Phenotypic Identification of the Redox Dye Methylene Blue as an Antagonist of Heat Shock Response Gene Expression in Metastatic Melanoma Cells


Abstract
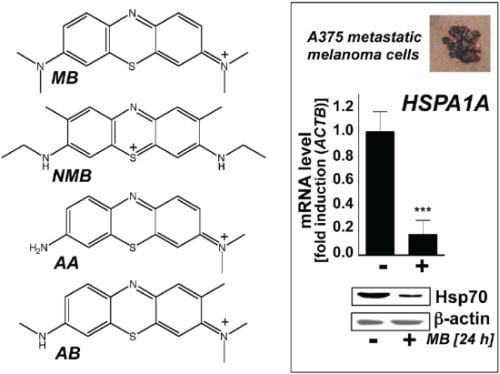
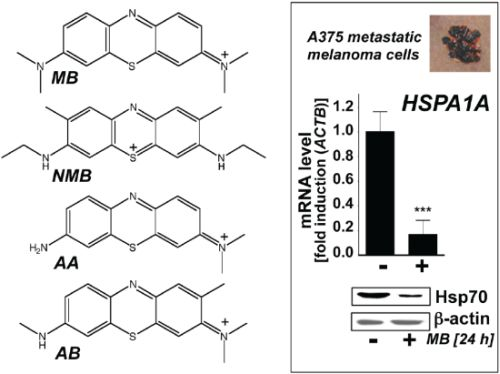
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
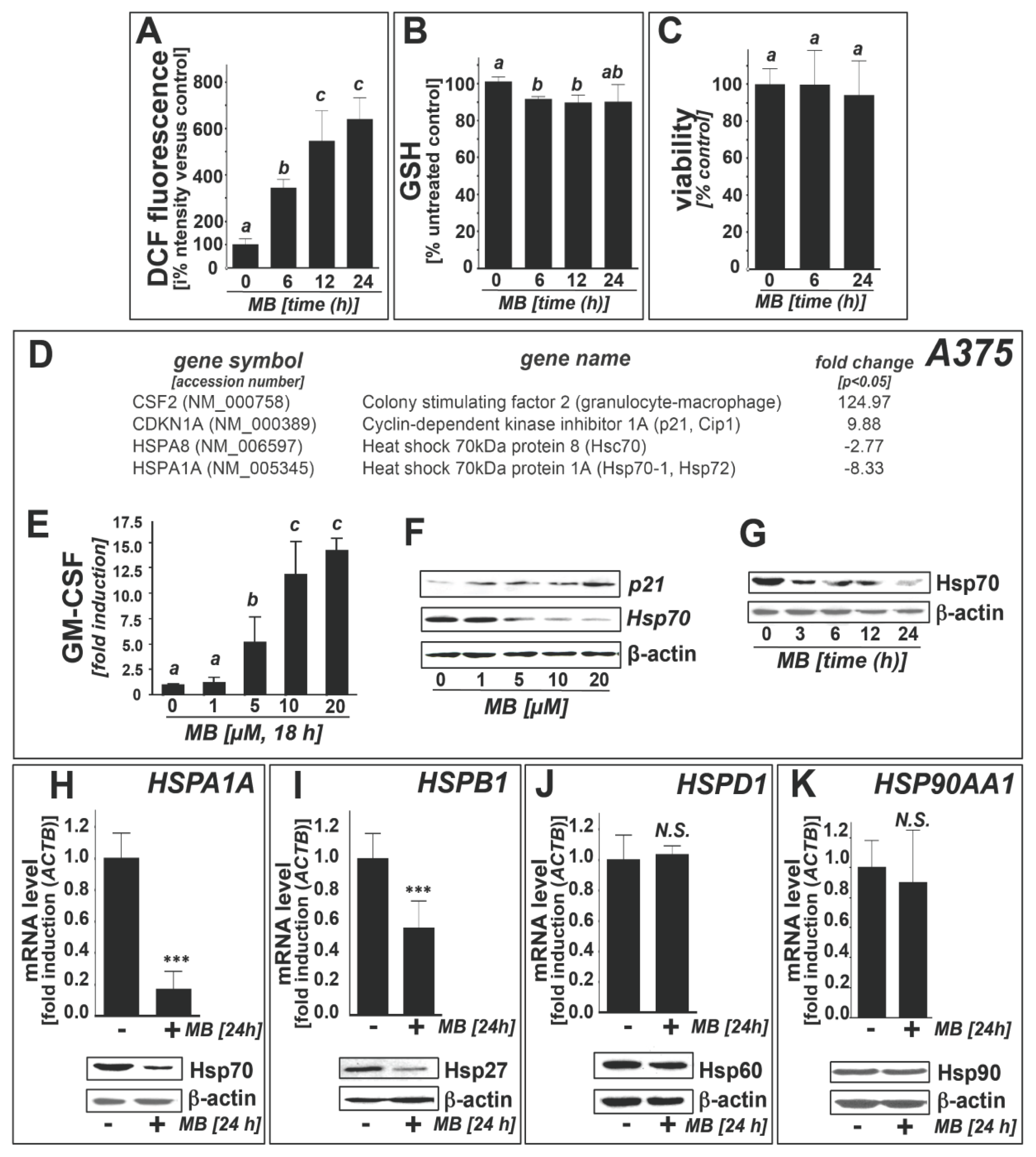
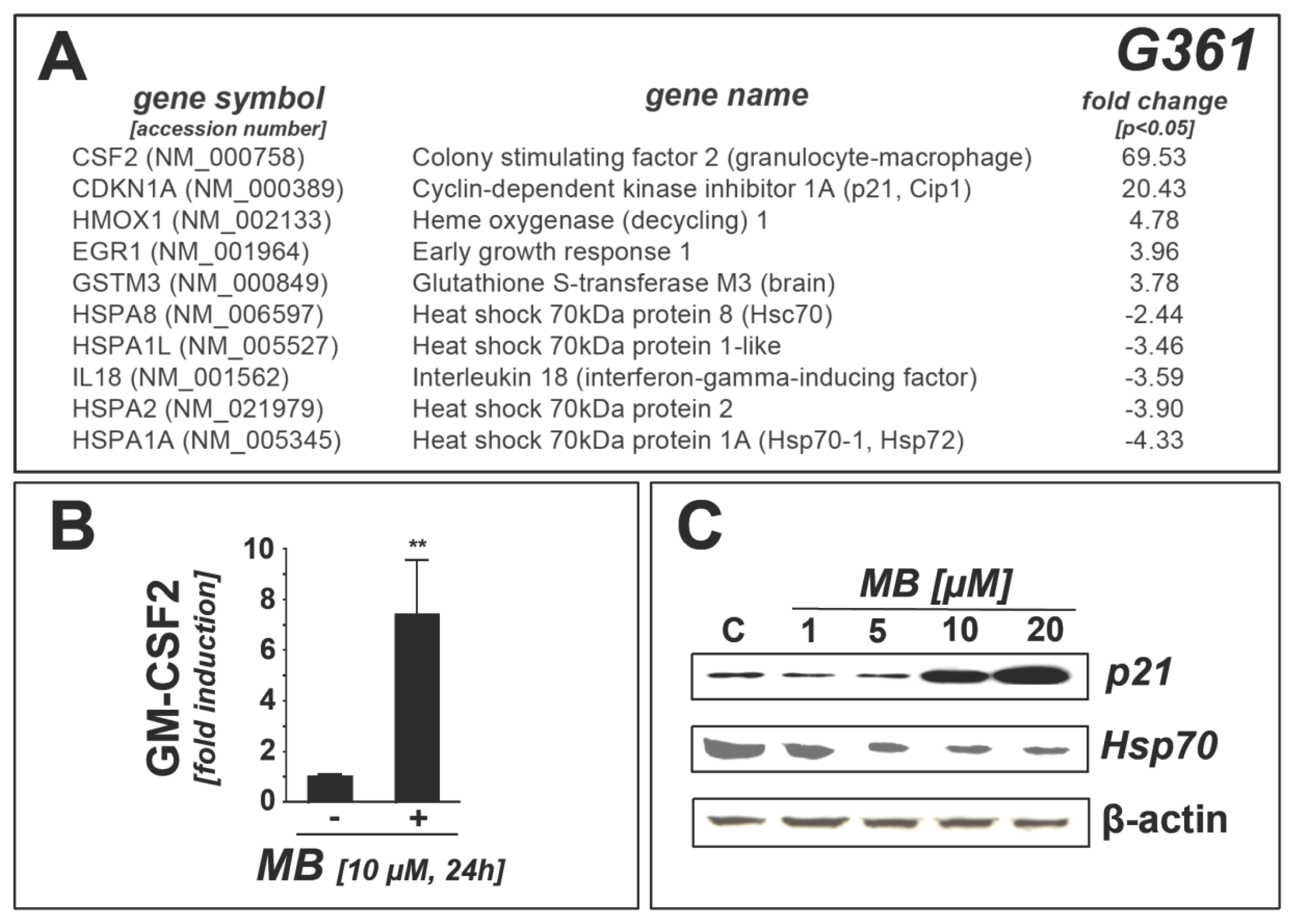
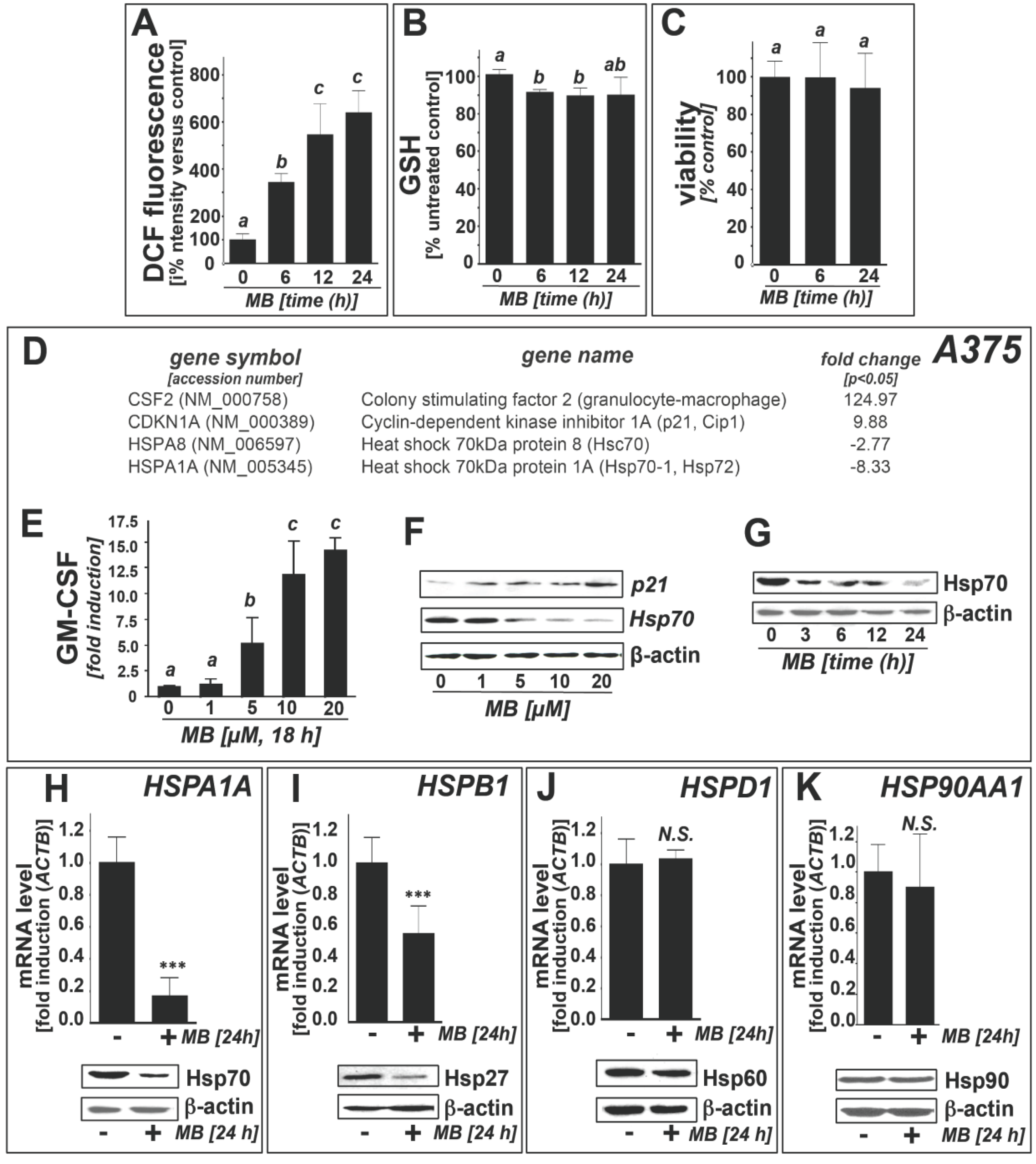
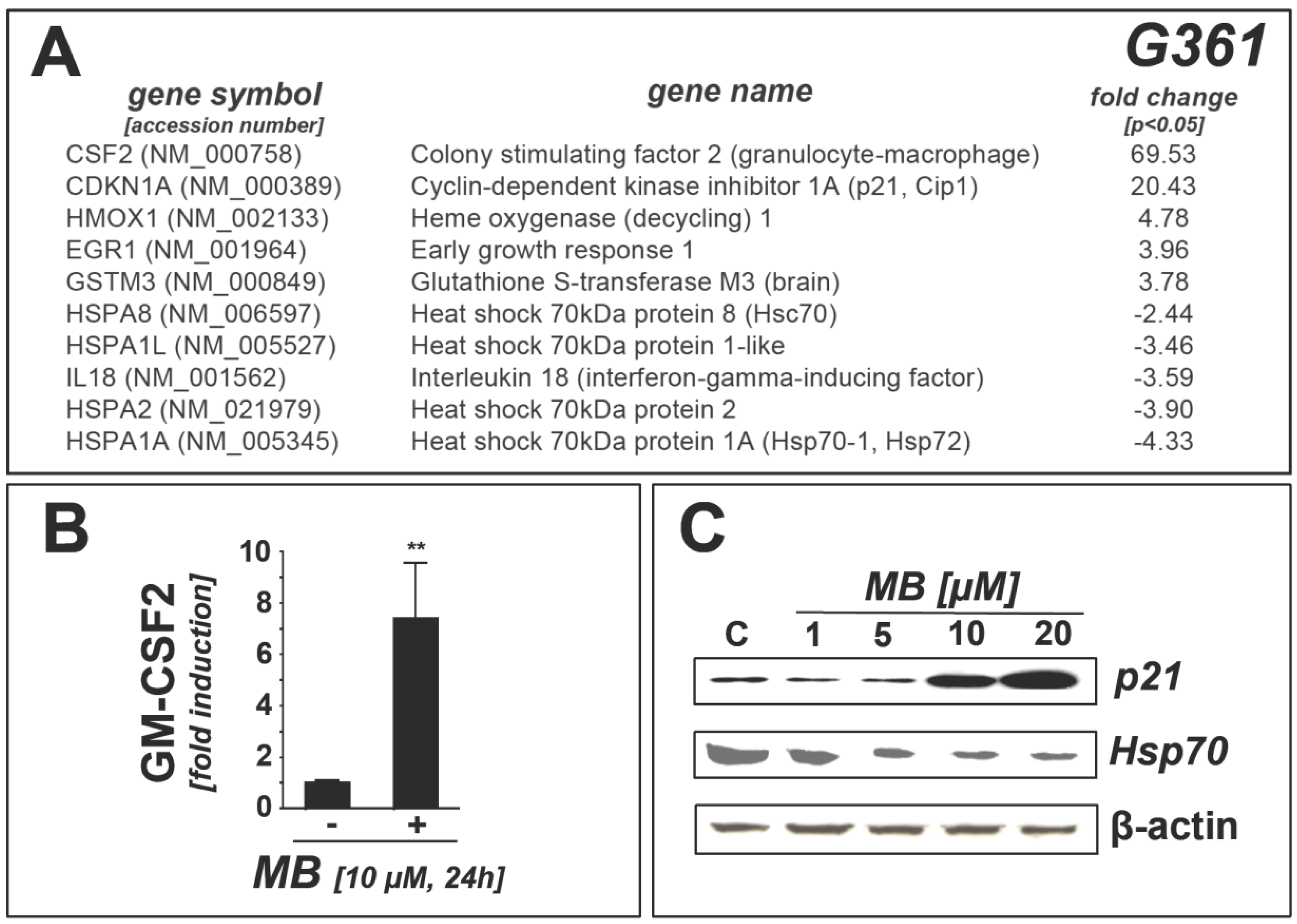
2.1. Downregulation of Heat Shock Response Gene Expression in Human A375 and G361 Metastatic Melanoma Cells Exposed to the Redox-Active Phenothiazine-Derivative Methylene Blue
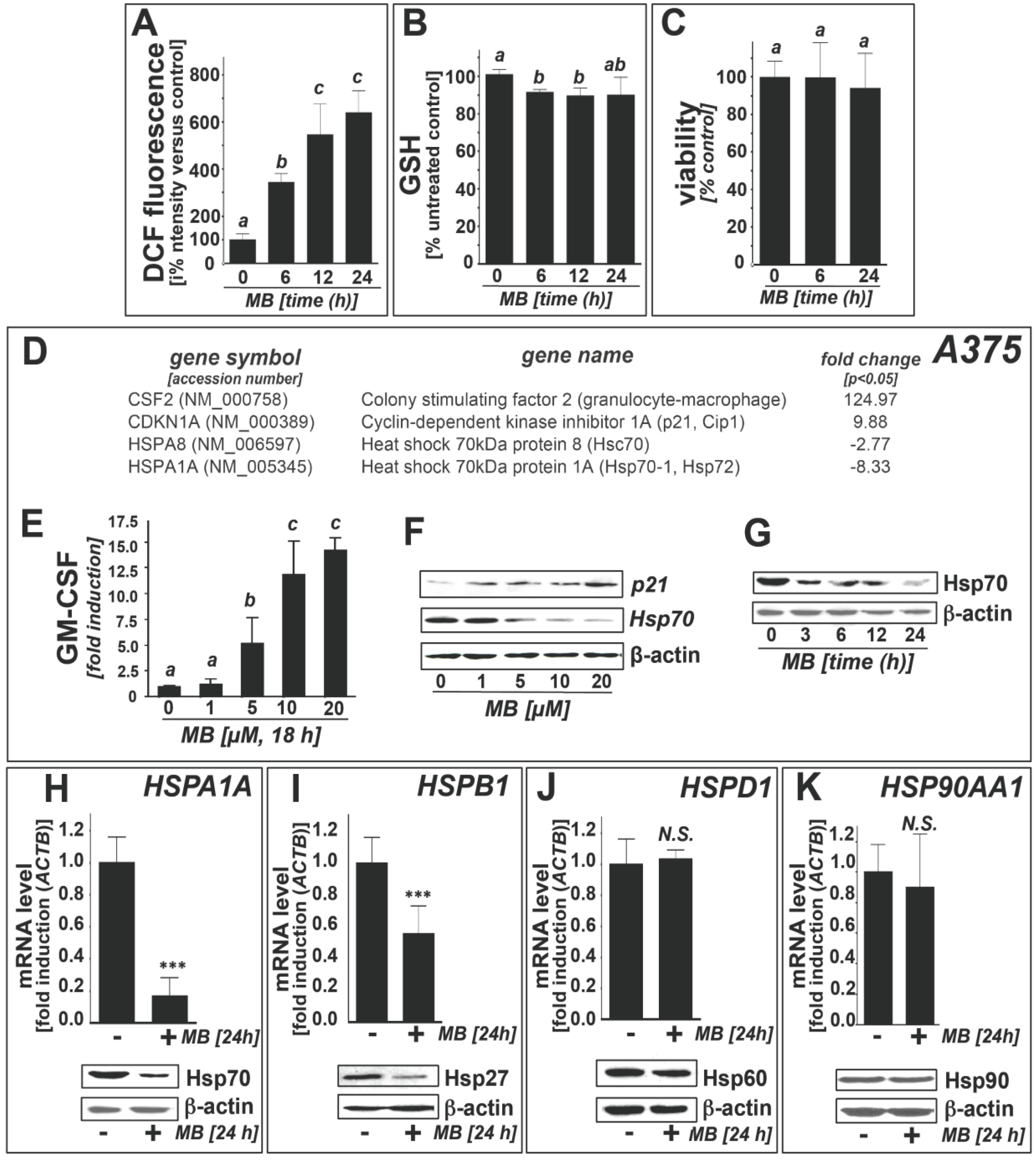
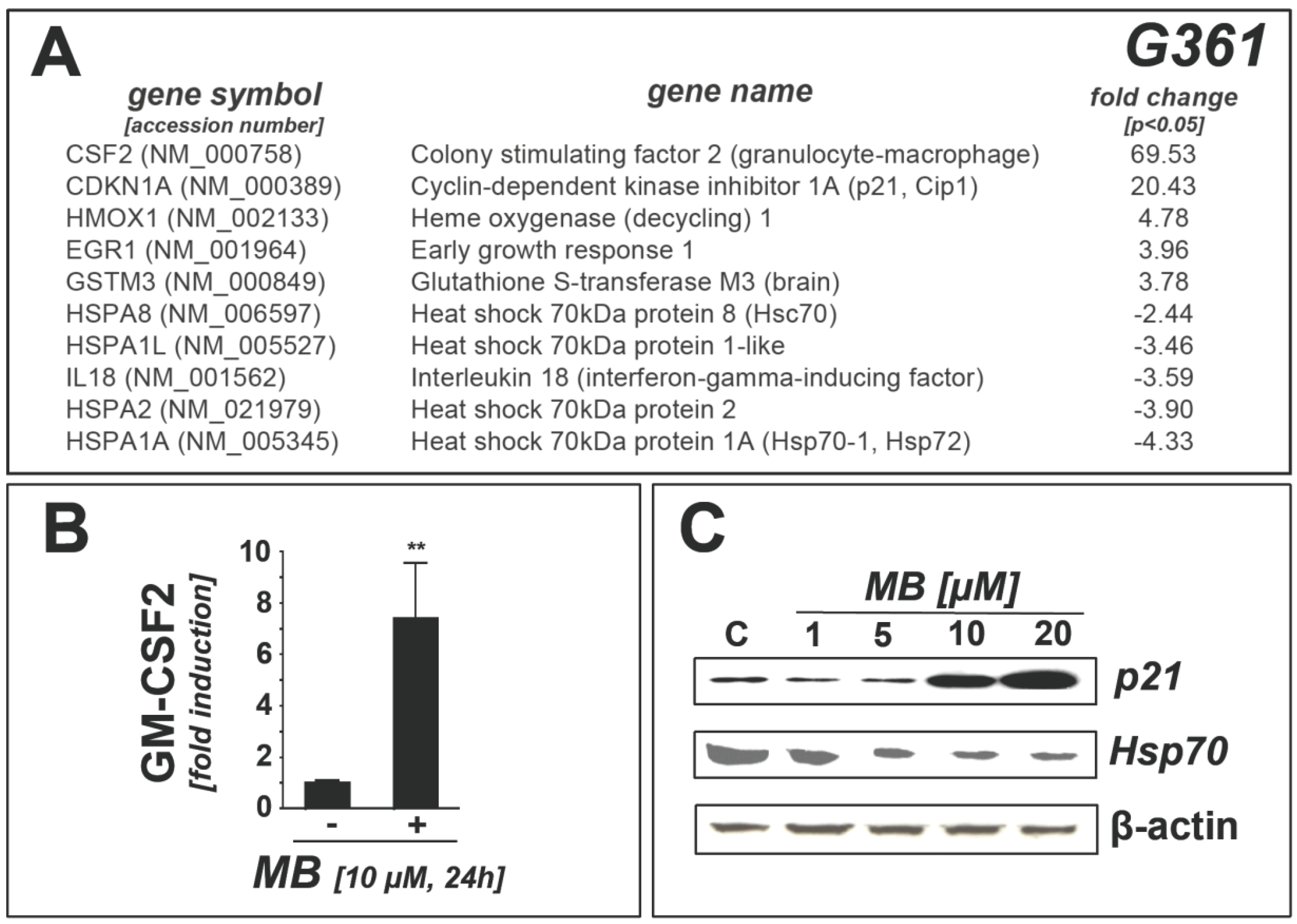
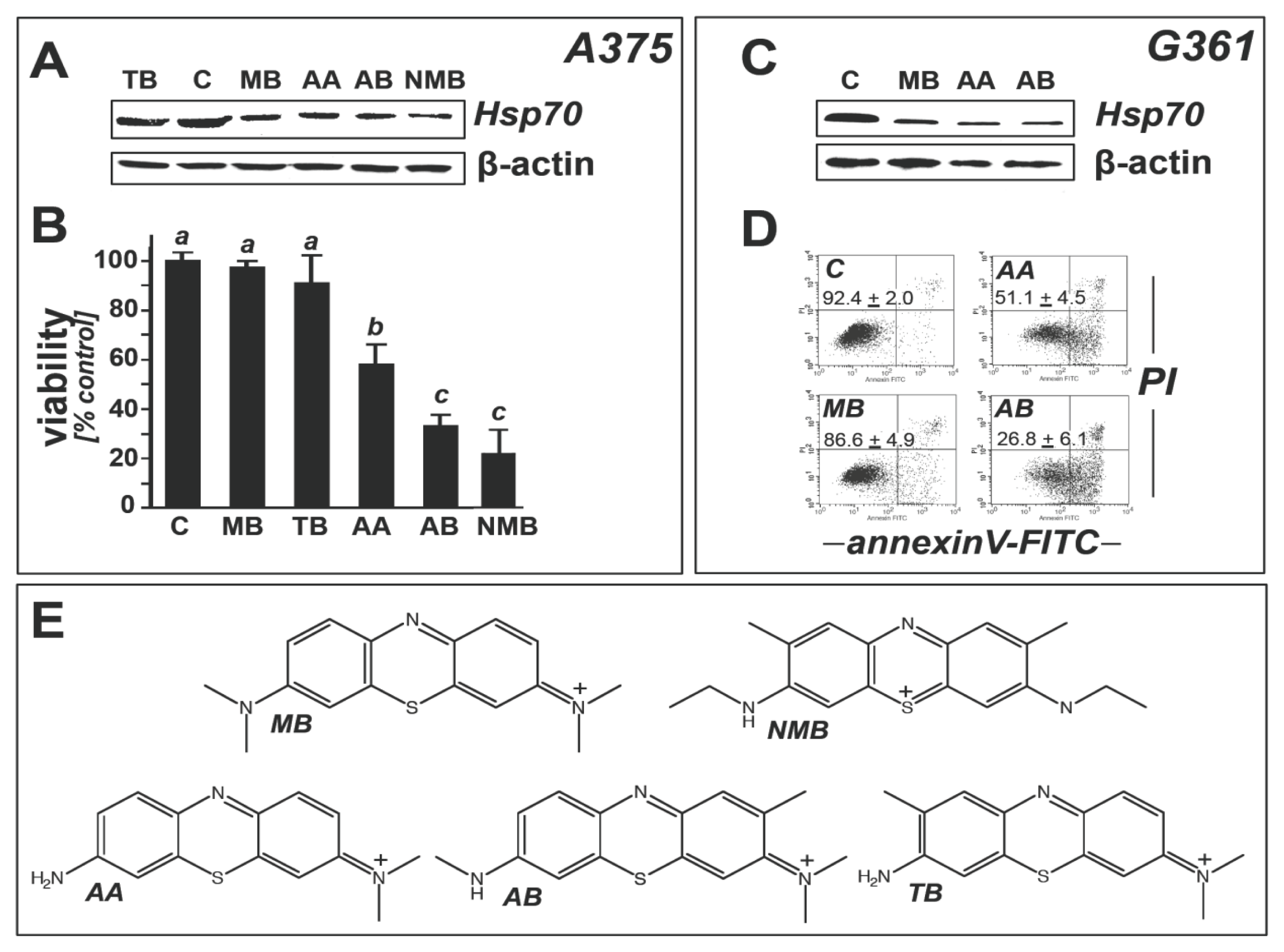
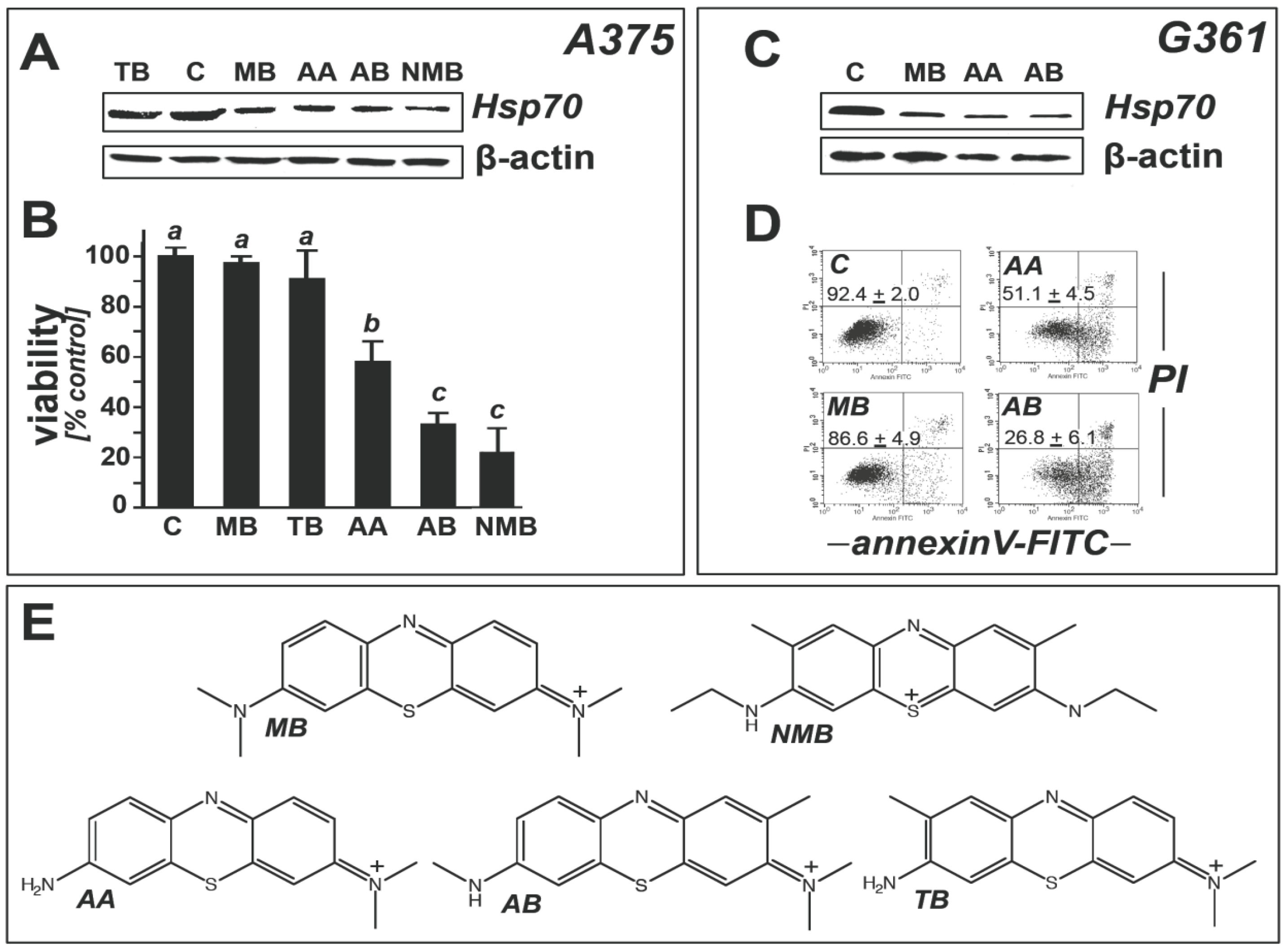
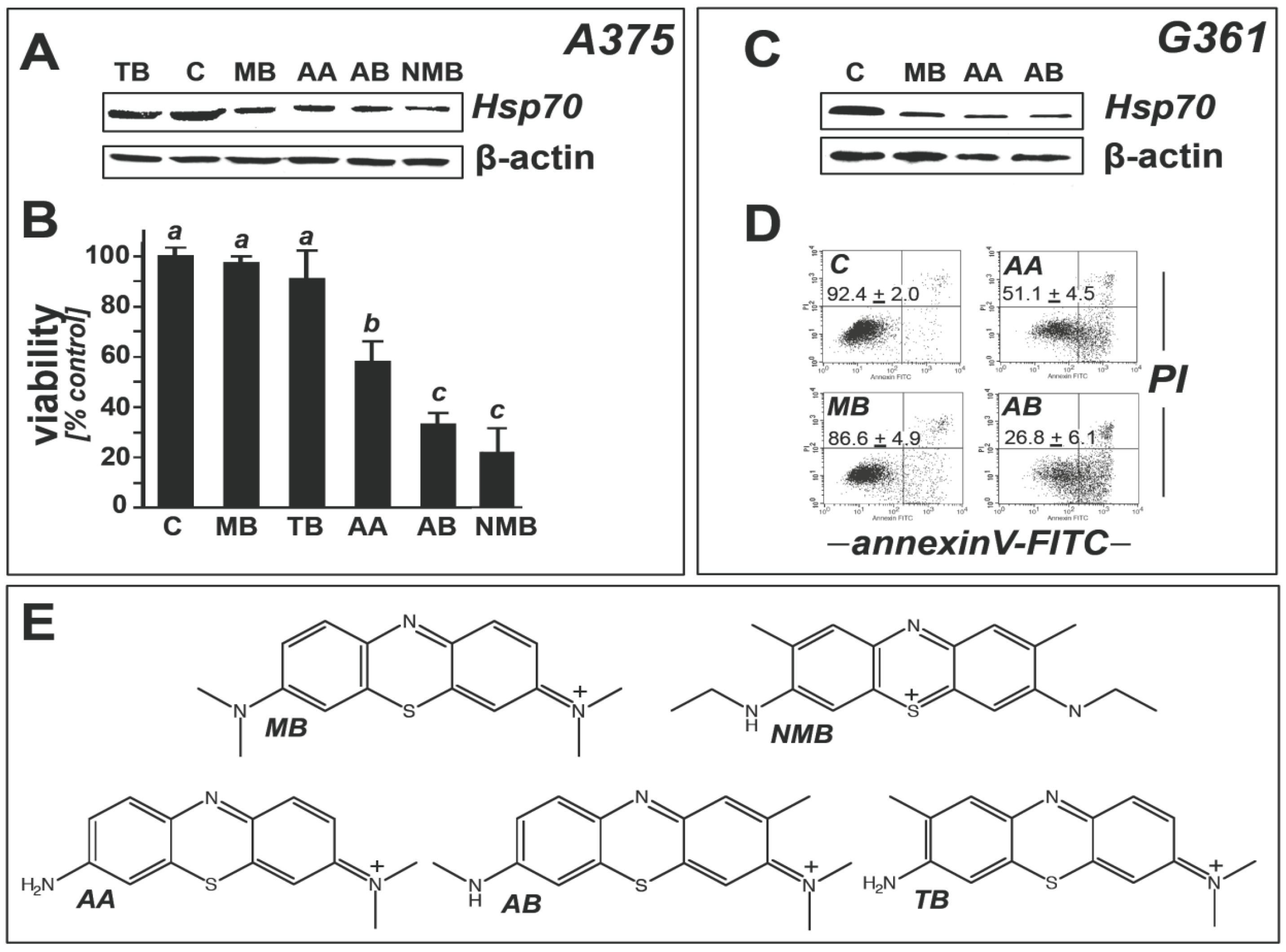
2.2. Modulation of Melanoma Cell Viability and Hsp70 Expression by MB-Related Phenothiazine-Derivatives
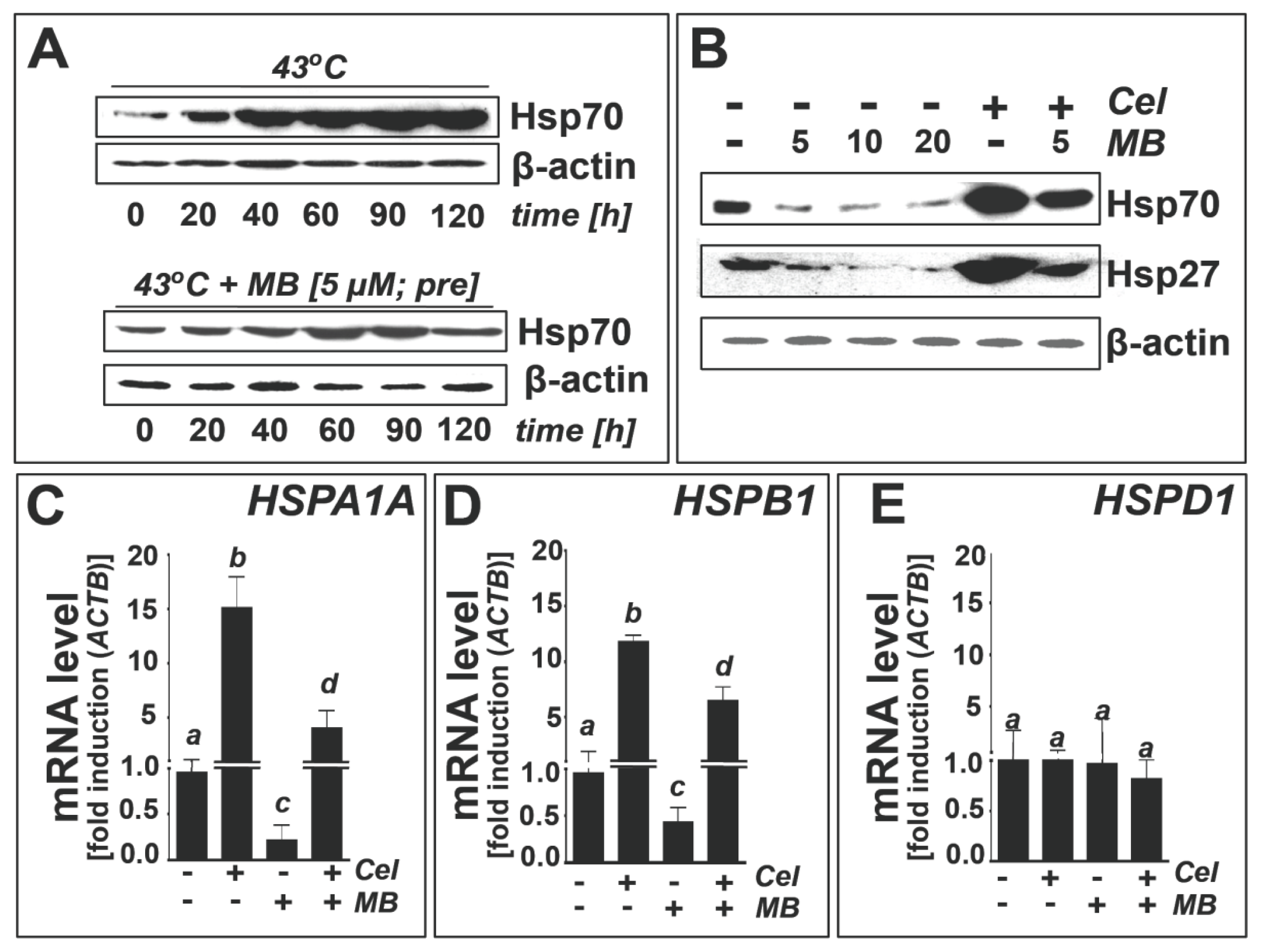
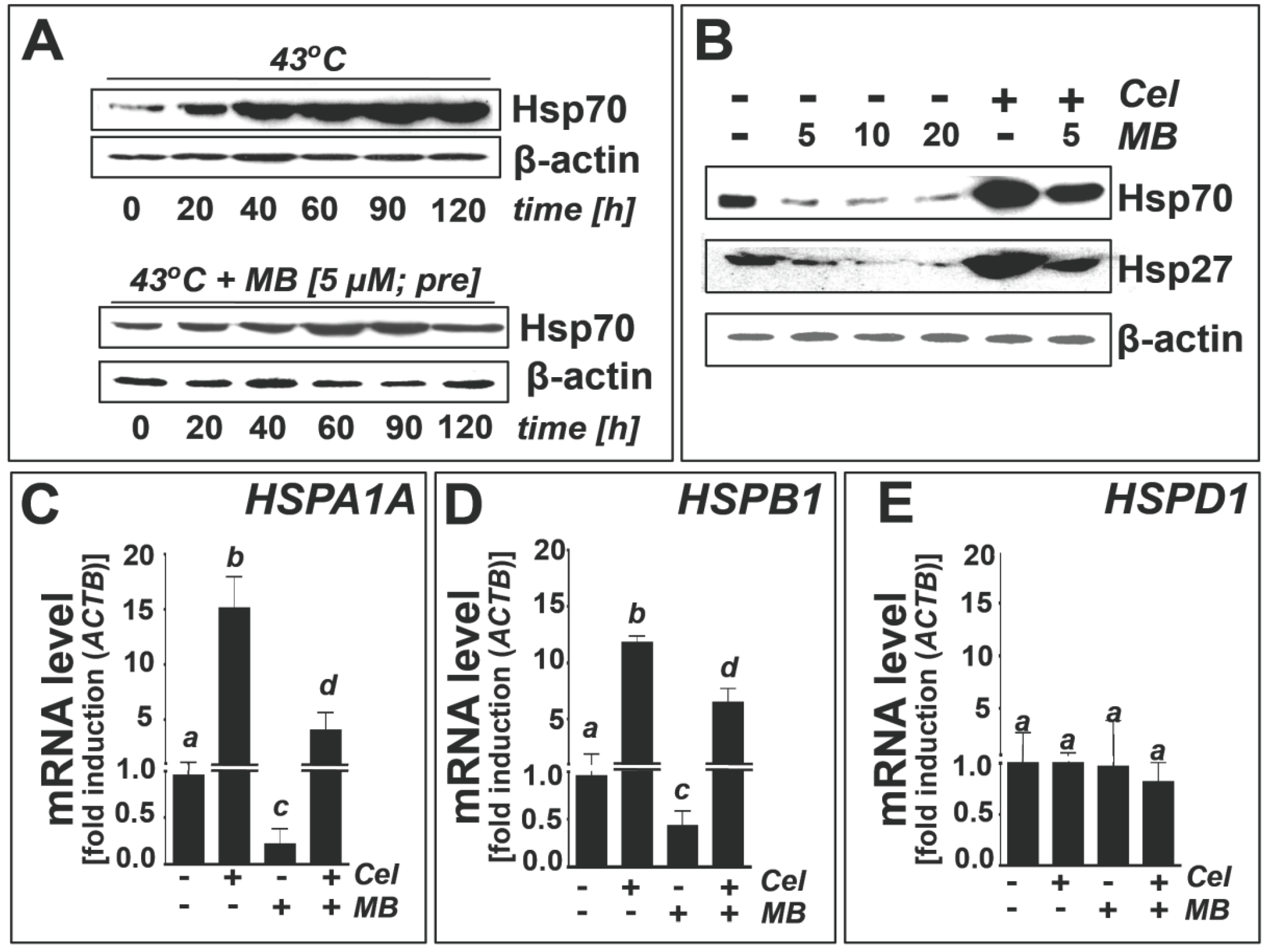
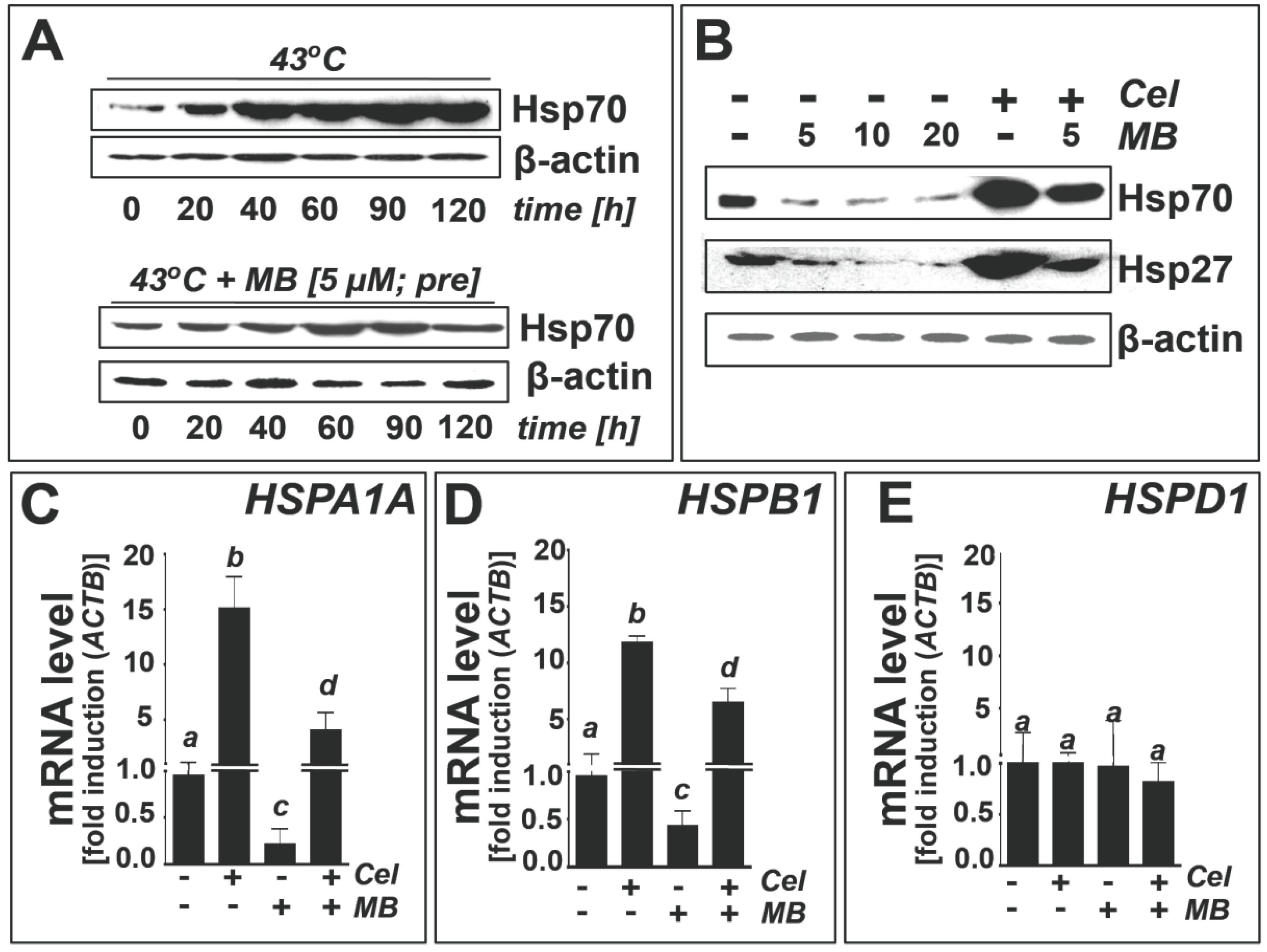
2.3. Methylene Blue Attenuates Thermal and Pharmacological Induction of Heat Shock Response Gene Expression
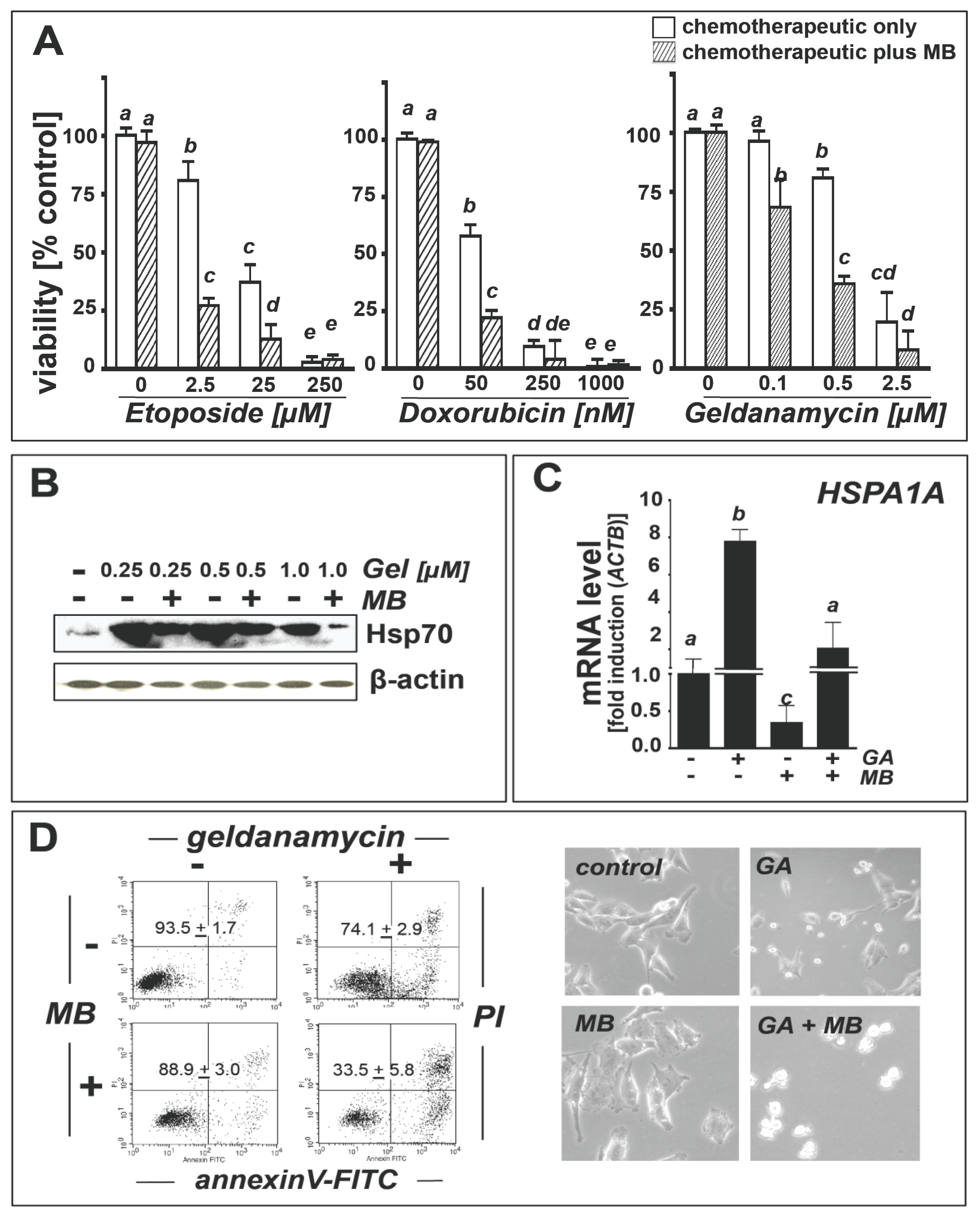
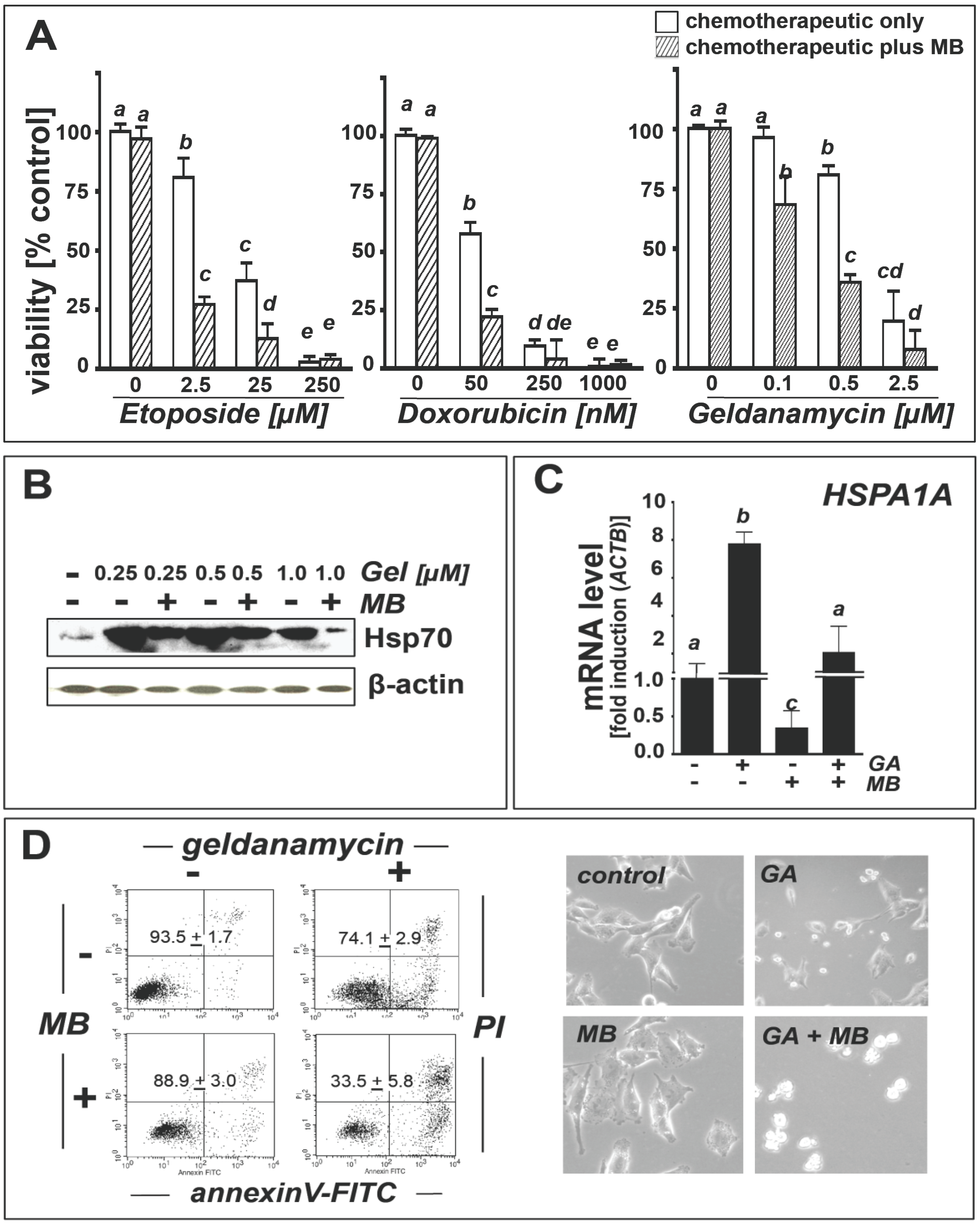
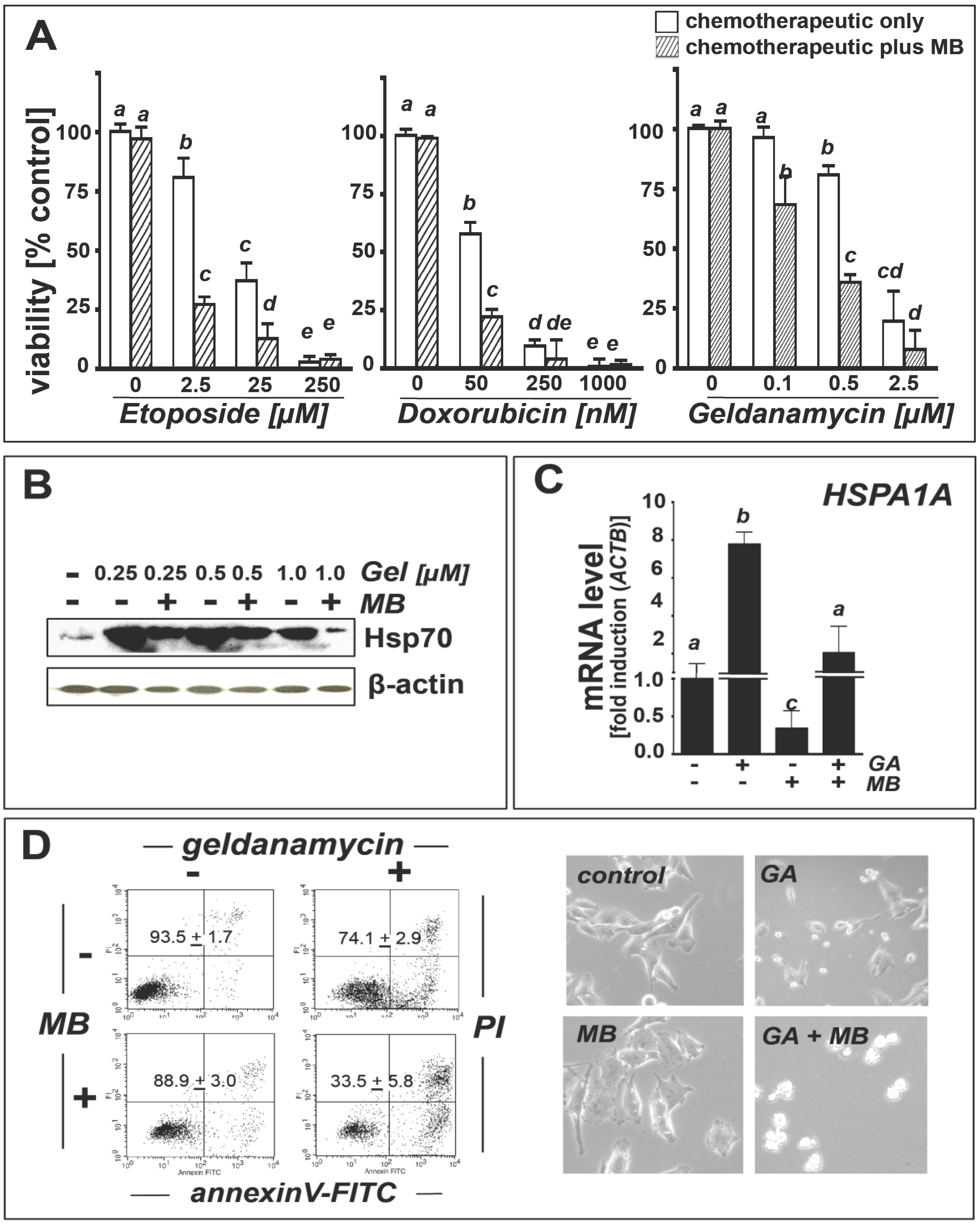
2.4. Methylene Blue Attenuates Geldanamycin-Induced HSPA1A Upregulation and Sensitizes Human A375 Metastatic Melanoma Cells to Geldanamycin-Induced Apoptosis
3. Experimental Section
3.1. Chemicals
3.2. Cell Culture
3.3. Human Stress and Toxicity Pathfinder™ RT2 Profiler™ PCR Expression Array
3.4. HSPA1A, HSPB1, HSPD1, HSP90AA1 Expression Analysis by Real Time RT-PCR
3.5. Immunoblot Analysis
3.6. GM-CSF ELISA
3.7. Cell Death Analysis
3.8. Detection of Intracellular Oxidative Stress by Flow Cytometric Analysis
3.9. Determination of Reduced Cellular Glutathione Content
3.10. Statistical Analysis
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Healy, S.J.; Gorman, A.M.; Mousavi-Shafaei, P.; Gupta, S.; Samali, A. Targeting the endoplasmic reticulum-stress response as an anticancer strategy. Eur. J. Pharmacol. 2009, 625, 234–246. [Google Scholar] [CrossRef]
