The Transcriptome of Brassica napus L. Roots under Waterlogging at the Seedling Stage

Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
2.1.1. Analysis of DGE Libraries
2.1.2. Changes in Global Gene Transcription under Waterlogging
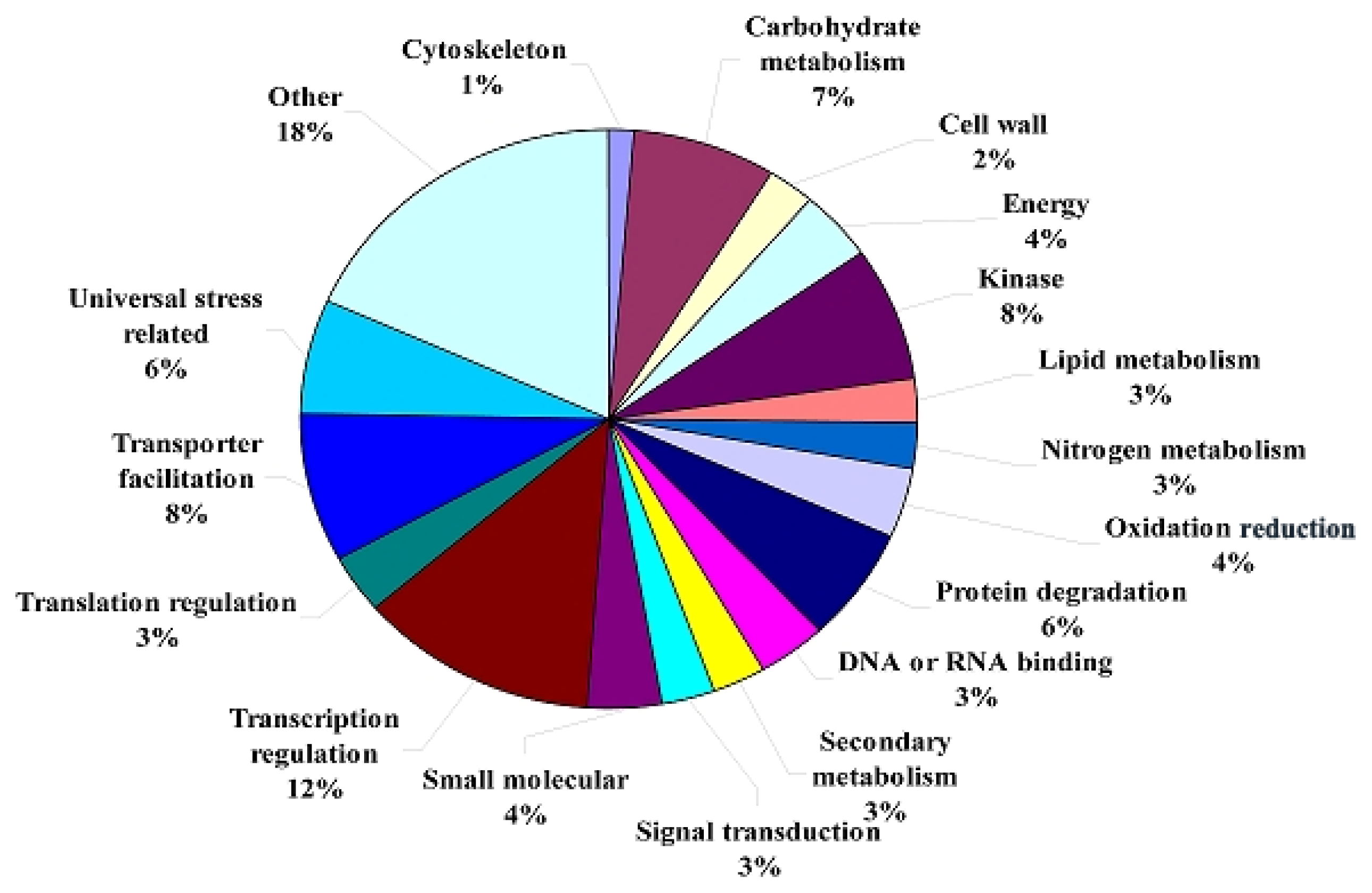
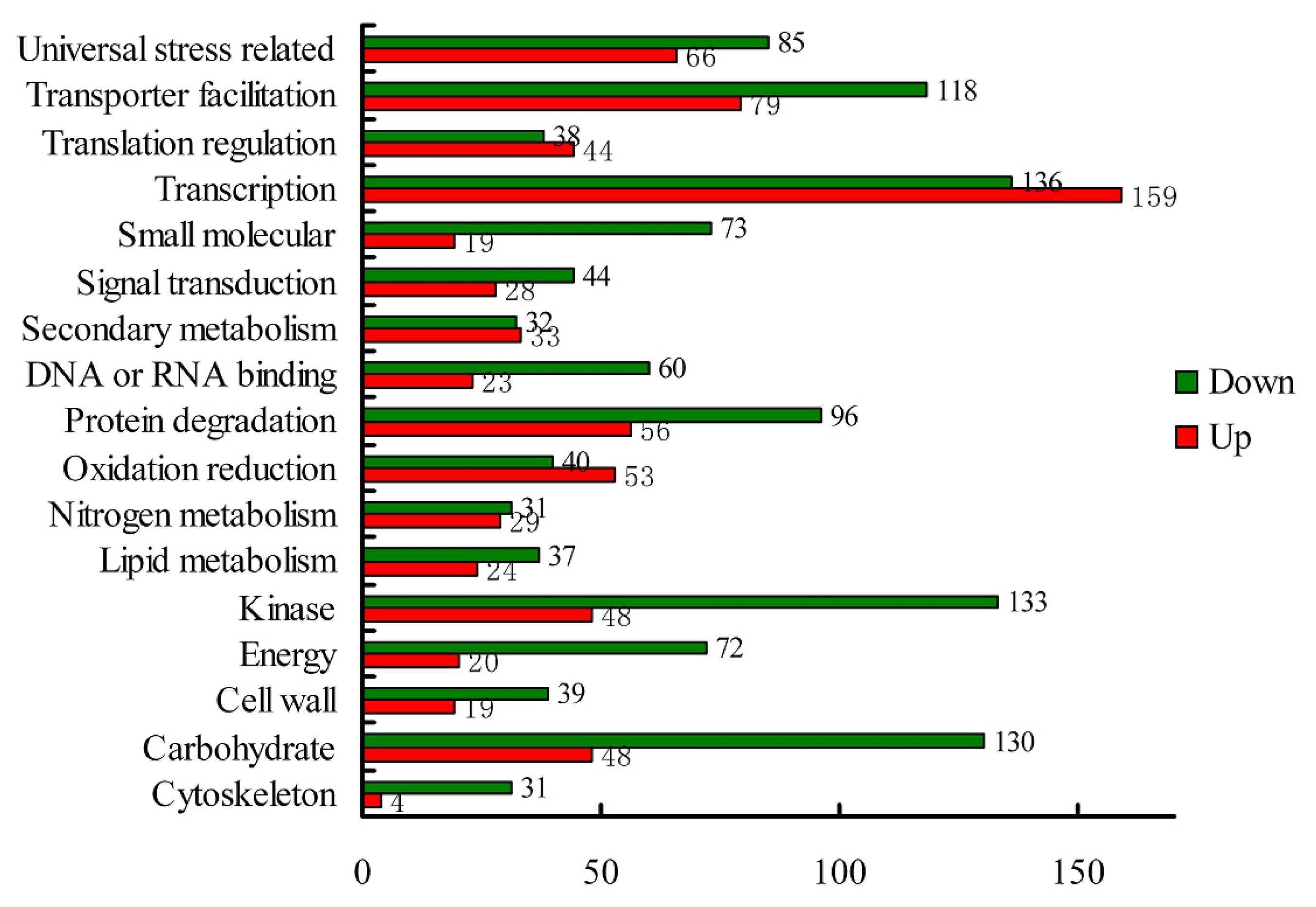
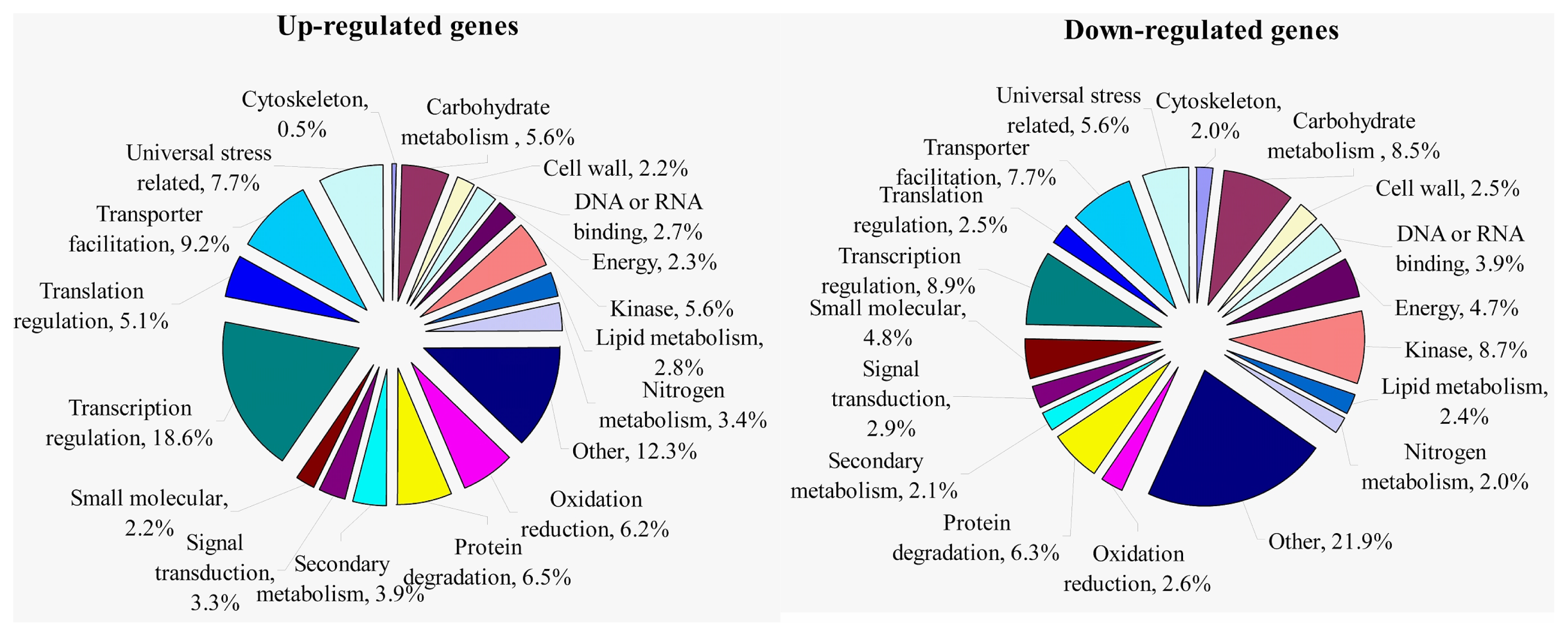
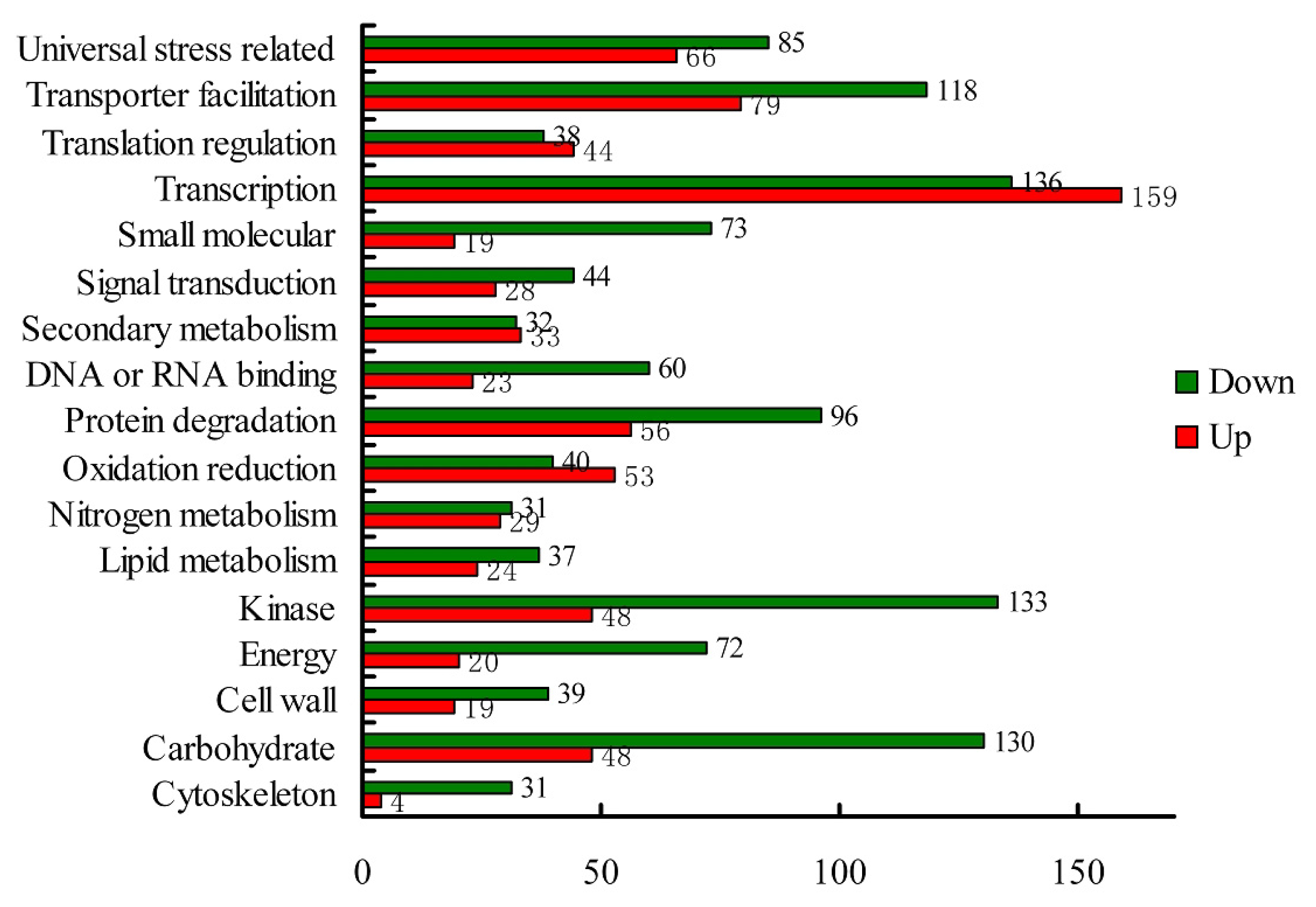
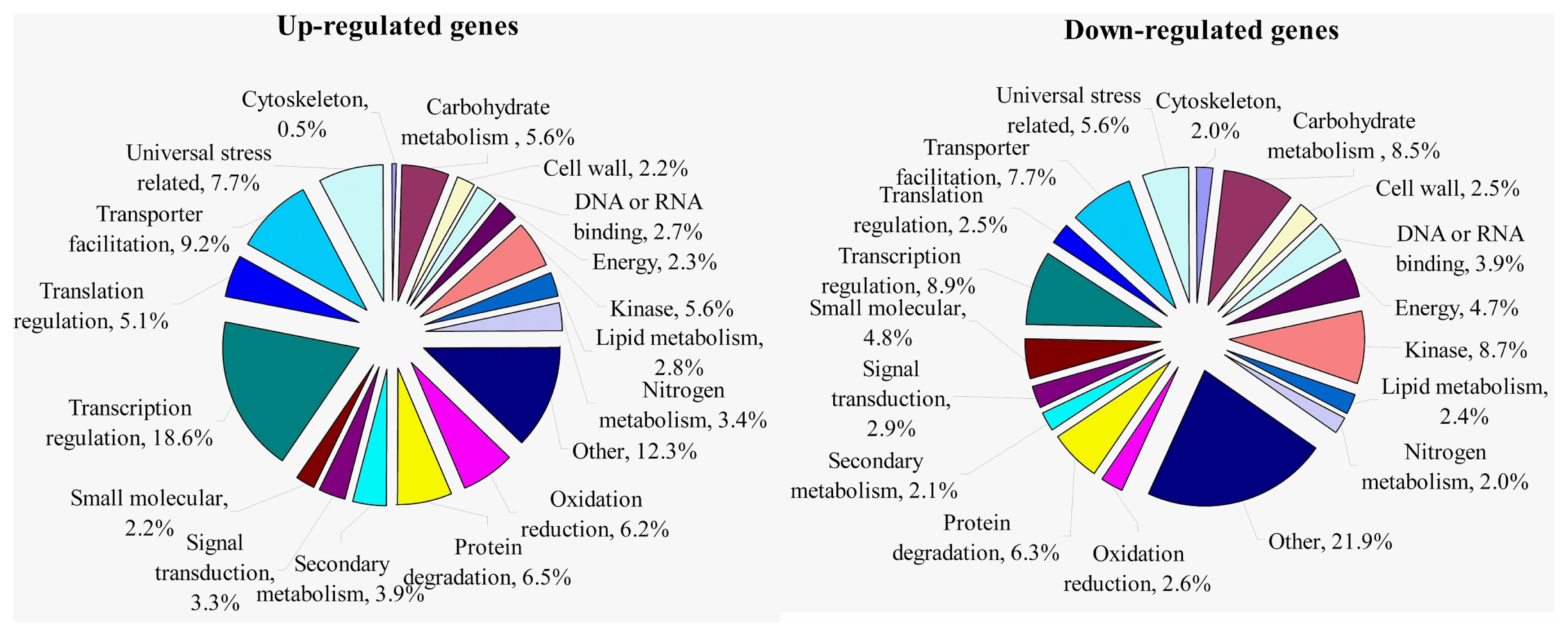
2.1.3. Transcriptomic Comparison of the Roots under Waterlogging Treatment and the Control Using DGE Tag Profiling
2.1.4. Expression Levels of Differentially Expressed Genes under the Control and Waterlogging Stress
2.1.5. Verification of The DGE Data by Real-Time Quantitative RT-PCR
2.2. Discussion
2.2.1. Protein Degradation May Be Involved in Negative Regulation in the Response to Waterlogging
2.2.2. Multiple Levels of Regulation Were Involved in the Response to Waterlogging
3. Experimental Section
3.1. Plant Materials and Waterlogging Treatment
3.2. RNA Isolation
3.3. DGE-Tag Profiling
3.4. Identification of Differentially Expressed Genes
3.5. Quantitative Real-Time PCR Analysis
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Subbaiah, C.C.; Sachs, M.M. Molecular and cellular adaptations of maize to flooding stress. Ann. Bot 2003, 91, 119–127. [Google Scholar]
- Dat, J.F.; Capelli, N.; Folzer, H.; Bourgeade, P.; Badot, P.M. Sensing and signalling during plant flooding. Plant Physiol. Biochem 2004, 42, 273–282. [Google Scholar]
- Jackson, M.B.; Colmer, T.D. Response and adaptation by plants to flooding stress. Ann. Bot 2005, 96, 501–505. [Google Scholar]
- Liu, F.; Vantoai, T.; Moy, L.P.; Bock, G.; Linford, L.D.; Quackenbush, J. Global transcription profiling reveals comprehensive insights into hypoxic response in Arabidopsis. Plant Physiol 2005, 137, 1115–1129. [Google Scholar]
