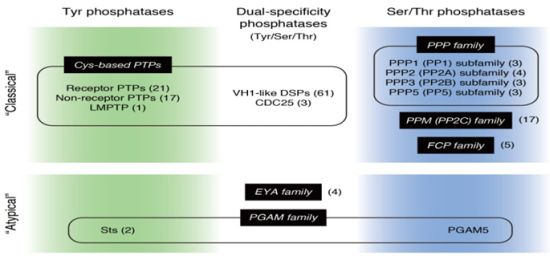
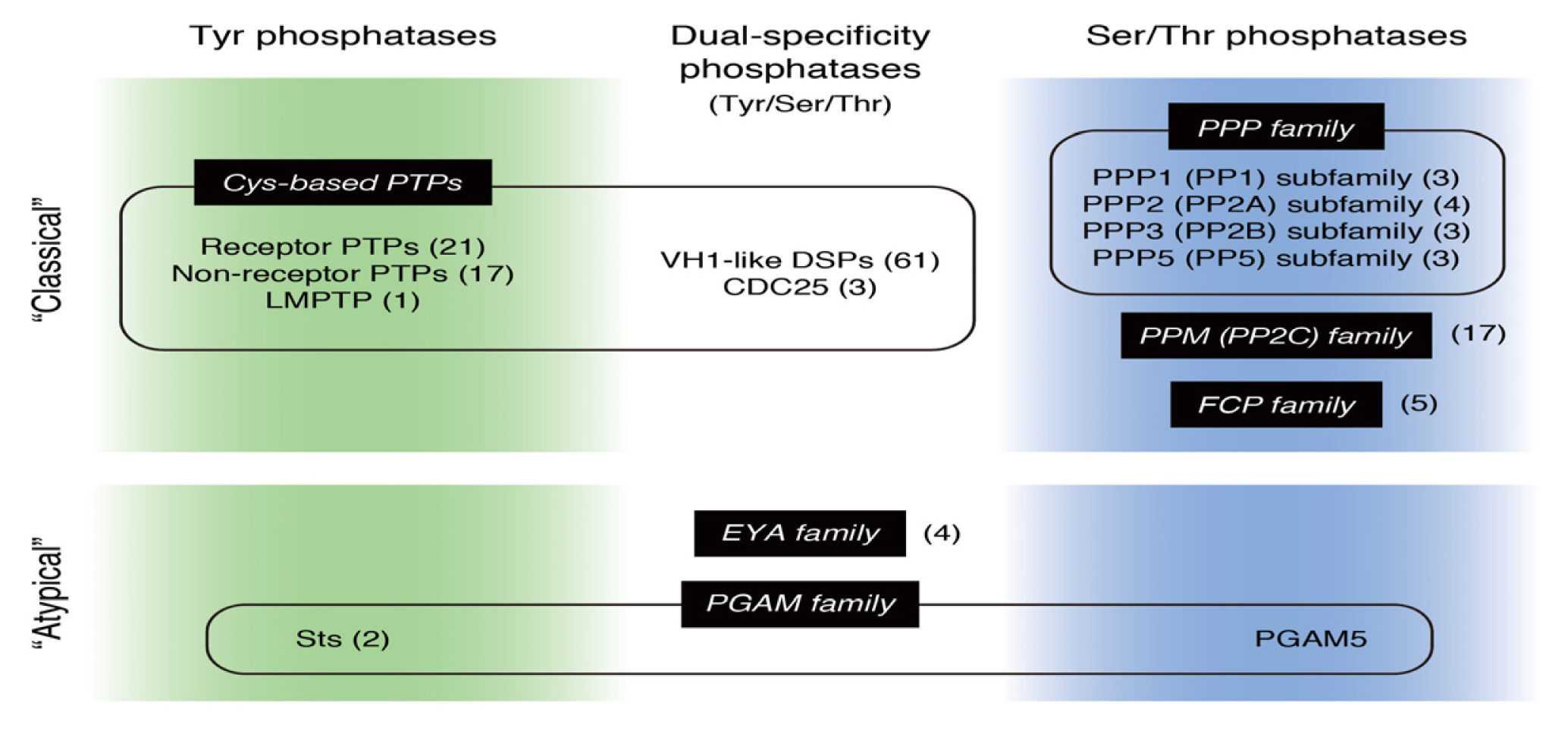
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. EYA: A New Type of Dual-Specificity Protein Phosphatase
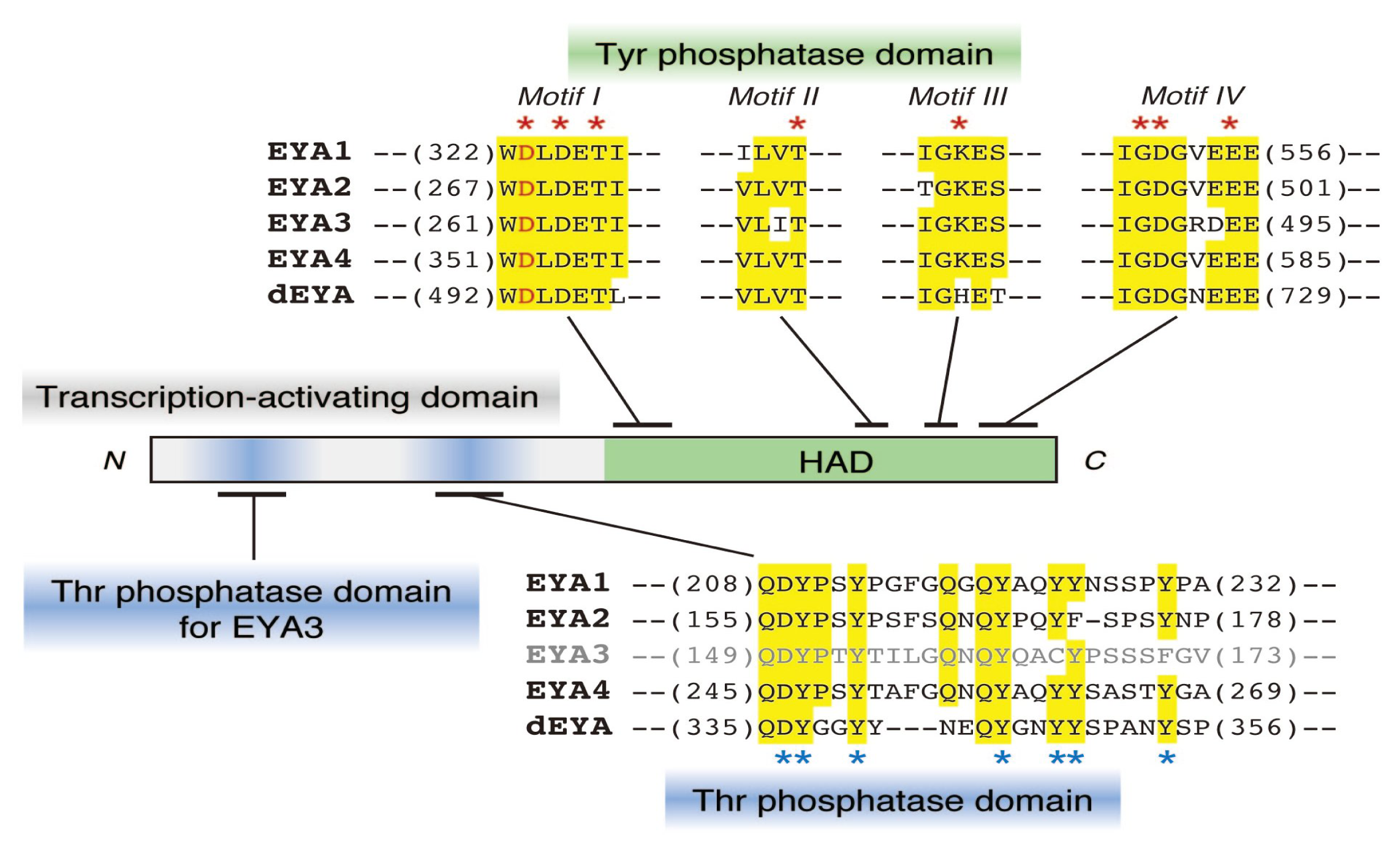
2.1. Protein Tyr Phosphatase Activity of EYA
2.2. EYA Also Functions as a Thr Phosphatase
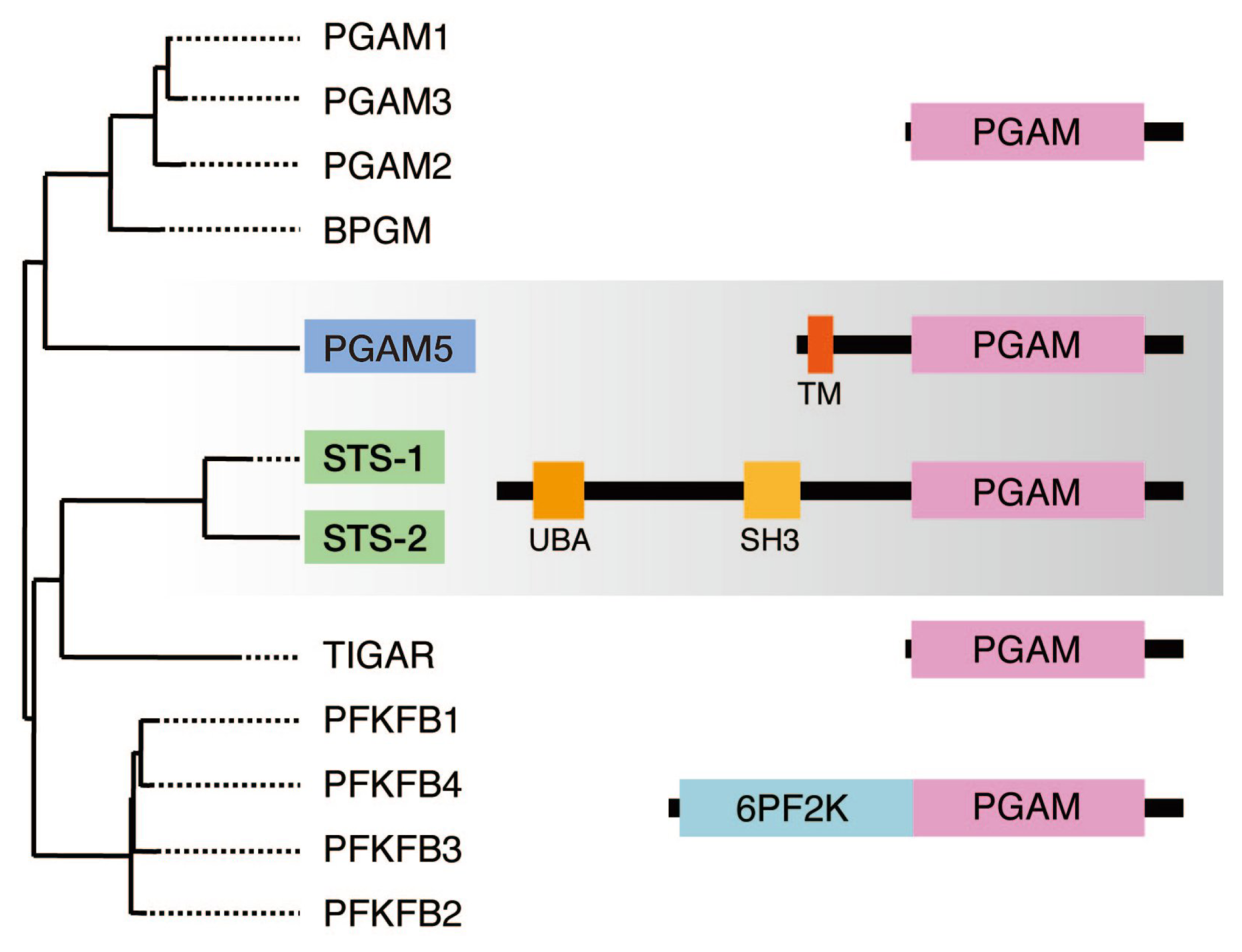
3. Sts-1 and Sts-2: Members of the PGAM Family that Function as Protein Tyr Phosphatases
3.1. His-Based Tyr Phosphatase Activity of Sts-1/2 and TCR Signaling
3.2. Regulation of Protein Tyr Kinase Signaling by Sts-1/2
4. PGAM5: Another Member of the PGAM Family that Functions as a Protein Ser/Thr Phosphatase
4.1. PGAM5 is a His-Based Protein Ser/Thr Phosphatase that Activates the Stress-Activated MAP Kinase Pathways
4.2. PGAM5, Together with Sts-1/2, Constitutes a Novel Family of Protein Phosphatases
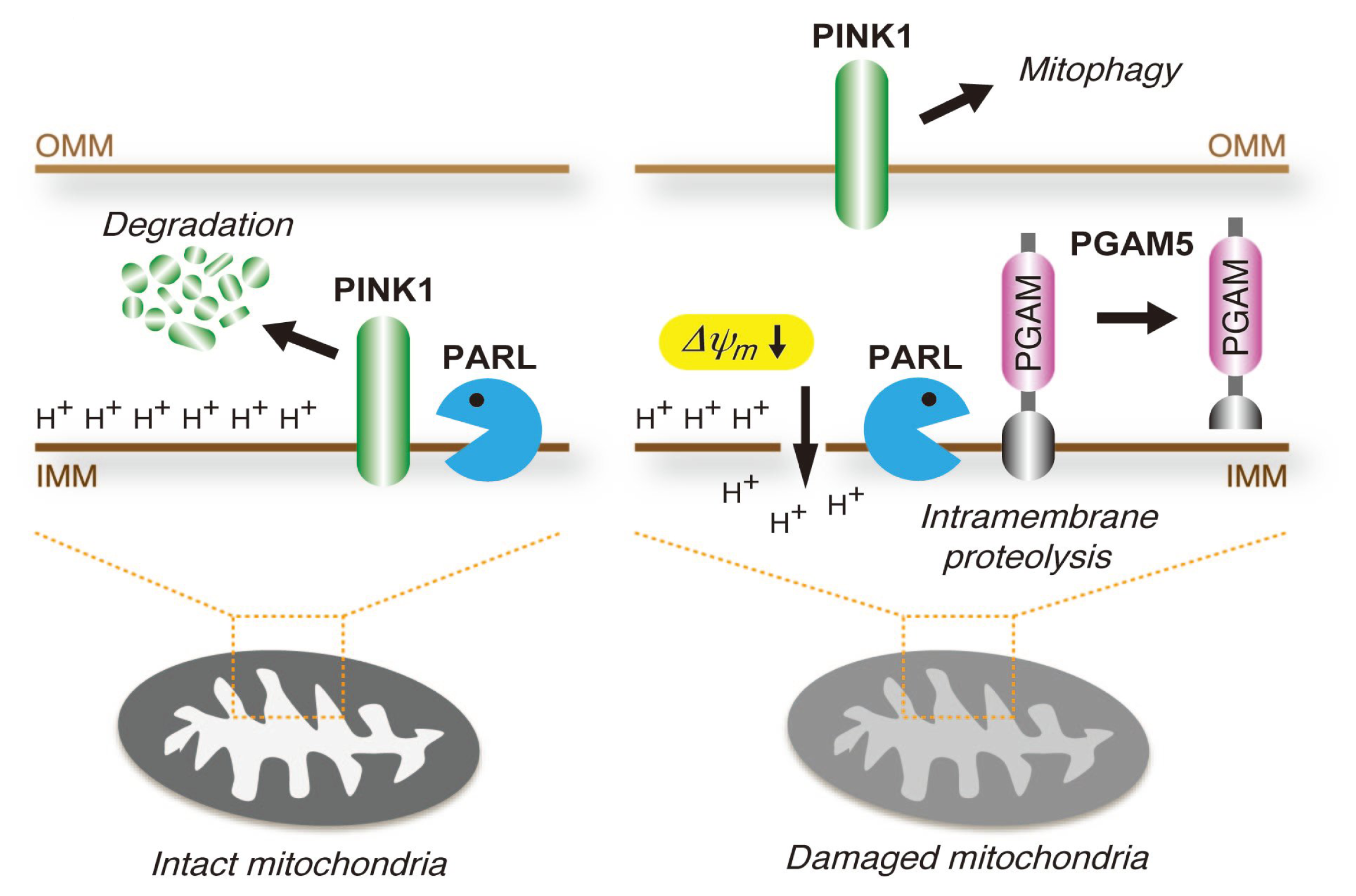
4.3. PGAM5 May Be Involved in the Mitochondrial Quality Control System
4.4. PGAM5 Regulates Cell Death in a Context-Dependent Manner
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar]
- Alonso, A.; Sasin, J.; Bottini, N.; Friedberg, I.; Osterman, A.; Godzik, A.; Hunter, T.; Dixon, J.; Mustelin, T. Protein tyrosine phosphatases in the human genome. Cell 2004, 117, 699–711. [Google Scholar]
- Shi, Y. Serine/threonine phosphatases: Mechanism through structure. Cell 2009, 139, 468–484. [Google Scholar]
- Andersen, J.N.; Tonks, N.K. Protein tyrosine phosphatase-based therapeutics: Lessons from PTP1B. In Topics in Current Genetics, Vol. 5, Protein phosphatases; Arino, J., Alexander, D.R., Eds.; Springer-Verlag: Berlin Heidelberg: Berlin, Germany, 2004; pp. 201–230. [Google Scholar]
- Cohen, P.T.W. Overview of protein serine/threonine phosphatases. In Topics in Current Genetics, Vol. 5, Protein phosphatases; Arino, J., Alexander, D.R., Eds.; Springer-Verlag: Berlin Heidelberg: Berlin, Germany, 2004; pp. 1–20. [Google Scholar]
- Bonini, N.M.; Leiserson, W.M.; Benzer, S. The eyes absent gene: Genetic control of cell survival and differentiation in the developing Drosophila eye. Cell 1993, 72, 379–395. [Google Scholar]
- Jemc, J.; Rebay, I. The eyes absent family of phosphotyrosine phosphatases: Properties and roles in developmental regulation of transcription. Annu. Rev. Biochem 2007, 76, 513–538. [Google Scholar]