- Wondrak, G.T. Redox-directed cancer therapeutics: Molecular mechanisms and opportunities. Antioxid. Redox Signal. 2009, 11, 3013–3069. [Google Scholar] [CrossRef]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar]
- Parcellier, A.; Schmitt, E.; Brunet, M.; Hammann, A.; Solary, E.; Garrido, C. Small heat shock proteins HSP27 and alphaB-crystallin: Cytoprotective and oncogenic functions. Antioxid. Redox Signal. 2005, 7, 404–413. [Google Scholar] [CrossRef]
- Goloudina, A.R.; Demidov, O.N.; Garrido, C. Inhibition of HSP70: A challenging anti-cancer strategy. Cancer Lett. 2012, 325, 117–124. [Google Scholar] [CrossRef]
- Trepel, J.; Mollapour, M.; Giaccone, G.; Neckers, L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 2010, 10, 537–549. [Google Scholar]
- Guo, F.; Rocha, K.; Bali, P.; Pranpat, M.; Fiskus, W.; Boyapalle, S.; Kumaraswamy, S.; Balasis, M.; Greedy, B.; Armitage, E.S.; et al. Abrogation of heat shock protein 70 induction as a strategy to increase antileukemia activity of heat shock protein 90 inhibitor 17-allylamino-demethoxy geldanamycin. Cancer Res. 2005, 65, 10536–10544. [Google Scholar]
- Whitesell, L.; Lindquist, S. Inhibiting the transcription factor HSF1 as an anticancer strategy. Expert Opin. Ther. Targets 2009, 13, 469–478. [Google Scholar] [CrossRef]
- Leu, J.I.; Pimkina, J.; Frank, A.; Murphy, M.E.; George, D.L. A small molecule inhibitor of inducible heat shock protein 70. Mol. Cell. 2009, 36, 15–27. [Google Scholar] [CrossRef]
- Leu, J.I.; Pimkina, J.; Pandey, P.; Murphy, M.E.; George, D.L. HSP70 inhibition by the small-molecule 2-phenylethynesulfonamide impairs protein clearance pathways in tumor cells. Mol. Cancer Res. 2011, 9, 936–947. [Google Scholar] [CrossRef]
- Taldone, T.; Gozman, A.; Maharaj, R.; Chiosis, G. Targeting Hsp90: Small-molecule inhibitors and their clinical development. Curr. Opin. Pharmacol. 2008, 8, 370–374. [Google Scholar] [CrossRef]