- Gonzali, S.; Loreti, E.; Novi, G.; Poggi, A.; Alpi, A.; Perata, P. The use of microarrays to study the anaerobic response in Arabidopsis. Ann. Bot 2005, 96, 661–668. [Google Scholar]
- Hsu, F.C.; Chou, M.Y.; Peng, H.P.; Chou, S.J.; Shih, M.C. Insights into hypoxic systemic responses based on analyses of transcriptional regulation in Arabidopsis. PLoS One 2011, 6, e28888. [Google Scholar]
- Van Dongen, J.T.; Frohlich, A.; Ramirez-Aguilar, S.J.; Schauer, N.; Fernie, A.R.; Erban, A.; Kopka, J.; Clark, J.; Langer, A.; Geigenberger, P. Transcript and metabolite profiling of the adaptive response to mild decreases in oxygen concentration in the roots of arabidopsis plants. Ann. Bot 2009, 103, 269–280. [Google Scholar]
- Zou, X.; Jiang, Y.; Liu, L.; Zhang, Z.; Zheng, Y. Identification of transcriptome induced in roots of maize seedlings at the late stage of waterlogging. BMC Plant Biol 2010, 10, 189. [Google Scholar]
- Peschke, V.M.; Sachs, M.M. Characterization and expression of transcripts induced by oxygen deprivation in maize (Zea mays L.). Plant Physiol 1994, 104, 387–394. [Google Scholar]
- Agarwal, S.; Grover, A. Isolation and transcription profiling of low-O2 stress-associated cDNA clones from the flooding-stress-tolerant FR13A rice genotype. Ann. Bot 2005, 96, 831–844. [Google Scholar]
- Lasanthi-Kudahettige, R.; Magneschi, L.; Loreti, E.; Gonzali, S.; Licausi, F.; Novi, G.; Beretta, O.; Vitulli, F.; Alpi, A.; Perata, P. Transcript profiling of the anoxic rice coleoptile. Plant Physiol 2007, 144, 218–231. [Google Scholar]
- Christianson, J.A.; Llewellyn, D.J.; Dennis, E.S.; Wilson, I.W. Global gene expression responses to waterlogging in roots and leaves of cotton (Gossypium hirsutum L.). Plant Cell Physiol 2010, 51, 21–37. [Google Scholar]