- Abdelhak, S.; Kalatzis, V.; Heilig, R.; Compain, S.; Samson, D.; Vincent, C.; Weil, D.; Cruaud, C.; Sahly, I.; Leibovici, M.; et al. A human homologue of the Drosophila eyes absent gene underlies branchio-oto-renal (BOR) syndrome and identifies a novel gene family. Nat. Genet. 1997, 15, 157–164. [Google Scholar]
- Vincent, C.; Kalatzis, V.; Abdelhak, S.; Chaib, H.; Compain, S.; Helias, J.; Vaneecloo, F.M.; Petit, C. BOR and BO syndromes are allelic defects of EYA1. Eur. J. Hum. Genet 1997, 5, 242–246. [Google Scholar]
- Xu, P.X.; Adams, J.; Peters, H.; Brown, M.C.; Heaney, S.; Maas, R. Eya1-deficient mice lack ears and kidneys and show abnormal apoptosis of organ primordia. Nat. Genet 1999, 23, 113–117. [Google Scholar]
- Soker, T.; Dalke, C.; Puk, O.; Floss, T.; Becker, L.; Bolle, I.; Favor, J.; Hans, W.; Holter, S.M.; Horsch, M.; et al. Pleiotropic effects in Eya3 knockout mice. BMC Dev. Biol. 2008, 8, 118. [Google Scholar]
- Depreux, F.F.; Darrow, K.; Conner, D.A.; Eavey, R.D.; Liberman, M.C.; Seidman, C.E.; Seidman, J.G. Eya4-deficient mice are a model for heritable otitis media. J. Clin. Invest 2008, 118, 651–658. [Google Scholar]
- Schonberger, J.; Wang, L.; Shin, J.T.; Kim, S.D.; Depreux, F.F.; Zhu, H.; Zon, L.; Pizard, A.; Kim, J.B.; Macrae, C.A.; et al. Mutation in the transcriptional coactivator EYA4 causes dilated cardiomyopathy and sensorineural hearing loss. Nat. Genet. 2005, 37, 418–422. [Google Scholar]
- O’Neill, M.E.; Marietta, J.; Nishimura, D.; Wayne, S.; van Camp, G.; van Laer, L.; Negrini, C.; Wilcox, E.R.; Chen, A.; Fukushima, K.; et al. A gene for autosomal dominant late-onset progressive non-syndromic hearing loss, DFNA10, maps to chromosome 6. Hum. Mol. Genet. 1996, 5, 853–856. [Google Scholar]
- Pfister, M.; Toth, T.; Thiele, H.; Haack, B.; Blin, N.; Zenner, H.P.; Sziklai, I.; Nurnberg, P.; Kupka, S. A 4-bp insertion in the eya-homologous region (eyaHR) of EYA4 causes hearing impairment in a Hungarian family linked to DFNA10. Mol. Med 2002, 8, 607–611. [Google Scholar]
- Wayne, S.; Robertson, N.G.; DeClau, F.; Chen, N.; Verhoeven, K.; Prasad, S.; Tranebjarg, L.; Morton, C.C.; Ryan, A.F.; van Camp, G.; et al. Mutations in the transcriptional activator EYA4 cause late-onset deafness at the DFNA10 locus. Hum. Mol. Genet. 2001, 10, 195–200. [Google Scholar]