- Porter, J.R.; Fritz, C.C.; Depew, K.M. Discovery and development of Hsp90 inhibitors: A promising pathway for cancer therapy. Curr. Opin. Chem. Biol. 2010, 14, 412–420. [Google Scholar] [CrossRef]
- Garbe, C.; Eigentler, T.K.; Keilholz, U.; Hauschild, A.; Kirkwood, J.M. Systematic review of medical treatment in melanoma: Current status and future prospects. Oncologist 2011, 16, 5–24. [Google Scholar] [CrossRef]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef]
- Coss, R.A.; Storck, C.W.; Daskalakis, C.; Berd, D.; Wahl, M.L. Intracellular acidification abrogates the heat shock response and compromises survival of human melanoma cells. Mol. Cancer Ther. 2003, 2, 383–388. [Google Scholar]
- Carta, F.; Demuro, P.P.; Zanini, C.; Santona, A.; Castiglia, D.; D’Atri, S.; Ascierto, P.A.; Napolitano, M.; Cossu, A.; Tadolini, B.; et al. Analysis of candidate genes through a proteomics-based approach in primary cell lines from malignant melanomas and their metastases. Melanoma Res. 2005, 15, 235–244. [Google Scholar] [CrossRef]
- Ciocca, D.R.; Calderwood, S.K. Heat shock proteins in cancer: Diagnostic, prognostic, predictive, and treatment implications. Cell. Stress Chaperones 2005, 10, 86–103. [Google Scholar] [CrossRef]
- Kalogeraki, A.; Garbagnati, F.; Darivianaki, K.; Delides, G.S.; Santinami, M.; Stathopoulos, E.N.; Zoras, O. HSP-70, C-myc and HLA-DR expression in patients with cutaneous malignant melanoma metastatic in lymph nodes. Anticancer Res. 2006, 26, 3551–3554. [Google Scholar]
- Bair, W.B., III; Cabello, C.M.; Uchida, K.; Bause, A.S.; Wondrak, G.T. GLO1 overexpression in human malignant melanoma. Melanoma Res. 2010, 20, 85–96. [Google Scholar] [CrossRef]
- de Ridder, G.G.; Ray, R.; Pizzo, S.V. A murine monoclonal antibody directed against the carboxyl-terminal domain of GRP78 suppresses melanoma growth in mice. Melanoma Res. 2012, 22, 225–235. [Google Scholar] [CrossRef]
- Straume, O.; Shimamura, T.; Lampa, M.J.; Carretero, J.; Oyan, A.M.; Jia, D.; Borgman, C.L.; Soucheray, M.; Downing, S.R.; Short, S.M.; et al. Suppression of heat shock protein 27 induces long-term dormancy in human breast cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 8699–8704. [Google Scholar]