- Nanjo, Y.; Maruyama, K.; Yasue, H.; Yamaguchi-Shinozaki, K.; Shinozaki, K.; Komatsu, S. Transcriptional responses to flooding stress in roots including hypocotyl of soybean seedlings. Plant Mol. Biol 2011, 77, 129–144. [Google Scholar]
- Qi, X.H.; Xu, X.W.; Lin, X.J.; Zhang, W.J.; Chen, X.H. Identification of differentially expressed genes in cucumber (Cucumis sativus L.) root under waterlogging stress by digital gene expression profile. Genomics 2012, 99, 160–168. [Google Scholar]
- Subbaiah, C.C.; Kollipara, K.P.; Sachs, M.M. A Ca2+-dependent cysteine protease is associated with anoxia-induced root tip death in maize. J. Exp. Bot 2000, 51, 721–730. [Google Scholar]
- Baxter-Burrell, A.; Yang, Z.; Springer, P.S.; Bailey-Serres, J. RopGAP4-dependent Rop GTPase rheostat control of Arabidopsis oxygen deprivation tolerance. Science 2002, 296, 2026–2028. [Google Scholar]
- Bailey-Serres, J.; Chang, R. Sensing and signalling in response to oxygen deprivation in plants and other organisms. Ann. Bot 2005, 96, 507–518. [Google Scholar]
- Manjunath, S.; Williams, A.J.; Bailey-Serres, J. Oxygen deprivation stimulates Ca2+-mediated phosphorylation of mRNA cap-binding protein eIF4E in maize roots. Plant J 1999, 19, 21–30. [Google Scholar]
- Barnawal, D.; Bharti, N.; Maji, D.; Chanotiya, C.S.; Kalra, A. 1-Aminocyclopropane-1-carboxylic acid (ACC) deaminase-containing rhizobacteria protect Ocimum sanctum plants during waterlogging stress via reduced ethylene generation. Plant Physiol. Biochem 2012, 58, 227–235. [Google Scholar]
- Hess, N.; Klode, M.; Anders, M.; Sauter, M. The hypoxia responsive transcription factor genes ERF71/HRE2 and ERF73/HRE1 of Arabidopsis are differentially regulated by ethylene. Physiol. Plant 2011, 143, 41–49. [Google Scholar]
- Yang, S.H.; Choi, D. Characterization of genes encoding ABA 8′-hydroxylase in ethylene-induced stem growth of deepwater rice (Oryza sativa L.). Biochem. Biophys. Res. Commun 2006, 350, 685–690. [Google Scholar]