- Rayapureddi, J.P.; Kattamuri, C.; Steinmetz, B.D.; Frankfort, B.J.; Ostrin, E.J.; Mardon, G.; Hegde, R.S. Eyes absent represents a class of protein tyrosine phosphatases. Nature 2003, 426, 295–298. [Google Scholar]
- Tootle, T.L.; Silver, S.J.; Davies, E.L.; Newman, V.; Latek, R.R.; Mills, I.A.; Selengut, J.D.; Parlikar, B.E.; Rebay, I. The transcription factor Eyes absent is a protein tyrosine phosphatase. Nature 2003, 426, 299–302. [Google Scholar]
- Li, X.; Oghi, K.A.; Zhang, J.; Krones, A.; Bush, K.T.; Glass, C.K.; Nigam, S.K.; Aggarwal, A.K.; Maas, R.; Rose, D.W.; et al. Eya protein phosphatase activity regulates Six1-Dach-Eya transcriptional effects in mammalian organogenesis. Nature 2003, 426, 247–254. [Google Scholar]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., 3rd; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar]
- Stokes, M.P.; Rush, J.; Macneill, J.; Ren, J.M.; Sprott, K.; Nardone, J.; Yang, V.; Beausoleil, S.A.; Gygi, S.P.; Livingstone, M.; et al. Profiling of UV-induced ATM/ATR signaling pathways. Proc. Natl. Acad. Sci. U S A 2007, 104, 19855–19860. [Google Scholar]
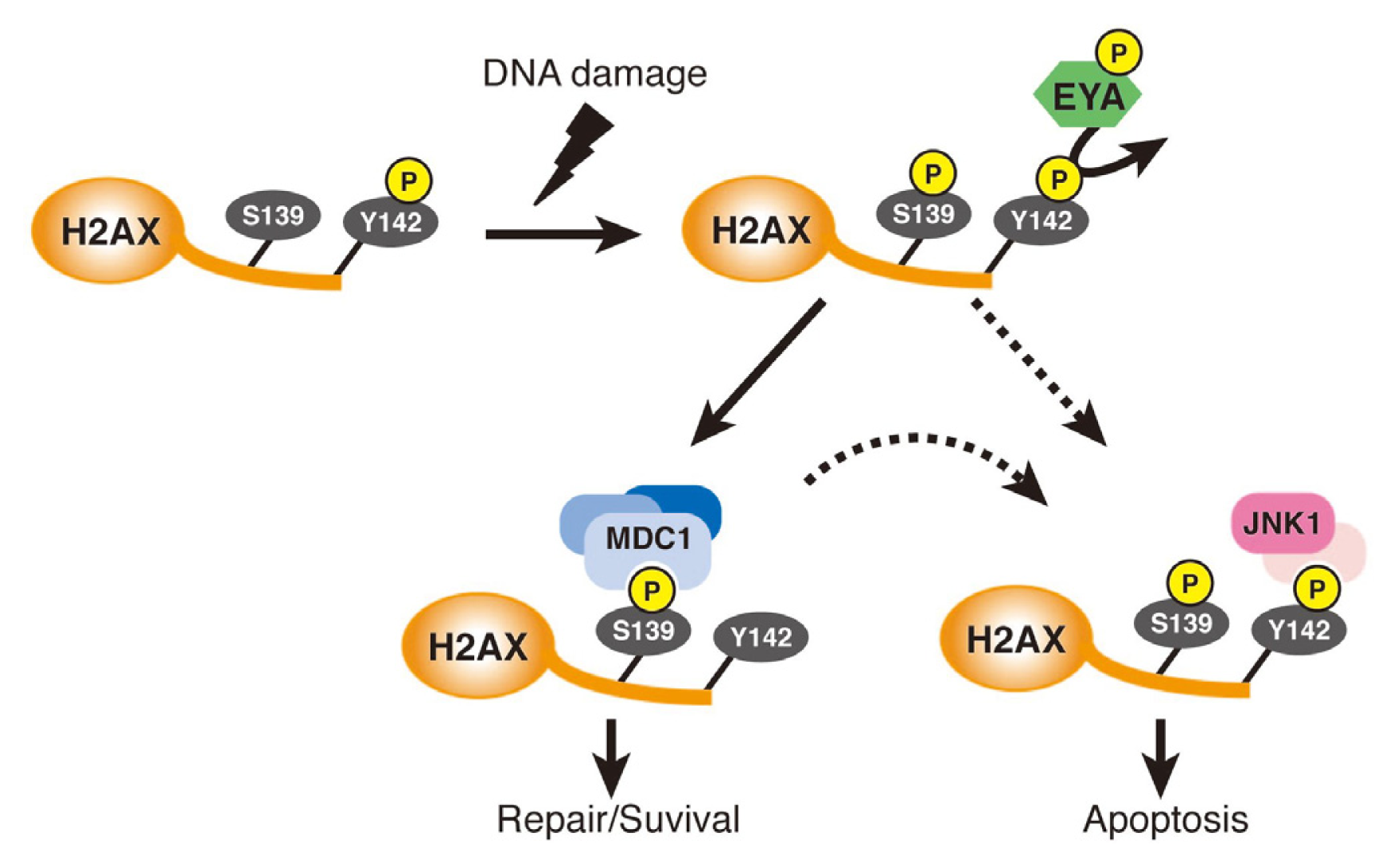
- Cook, P.J.; Ju, B.G.; Telese, F.; Wang, X.; Glass, C.K.; Rosenfeld, M.G. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature 2009, 458, 591–596. [Google Scholar]
- Krishnan, N.; Jeong, D.G.; Jung, S.K.; Ryu, S.E.; Xiao, A.; Allis, C.D.; Kim, S.J.; Tonks, N.K. Dephosphorylation of the C-terminal tyrosyl residue of the DNA damage-related histone H2A.X is mediated by the protein phosphatase eyes absent. J. Biol. Chem 2009, 284, 16066–16070. [Google Scholar]
- Okabe, Y.; Sano, T.; Nagata, S. Regulation of the innate immune response by threonine-phosphatase of Eyes absent. Nature 2009, 460, 520–524. [Google Scholar]
- Miller, S.J.; Lan, Z.D.; Hardiman, A.; Wu, J.; Kordich, J.J.; Patmore, D.M.; Hegde, R.S.; Cripe, T.P.; Cancelas, J.A.; Collins, M.H.; Ratner, N. Inhibition of Eyes Absent Homolog 4 expression induces malignant peripheral nerve sheath tumor necrosis. Oncogene 2010, 29, 368–379. [Google Scholar]
- Farabaugh, S.M.; Micalizzi, D.S.; Jedlicka, P.; Zhao, R.; Ford, H.L. Eya2 is required to mediate the pro-metastatic functions of Six1 via the induction of TGF-beta signaling, epithelial-mesenchymal transition, and cancer stem cell properties. Oncogene 2012, 31, 552–562. [Google Scholar]