- Deichmann, M.; Polychronidis, M.; Benner, A.; Kleist, C.; Thome, M.; Kahle, B.; Helmke, B.M. Expression of the heat shock cognate protein HSP73 correlates with tumour thickness of primary melanomas and is enhanced in melanoma metastases. Int. J. Oncol. 2004, 25, 259–268. [Google Scholar]
- Knorr, C.; Pelz, J.O.; Gohl, J.; Hohenberger, W.; Meyer, T. Expression of chemoresistance-related genes and heat shock protein 72 in hyperthermic isolated limb perfusion of malignant melanoma: An experimental study. J. Oncol. 2010, 2010, 138758. [Google Scholar]
- Wondrak, G.T. NQO1-activated phenothiazinium redox cyclers for the targeted bioreductive induction of cancer cell apoptosis. Free Radic. Biol. Med. 2007, 43, 178–190. [Google Scholar] [CrossRef]
- Cabello, C.M.; Bair, W.B., III; Bause, A.S.; Wondrak, G.T. Antimelanoma activity of the redox dye DCPIP (2,6-dichlorophenolindophenol) is antagonized by NQO1. Biochem. Pharmacol. 2009, 78, 344–354. [Google Scholar]
- Meissner, P.E.; Mandi, G.; Coulibaly, B.; Witte, S.; Tapsoba, T.; Mansmann, U.; Rengelshausen, J.; Schiek, W.; Jahn, A.; Walter-Sack, I.; et al. Methylene blue for malaria in Africa: Results from a dose-finding study in combination with chloroquine. Malar. J. 2006, 5, 84. [Google Scholar] [CrossRef]
- Skold, A.; Cosco, D.L.; Klein, R. Methemoglobinemia: Pathogenesis, diagnosis, and management. South. Med. J. 2011, 104, 757–761. [Google Scholar] [CrossRef]
- Ginimuge, P.R.; Jyothi, S.D. Methylene blue: Revisited. J. Anaesthesiol. Clin. Pharmacol. 2010, 26, 517–520. [Google Scholar]
- Mayer, B.; Brunner, F.; Schmidt, K. Inhibition of nitric oxide synthesis by methylene blue. Biochem. Pharmacol. 1993, 45, 367–374. [Google Scholar] [CrossRef]
- Ramsay, R.R.; Dunford, C.; Gillman, P.K. Methylene blue and serotonin toxicity: Inhibition of monoamine oxidase A (MAO A) confirms a theoretical prediction. Br. J. Pharmacol. 2007, 152, 946–951. [Google Scholar] [CrossRef]