- De Bruxelles, G.L.; Peacock, W.J.; Dennis, E.S.; Dolferus, R. Abscisic acid induces the alcohol dehydrogenase gene in Arabidopsis. Plant Physiol 1996, 111, 381–391. [Google Scholar]
- Else, M.A.; Davies, W.J.; Malone, M.; Jackson, M.B. A negative hydraulic message from oxygen-deficient roots of tomato plants? (influence of soil flooding on leaf water potential, leaf expansion, and synchrony between stomatal conductance and root hydraulic conductivity). Plant Physiol 1995, 109, 1017–1024. [Google Scholar]
- Neuman, D.S.; Smit, B.A. The influence of leaf water status and aba on leaf growth and stomata of phaseolus seedlings with hypoxic roots. J. Exp. Bot 1991, 42, 1499–1506. [Google Scholar]
- Dennis, E.S.; Dolferus, R.; Ellis, M.; Rahman, M.; Wu, Y.; Hoeren, F.U.; Grover, A.; Ismond, K.P.; Good, A.G.; Peacock, W.J. Molecular strategies for improving waterlogging tolerance in plants. J. Exp. Bot 2000, 51, 89–97. [Google Scholar]
- He, C.J.; Morgan, P.W.; Drew, M.C. Transduction of an ethylene signal is required for cell death and lysis in the root cortex of maize during aerenchyma formation induced by hypoxia. Plant Physiol 1996, 112, 463–472. [Google Scholar]
- Klok, E.J.; Wilson, I.W.; Wilson, D.; Chapman, S.C.; Peacock, W.J.; Dolferus, R.; Dennis, E.S. Expression profile analysis of the low-oxygen response in Arabidopsis root cultures. Plant Cell 2002, 14, 2481–2494. [Google Scholar]
- Visser, E.; Cohen, J.D.; Barendse, G.; Blom, C.; Voesenek, L. An ethylene-mediated increase in sensitivity to auxin induces adventitious root formation in flooded Rumex palustris Sm. J. Exp. Bot 1996, 112, 1687–1692. [Google Scholar]
- Chen, J.; Zhang, X.; Chen, L.; Chao, G.; Li, J. Evaluation of waterlogging tolerance germplasm in rapeseed (Brassica napus L.) with germinated seeds with anoxic stress. Chin. J. Oil Crop Sci 2006, 28, 138–143. [Google Scholar]