- Zhang, L.; Yang, N.; Huang, J.; Buckanovich, R.J.; Liang, S.; Barchetti, A.; Vezzani, C.; O’Brien-Jenkins, A.; Wang, J.; Ward, M.R.; et al. Transcriptional coactivator Drosophila eyes absent homologue 2 is up-regulated in epithelial ovarian cancer and promotes tumor growth. Cancer Res. 2005, 65, 925–932. [Google Scholar]
- Pandey, R.N.; Rani, R.; Yeo, E.J.; Spencer, M.; Hu, S.; Lang, R.A.; Hegde, R.S. The Eyes Absent phosphatase-transactivator proteins promote proliferation, transformation, migration, and invasion of tumor cells. Oncogene 2010, 29, 3715–3722. [Google Scholar]
- Tadjuidje, E.; Wang, T.S.; Pandey, R.N.; Sumanas, S.; Lang, R.A.; Hegde, R.S. The EYA tyrosine phosphatase activity is pro-angiogenic and is inhibited by benzbromarone. PLoS One 2012, 7, e34806. [Google Scholar]
- Sano, T.; Nagata, S. Characterization of the threonine-phosphatase of mouse eyes absent 3. FEBS Lett 2011, 585, 2714–2719. [Google Scholar]
- Liu, X.; Sano, T.; Guan, Y.; Nagata, S.; Hoffmann, J.A.; Fukuyama, H. Drosophila EYA regulates the immune response against DNA through an evolutionarily conserved threonine phosphatase motif. PLoS One 2012, 7, e42725. [Google Scholar]
- Ganesan, S.; Aggarwal, K.; Paquette, N.; Silverman, N. NF-kappaB/Rel proteins and the humoral immune responses of Drosophila melanogaster. Curr. Top. Microbiol. Immunol 2011, 349, 25–60. [Google Scholar]
- Carpino, N.; Kobayashi, R.; Zang, H.; Takahashi, Y.; Jou, S.T.; Feng, J.; Nakajima, H.; Ihle, J.N. Identification, cDNA cloning, and targeted deletion of p70, a novel, ubiquitously expressed SH3 domain-containing protein. Mol. Cell Biol 2002, 22, 7491–7500. [Google Scholar]
- Kowanetz, K.; Crosetto, N.; Haglund, K.; Schmidt, M.H.; Heldin, C.H.; Dikic, I. Suppressors of T-cell receptor signaling Sts-1 and Sts-2 bind to Cbl and inhibit endocytosis of receptor tyrosine kinases. J. Biol. Chem 2004, 279, 32786–32795. [Google Scholar]
- Feshchenko, E.A.; Smirnova, E.V.; Swaminathan, G.; Teckchandani, A.M.; Agrawal, R.; Band, H.; Zhang, X.; Annan, R.S.; Carr, S.A.; Tsygankov, A.Y. TULA: An SH3- and UBA-containing protein that binds to c-Cbl and ubiquitin. Oncogene 2004, 23, 4690–4706. [Google Scholar]
- Jedrzejas, M.J. Structure, function, and evolution of phosphoglycerate mutases: Comparison with fructose-2,6-bisphosphatase, acid phosphatase, and alkaline phosphatase. Prog. Biophys. Mol. Biol 2000, 73, 263–287. [Google Scholar]