- Wischik, C.M.; Edwards, P.C.; Lai, R.Y.; Roth, M.; Harrington, C.R. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc. Natl. Acad. Sci. USA 1996, 93, 11213–11218. [Google Scholar]
- Hattori, M.; Sugino, E.; Minoura, K.; In, Y.; Sumida, M.; Taniguchi, T.; Tomoo, K.; Ishida, T. Different inhibitory response of cyanidin and methylene blue for filament formation of tau microtubule-binding domain. Biochem. Biophys. Res. Commun. 2008, 374, 158–163. [Google Scholar] [CrossRef]
- Gura, T. Hope in Alzheimer’s fight emerges from unexpected places. Nat. Med. 2008, 14, 894. [Google Scholar] [CrossRef]
- Oz, M.; Lorke, D.E.; Petroianu, G.A. Methylene blue and Alzheimer’s disease. Biochem. Pharmacol. 2009, 78, 927–932. [Google Scholar] [CrossRef]
- Wen, Y.; Li, W.; Poteet, E.C.; Xie, L.; Tan, C.; Yan, L.J.; Ju, X.; Liu, R.; Qian, H.; Marvin, M.A.; et al. Alternative mitochondrial electron transfer as a novel strategy for neuroprotection. J. Biol. Chem. 2011, 286, 16504–16515. [Google Scholar]
- Lin, A.L.; Poteet, E.; Du, F.; Gourav, R.C.; Liu, R.; Wen, Y.; Bresnen, A.; Huang, S.; Fox, P.T.; Yang, S.H.; et al. Methylene blue as a cerebral metabolic and hemodynamic enhancer. PLoS One 2012, 7, e46585. [Google Scholar]
- Medina, D.X.; Caccamo, A.; Oddo, S. Methylene blue reduces abeta levels and rescues early cognitive deficit by increasing proteasome activity. Brain Pathol. 2011, 21, 140–149. [Google Scholar] [CrossRef]
- Atamna, H.; Nguyen, A.; Schultz, C.; Boyle, K.; Newberry, J.; Kato, H.; Ames, B.N. Methylene blue delays cellular senescence and enhances key mitochondrial biochemical pathways. FASEB J. 2008, 22, 703–712. [Google Scholar]
- Cabello, C.M.; Lamore, S.D.; Bair, W.B., III; Qiao, S.; Azimian, S.; Lesson, J.L.; Wondrak, G.T. The redox antimalarial dihydroartemisinin targets human metastatic melanoma cells but not primary melanocytes with induction of NOXA-dependent apoptosis. Invest. New Drugs 2012, 30, 1289–1301. [Google Scholar] [CrossRef]
- Cabello, C.M.; Bair, W.B., III; Ley, S.; Lamore, S.D.; Azimian, S.; Wondrak, G.T. The experimental chemotherapeutic N(6)-furfuryladenosine (kinetin-riboside) induces rapid ATP depletion, genotoxic stress, and CDKN1A (p21) upregulation in human cancer cell lines. Biochem. Pharmacol. 2009, 77, 1125–1138. [Google Scholar] [CrossRef]