- Cheng, Y.; Gu, M.; Cong, Y.; Zou, C.; Zhang, X.; Wang, H. Combining ability and genetic effects of germination traits of Brassica napus L. under waterlogging stress condition. Agric. Sci. China 2010, 9, 101–105. [Google Scholar]
- Hattori, Y.; Nagai, K.; Furukawa, S.; Song, X.J.; Kawano, R.; Sakakibara, H.; Wu, J.; Matsumoto, T.; Yoshimura, A.; Kitano, H. The ethylene response factors SNORKEL1 and SNORKEL2 allow rice to adapt to deep water. Nature 2009, 460, 1026–1030. [Google Scholar]
- Xu, K.; Xu, X.; Fukao, T.; Canlas, P.; Maghirang-Rodriguez, R.; Heuer, S.; Ismail, A.M.; Bailey-Serres, J.; Ronald, P.C.; Mackill, D.J. Sub1A is an ethylene-response-factor-like gene that confers submergence tolerance to rice. Nature 2006, 442, 705–708. [Google Scholar]
- Sasidharan, R.; Mustroph, A. Plant oxygen sensing is mediated by the N-end rule pathway: A milestone in plant anaerobiosis. Plant Cell 2011, 23, 4173–4183. [Google Scholar]
- Linsen, S.E.; Cuppen, E. Methods for small RNA preparation for digital gene expression profiling by next-generation sequencing. Methods Mol. Biol 2012, 822, 205–217. [Google Scholar]
- Hong, L.Z.; Li, J.; Schmidt-Kuntzel, A.; Warren, W.C.; Barsh, G.S. Digital gene expression for non-model organisms. Genome Res 2011, 21, 1905–1915. [Google Scholar]
- Pucciariello, C.; Parlanti, S.; Banti, V.; Novi, G.; Perata, P. Reactive oxygen species-driven transcription in Arabidopsis under oxygen deprivation. Plant Physiol 2012, 159, 184–196. [Google Scholar]
- Banti, V.; Mafessoni, F.; Loreti, E.; Alpi, A.; Perata, P. The heat-inducible transcription factor HsfA2 enhances anoxia tolerance in Arabidopsis. Plant Physiol 2010, 152, 1471–1483. [Google Scholar]