- Rigden, D.J. The histidine phosphatase superfamily: Structure and function. Biochem. J 2008, 409, 333–348. [Google Scholar]
- Hoeller, D.; Crosetto, N.; Blagoev, B.; Raiborg, C.; Tikkanen, R.; Wagner, S.; Kowanetz, K.; Breitling, R.; Mann, M.; Stenmark, H.; et al. Regulation of ubiquitin-binding proteins by monoubiquitination. Nat. Cell Biol. 2006, 8, 163–169. [Google Scholar]
- Carpino, N.; Turner, S.; Mekala, D.; Takahashi, Y.; Zang, H.; Geiger, T.L.; Doherty, P.; Ihle, J.N. Regulation of ZAP-70 activation and TCR signaling by two related proteins, Sts-1 and Sts-2. Immunity 2004, 20, 37–46. [Google Scholar]
- Wattenhofer, M.; Shibuya, K.; Kudoh, J.; Lyle, R.; Michaud, J.; Rossier, C.; Kawasaki, K.; Asakawa, S.; Minoshima, S.; Berry, A.; et al. Isolation and characterization of the UBASH3A gene on 21q22.3 encoding a potential nuclear protein with a novel combination of domains. Human genetics 2001, 108, 140–147. [Google Scholar]
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T cell activation. Annu. Rev. Immunol 2009, 27, 591–619. [Google Scholar]
- Au-Yeung, B.B.; Deindl, S.; Hsu, L.Y.; Palacios, E.H.; Levin, S.E.; Kuriyan, J.; Weiss, A. The structure, regulation, and function of ZAP-70. Immunol. Rev 2009, 228, 41–57. [Google Scholar]
- Mikhailik, A.; Ford, B.; Keller, J.; Chen, Y.; Nassar, N.; Carpino, N. A phosphatase activity of Sts-1 contributes to the suppression of TCR signaling. Mol. Cell 2007, 27, 486–497. [Google Scholar]
- Tsygankov, A.Y. TULA-family proteins: An odd couple. Cell Mol. Life Sci 2009, 66, 2949–2952. [Google Scholar]
- San Luis, B.; Sondgeroth, B.; Nassar, N.; Carpino, N. Sts-2 Is a phosphatase that negatively regulates zeta-associated protein (ZAP)-70 and T cell receptor signaling pathways. J. Biol. Chem 2011, 286, 15943–15954. [Google Scholar]
- Raguz, J.; Wagner, S.; Dikic, I.; Hoeller, D. Suppressor of T-cell receptor signalling 1 and 2 differentially regulate endocytosis and signalling of receptor tyrosine kinases. FEBS Lett 2007, 581, 4767–4772. [Google Scholar]
- Bertelsen, V.; Breen, K.; Sandvig, K.; Stang, E.; Madshus, I.H. The Cbl-interacting protein TULA inhibits dynamin-dependent endocytosis. Exp. Cell Res 2007, 313, 1696–1709. [Google Scholar]