- Soiffer, R.; Hodi, F.S.; Haluska, F.; Jung, K.; Gillessen, S.; Singer, S.; Tanabe, K.; Duda, R.; Mentzer, S.; Jaklitsch, M.; et al. Vaccination with irradiated, autologous melanoma cells engineered to secrete granulocyte-macrophage colony-stimulating factor by adenoviral-mediated gene transfer augments antitumor immunity in patients with metastatic melanoma. J. Clin. Oncol. 2003, 21, 3343–3350. [Google Scholar]
- Tarhini, A.A.; Leng, S.; Moschos, S.J.; Yin, Y.; Sander, C.; Lin, Y.; Gooding, W.E.; Kirkwood, J.M. Safety and immunogenicity of vaccination with MART-1 (26–35, 27L), gp100 (209–217, 210M), and tyrosinase (368–376, 370D) in adjuvant with PF-3512676 and GM-CSF in metastatic melanoma. J. Immunother. 2012, 35, 359–366. [Google Scholar] [CrossRef]
- Jinwal, U.K.; Miyata, Y.; Koren, J., III; Jones, J.R.; Trotter, J.H.; Chang, L.; O’Leary, J.; Morgan, D.; Lee, D.C.; Shults, C.L.; et al. Chemical manipulation of hsp70 ATPase activity regulates tau stability. J. Neurosci. 2009, 29, 12079–12088. [Google Scholar]
- Wang, A.M.; Morishima, Y.; Clapp, K.M.; Peng, H.M.; Pratt, W.B.; Gestwicki, J.E.; Osawa, Y.; Lieberman, A.P. Inhibition of hsp70 by methylene blue affects signaling protein function and ubiquitination and modulates polyglutamine protein degradation. J. Biol. Chem. 2010, 285, 15714–15723. [Google Scholar]
- Miyata, Y.; Rauch, J.N.; Jinwal, U.K.; Thompson, A.D.; Srinivasan, S.; Dickey, C.A.; Gestwicki, J.E. Cysteine reactivity distinguishes redox sensing by the heat-inducible and constitutive forms of heat shock protein 70. Chem. Biol. 2012, 19, 1391–1399. [Google Scholar] [CrossRef]
- Yang, F.; Xia, S.; Liu, Z.; Chen, J.; Lin, Y.; Qiu, B.; Chen, G. Analysis of methylene blue and its metabolites in blood by capillary electrophoresis/electrospray ionization mass spectrometry. Electrophoresis 2011, 32, 659–664. [Google Scholar] [CrossRef]