- Chang, R.; Jang, C.J.; Branco-Price, C.; Nghiem, P.; Bailey-Serres, J. Transient MPK6 activation in response to oxygen deprivation and reoxygenation is mediated by mitochondria and aids seedling survival in Arabidopsis. Plant Mol. Biol 2012, 78, 109–122. [Google Scholar]
- Branco-Price, C.; Kawaguchi, R.; Ferreira, R.B.; Bailey-Serres, J. Genome-wide analysis of transcript abundance and translation in Arabidopsis seedlings subjected to oxygen deprivation. Ann. Bot 2005, 96, 647–660. [Google Scholar]
- Ellis, M.H.; Dennis, E.S.; Peacock, W.J. Arabidopsis roots and shoots have different mechanisms for hypoxic stress tolerance. Plant Physiol 1999, 119, 57–64. [Google Scholar]
- Keller, T.; Damude, H.G.; Werner, D.; Doerner, P.; Dixon, R.A.; Lamb, C. A plant homolog of the neutrophil NADPH oxidase gp91phox subunit gene encodes a plasma membrane protein with Ca2+ binding motifs. Plant Cell 1998, 10, 255–266. [Google Scholar]
- Gibbs, D.J.; Lee, S.C.; Isa, N.M.; Gramuglia, S.; Fukao, T.; Bassel, G.W.; Correia, C.S.; Corbineau, F.; Theodoulou, F.L.; Bailey-Serres, J. Homeostatic response to hypoxia is regulated by the N-end rule pathway in plants. Nature 2011, 479, 415–418. [Google Scholar]
- Licausi, F.; Kosmacz, M.; Weits, D.A.; Giuntoli, B.; Giorgi, F.M.; Voesenek, L.A.C.J.; Perata, P.; van Dongen, J.T. Oxygen sensing in plants is mediated by an N-end rule pathway for protein destabilization. Nature 2011, 479, 419–422. [Google Scholar]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, M.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar]
- Hinz, M.; Wilson, I.W.; Yang, J.; Buerstenbinder, K.; Liewellyn, D.; Dennis, E.S.; Sauter, M.; Dolferus, R. Arabidopsis RAP2.2: An ethylene response transcription factor that is important for hypoxia survival. Plant Physiol 2010, 153, 757–772. [Google Scholar]