- Agrawal, R.; Carpino, N.; Tsygankov, A. TULA proteins regulate activity of the protein tyrosine kinase Syk. J. Cell Biochem 2008, 104, 953–964. [Google Scholar]
- Chen, X.; Ren, L.; Kim, S.; Carpino, N.; Daniel, J.L.; Kunapuli, S.P.; Tsygankov, A.Y.; Pei, D. Determination of the substrate specificity of protein-tyrosine phosphatase TULA-2 and identification of Syk as a TULA-2 substrate. J. Biol. Chem 2010, 285, 31268–31276. [Google Scholar]
- De Castro, R.O.; Zhang, J.; Groves, J.R.; Barbu, E.A.; Siraganian, R.P. Once phosphorylated, tyrosines in carboxyl terminus of protein-tyrosine kinase Syk interact with signaling proteins, including TULA-2, a negative regulator of mast cell degranulation. J. Biol. Chem 2012, 287, 8194–8204. [Google Scholar]
- Hammond, P.W.; Alpin, J.; Rise, C.E.; Wright, M.; Kreider, B.L. In vitro selection and characterization of Bcl-X(L)-binding proteins from a mix of tissue-specific mRNA display libraries. J. Biol. Chem 2001, 276, 20898–20906. [Google Scholar]
- Lo, S.C.; Hannink, M. PGAM5, a Bcl-XL-interacting protein, is a novel substrate for the redox-regulated Keap1-dependent ubiquitin ligase complex. J. Biol. Chem 2006, 281, 37893–37903. [Google Scholar]
- Padmanabhan, B.; Tong, K.I.; Ohta, T.; Nakamura, Y.; Scharlock, M.; Ohtsuji, M.; Kang, M.I.; Kobayashi, A.; Yokoyama, S.; Yamamoto, M. Structural basis for defects of Keap1 activity provoked by its point mutations in lung cancer. Mol. Cell 2006, 21, 689–700. [Google Scholar]
- Lo, S.C.; Li, X.; Henzl, M.T.; Beamer, L.J.; Hannink, M. Structure of the Keap1: Nrf2 interface provides mechanistic insight into Nrf2 signaling. EMBO J 2006, 25, 3605–3617. [Google Scholar]
- Lo, S.C.; Hannink, M. PGAM5 tethers a ternary complex containing Keap1 and Nrf2 to mitochondria. Exp. Cell Res 2008, 314, 1789–1803. [Google Scholar]
- Takeda, K.; Komuro, Y.; Hayakawa, T.; Oguchi, H.; Ishida, Y.; Murakami, S.; Noguchi, T.; Kinoshita, H.; Sekine, Y.; Iemura, S.I.; et al. Mitochondrial phosphoglycerate mutase 5 uses alternate catalytic activity as a protein serine/threonine phosphatase to activate ASK1. Proc. Natl. Acad. Sci. USA 2009, 106, 12301–12305. [Google Scholar]
- Ichijo, H.; Nishida, E.; Irie, K.; ten Dijke, P.; Saitoh, M.; Moriguchi, T.; Takagi, M.; Matsumoto, K.; Miyazono, K.; Gotoh, Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 1997, 275, 90–94. [Google Scholar]