- Westerheide, S.D.; Bosman, J.D.; Mbadugha, B.N.; Kawahara, T.L.; Matsumoto, G.; Kim, S.; Gu, W.; Devlin, J.P.; Silverman, R.B.; Morimoto, R.I. Celastrols as inducers of the heat shock response and cytoprotection. J. Biol. Chem. 2004, 279, 56053–56060. [Google Scholar]
- Trott, A.; West, J.D.; Klaic, L.; Westerheide, S.D.; Silverman, R.B.; Morimoto, R.I.; Morano, K.A. Activation of heat shock and antioxidant responses by the natural product celastrol: Transcriptional signatures of a thiol-targeted molecule. Mol. Biol. Cell. 2008, 19, 1104–1112. [Google Scholar]
- Klaic, L.; Morimoto, R.I.; Silverman, R.B. Celastrol analogues as inducers of the heat shock response. Design and synthesis of affinity probes for the identification of protein targets. ACS Chem. Biol. 2012, 7, 928–937. [Google Scholar] [CrossRef]
- Park, S.R.; Lee, K.D.; Kim, U.K.; Gil, Y.G.; Oh, K.S.; Park, B.S.; Kim, G.C. Pseudomonas aeruginosa exotoxin A reduces chemoresistance of oral squamous carcinoma cell via inhibition of heat shock proteins 70 (HSP70). Yonsei Med. J. 2010, 51, 708–716. [Google Scholar] [CrossRef]
- Gabai, V.L.; Sherman, M.Y.; Yaglom, J.A. HSP72 depletion suppresses gammaH2AX activation by genotoxic stresses via p53/p21 signaling. Oncogene 2010, 29, 1952–1962. [Google Scholar] [CrossRef]
- McCollum, A.K.; Lukasiewicz, K.B.; Teneyck, C.J.; Lingle, W.L.; Toft, D.O.; Erlichman, C. Cisplatin abrogates the geldanamycin-induced heat shock response. Mol. Cancer Ther. 2008, 7, 3256–3264. [Google Scholar] [CrossRef]
- Smith, V.; Sausville, E.A.; Camalier, R.F.; Fiebig, H.H.; Burger, A.M. Comparison of 17-dimethylaminoethylamino-17-demethoxy-geldanamycin (17DMAG) and 17-allylamino-17-demethoxygeldanamycin (17AAG) in vitro: Effects on Hsp90 and client proteins in melanoma models. Cancer Chemother. Pharmacol. 2005, 56, 126–137. [Google Scholar] [CrossRef]
- Lamore, S.D.; Cabello, C.M.; Wondrak, G.T. The topical antimicrobial zinc pyrithione is a heat shock response inducer that causes DNA damage and PARP-dependent energy crisis in human skin cells. Cell. Stress Chaperones 2010, 15, 309–322. [Google Scholar] [CrossRef]