- Zhai, L.; Liu, Z.; Zou, X.; Jiang, Y.; Qiu, F.; Zheng, Y.; Zhang, Z. Genome-wide identification and analysis of microRNA responding to long-term waterlogging in crown roots of maize seedlings. Physiol. Plant 2012, 147, 181–193. [Google Scholar]
- Zou, X.; Jiang, Y.; Zheng, Y.; Zhang, M.; Zhang, Z. Prolyl 4-hydroxylase genes are subjected to alternative splicing in roots of maize seedlings under waterlogging. Ann. Bot 2011, 108, 1323–1335. [Google Scholar]
- Moldovan, D.; Spriggs, A.; Yang, J.; Pogson, B.J.; Dennis, E.S.; Wilson, I.W. Hypoxia-responsive microRNAs and trans-acting small interfering RNAs in Arabidopsis. J. Exp. Bot 2010, 61, 165–177. [Google Scholar]
- Chang, W.W.; Huang, L.; Shen, M.; Webster, C.; Burlingame, A.L.; Roberts, J.K.M. Patterns of protein synthesis and tolerance of anoxia in root tips of maize seedlings acclimated to a low-oxygen environment, and identification of proteins by mass spectrometry. Plant Physiol 2000, 122, 295–318. [Google Scholar]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinforma 2012, 13, 134. [Google Scholar]
- Libourel, I.G.; van Bodegom, P.M.; Fricker, M.D.; Ratcliffe, R.G. Nitrite reduces cytoplasmic acidosis under anoxia. Plant Physiol 2006, 142, 1710–1717. [Google Scholar]
- Dixon, M.H.; Hill, S.A.; Jackson, M.B.; Ratcliffe, R.G.; Sweetlove, L.J. Physiological and metabolic adaptations of Potamogeton pectinatus L. tubers support rapid elongation of stem tissue in the absence of oxygen. Plant Cell Physiol 2006, 47, 128–140. [Google Scholar]
- Ratcliffe, R.G. In vivo NMR studies of the metabolic response of plant tissues to anoxia. Ann. Bot 1997, 79, 39–48. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| 0 h | 12 h | |||
|---|---|---|---|---|
| Raw Reads | 13,457,553 | 19,621,584 | ||
| Clean Reads | 13,283,443 | 19,364,949 | ||
| Mapped Reads | 8,138,298 | 12,591,493 | ||
| 61.27% (8,138,298/13,283,443) | 65.02% (12,591,493/19,364,949) | |||