- Takeda, K.; Noguchi, T.; Naguro, I.; Ichijo, H. Apoptosis signal-regulating kinase 1 in stress and immune response. Annu. Rev. Pharmacol. Toxicol 2008, 48, 199–225. [Google Scholar]
- Sekine, S.; Ichijo, H. The University of Tokyo, Tokyo, Japan; Takeda, K. Nagasaki University: Nagasaki, Japan, Unpublished work; 2013. [Google Scholar]
- Chuang, T.D.; Chen, S.J.; Lin, F.F.; Veeramani, S.; Kumar, S.; Batra, S.K.; Tu, Y.; Lin, M.F. Human prostatic acid phosphatase, an authentic tyrosine phosphatase, dephosphorylates ErbB-2 and regulates prostate cancer cell growth. J. Biol. Chem 2010, 285, 23598–23606. [Google Scholar]
- Sekine, S.; Kanamaru, Y.; Koike, M.; Nishihara, A.; Okada, M.; Kinoshita, H.; Kamiyama, M.; Maruyama, J.; Uchiyama, Y.; Ishihara, N.; et al. Rhomboid protease PARL mediates the mitochondrial membrane potential loss-induced cleavage of PGAM5. J. Biol. Chem. 2012, 287, 34635–34645. [Google Scholar]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J. Cell Biol 2010, 191, 933–942. [Google Scholar]
- Deas, E.; Plun-Favreau, H.; Gandhi, S.; Desmond, H.; Kjaer, S.; Loh, S.H.; Renton, A.E.; Harvey, R.J.; Whitworth, A.J.; Martins, L.M.; et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum. Mol. Genet. 2011, 20, 867–879. [Google Scholar]
- Shi, G.; Lee, J.R.; Grimes, D.A.; Racacho, L.; Ye, D.; Yang, H.; Ross, O.A.; Farrer, M.; McQuibban, G.A.; Bulman, D.E. Functional alteration of PARL contributes to mitochondrial dysregulation in Parkinson’s disease. Hum. Mol. Genet 2011, 20, 1966–1974. [Google Scholar]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 2010, 8, e1000298. [Google Scholar]
- Tanaka, A. Parkin-mediated selective mitochondrial autophagy, mitophagy: Parkin purges damaged organelles from the vital mitochondrial network. FEBS Lett 2010, 584, 1386–1392. [Google Scholar]
- Imai, Y.; Kanao, T.; Sawada, T.; Kobayashi, Y.; Moriwaki, Y.; Ishida, Y.; Takeda, K.; Ichijo, H.; Lu, B.; Takahashi, R. The loss of PGAM5 suppresses the mitochondrial degeneration caused by inactivation of PINK1 in Drosophila. PLoS Genet 2010, 6, e1001229. [Google Scholar]
- Ishida, Y.; Sekine, Y.; Oguchi, H.; Chihara, T.; Miura, M.; Ichijo, H.; Takeda, K. Prevention of apoptosis by mitochondrial phosphatase PGAM5 in the mushroom body is crucial for heat shock resistance in Drosophila melanogaster. PLoS One 2012, 7, e30265. [Google Scholar]