- Cabello, C.M.; Lamore, S.D.; Bair, W.B.; Davis, A.L.; Azimian, S.M.; Wondrak, G.T. DCPIP (2,6-dichlorophenolindophenol) as a genotype-directed redox chemotherapeutic targeting NQO1*2 breast carcinoma. Free Radic. Res. 2011, 45, 276–292. [Google Scholar] [CrossRef]
- Tran, P.L.; Kim, S.A.; Choi, H.S.; Yoon, J.H.; Ahn, S.G. Epigallocatechin-3-gallate suppresses the expression of HSP70 and HSP90 and exhibits anti-tumor activity in vitro and in vivo. BMC Cancer 2010, 10, 276. [Google Scholar] [CrossRef]
- Samadi, A.K.; Zhang, X.; Mukerji, R.; Donnelly, A.C.; Blagg, B.S.; Cohen, M.S. A novel C-terminal HSP90 inhibitor KU135 induces apoptosis and cell cycle arrest in melanoma cells. Cancer Lett. 2011, 312, 158–167. [Google Scholar] [CrossRef]
- Reaume, A.G. Drug repurposing through nonhypothesis driven phenotypic screening. Drug Discov. Today 2011, 8, 85–88. [Google Scholar] [CrossRef]
- Weir, S.J.; DeGennaro, L.J.; Austin, C.P. Repurposing approved and abandoned drugs for the treatment and prevention of cancer through public-private partnership. Cancer Res. 2012, 72, 1055–1058. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davis, A.L.; Cabello, C.M.; Qiao, S.; Azimian, S.; Wondrak, G.T. Phenotypic Identification of the Redox Dye Methylene Blue as an Antagonist of Heat Shock Response Gene Expression in Metastatic Melanoma Cells. Int. J. Mol. Sci. 2013, 14, 4185-4202. https://doi.org/10.3390/ijms14024185
Davis AL, Cabello CM, Qiao S, Azimian S, Wondrak GT. Phenotypic Identification of the Redox Dye Methylene Blue as an Antagonist of Heat Shock Response Gene Expression in Metastatic Melanoma Cells. International Journal of Molecular Sciences. 2013; 14(2):4185-4202. https://doi.org/10.3390/ijms14024185
Chicago/Turabian StyleDavis, Angela L., Christopher M. Cabello, Shuxi Qiao, Sara Azimian, and Georg T. Wondrak. 2013. "Phenotypic Identification of the Redox Dye Methylene Blue as an Antagonist of Heat Shock Response Gene Expression in Metastatic Melanoma Cells" International Journal of Molecular Sciences 14, no. 2: 4185-4202. https://doi.org/10.3390/ijms14024185