- Niture, S.K.; Jaiswal, A.K. Inhibitor of Nrf2 (INrf2 or Keap1) protein degrades Bcl-xL via phosphoglycerate mutase 5 and controls cellular apoptosis. J. Biol. Chem 2011, 286, 44542–44556. [Google Scholar]
- Zhuang, M.; Guan, S.; Wand, H.; Burlingame, A.L.; Wells, J.A. Substrates of IAP ubiquitin ligases identified with a designed orthogonal E3 ligase, the NEDDylator. Mol. Cell 2013, 49, 273–282. [Google Scholar]
- Vaux, D.L.; Silke, J. IAPs, RINGs and ubiquitylation. Nat. Rev. Mol. Cell Biol 2005, 6, 287–297. [Google Scholar]
- Vucic, D.; Dixit, V.M.; Wertz, I.E. Ubiquitylation in apoptosis: A post-translational modification at the edge of life and death. Nat. Rev. Mol. Cell Biol 2011, 12, 439–452. [Google Scholar]
- Vandenabeele, P.; Galluzzi, L.; Vanden Berghe, T.; Kroemer, G. Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol 2010, 11, 700–714. [Google Scholar]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K. Phosphorylation-driven assembly of the RIP1–RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123. [Google Scholar]
- He, S.; Wang, L.; Miao, L.; Wang, T.; Du, F.; Zhao, L.; Wang, X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009, 137, 1100–1111. [Google Scholar]
- Zhang, D.W.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.J.; Lin, S.C.; Dong, M.Q.; Han, J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009, 325, 332–336. [Google Scholar]
- Wang, Z.; Jiang, H.; Chen, S.; Du, F.; Wang, X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 2012, 148, 228–243. [Google Scholar]




© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sadatomi, D.; Tanimura, S.; Ozaki, K.-i.; Takeda, K. Atypical Protein Phosphatases: Emerging Players in Cellular Signaling. Int. J. Mol. Sci. 2013, 14, 4596-4612. https://doi.org/10.3390/ijms14034596
Sadatomi D, Tanimura S, Ozaki K-i, Takeda K. Atypical Protein Phosphatases: Emerging Players in Cellular Signaling. International Journal of Molecular Sciences. 2013; 14(3):4596-4612. https://doi.org/10.3390/ijms14034596
Chicago/Turabian StyleSadatomi, Daichi, Susumu Tanimura, Kei-ichi Ozaki, and Kohsuke Takeda. 2013. "Atypical Protein Phosphatases: Emerging Players in Cellular Signaling" International Journal of Molecular Sciences 14, no. 3: 4596-4612. https://doi.org/10.3390/ijms14034596