| Perfect match | 4,221,453 | 6,684,936 | ||
| 31.78% a | 51.87% b | 34.52% a | 53.09% b | |
| ≤2bp mismatch | 3,916,845 | 5,906,557 | ||
| 29.49% a | 48.13% b | 30.5% a | 46.91% b | |
| Unique match | 7,614,486 | 11,679,339 | ||
| 57.32% a | 93.56% b | 60.31% a | 92.76% b | |
| Multi-position match | 523,812 | 912,154 | ||
| 3.94% a | 6.44% b | 4.71% a | 7.24% b | |
| Unmapped Reads | 5,145,145 | 6,773,456 | ||
| 38.73% a | 34.98% a | |||
| Categories | Control-total | % a | Control-down | % b | Control-up | % c | Waterlogged-total | % d | Waterlogged-up | % e | Waterlogged-down |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Unknown function | 79 | 39.5% | 60 | 41.7% | 19 | 33.9% | 84 | 42.0% | 80 | 41.9% | 4 |
| Universal stress related | 17 | 8.5% | 10 | 6.9% | 7 | 12.5% | 20 | 10.0% | 18 | 9.4% | 2 |
| Carbohydrate metabolism | 13 | 6.5% | 8 | 5.6% | 5 | 8.9% | 12 | 6.0% | 12 | 6.3% | 0 |
| Transporter facilitation | 10 | 5.0% | 7 | 4.9% | 3 | 5.4% | 11 | 5.5% | 10 | 5.2% | 1 |
| Other | 15 | 7.5% | 12 | 8.3% | 3 | 5.4% | 9 | 4.5% | 9 | 4.7% | 0 |
| Translation regulation | 8 | 4.0% | 2 | 1.4% | 6 | 10.7% | 8 | 4.0% | 8 | 4.2% | 0 |
| Protein degradation | 8 | 4.0% | 5 | 3.5% | 3 | 5.4% | 8 | 4.0% | 8 | 4.2% | 0 |
| Oxidation reduction | 6 | 3.0% | 4 | 2.8% | 2 | 3.6% | 7 | 3.5% | 7 | 3.7% | 0 |
| No homology | 4 | 2.0% | 1 | 0.7% | 3 | 5.4% | 7 | 3.5% | 6 | 3.1% | 1 |
| Transcription regulation | 4 | 2.0% | 3 | 2.1% | 1 | 1.8% | 6 | 3.0% | 6 | 3.1% | 0 |
| Nitrogen metabolism | 5 | 2.5% | 4 | 2.8% | 1 | 1.8% | 6 | 3.0% | 6 | 3.1% | 0 |
| Lipid metabolism | 3 | 1.5% | 2 | 1.4% | 1 | 1.8% | 5 | 2.5% | 5 | 2.6% | 0 |
| Kinase | 3 | 1.5% | 3 | 2.1% | 0 | 0.0% | 4 | 2.0% | 4 | 2.1% | 0 |
| Signal transduction | 5 | 2.5% | 5 | 3.5% | 0 | 0.0% | 3 | 1.5% | 2 | 1.0% | 1 |
| Secondary metabolism | 5 | 2.5% | 4 | 2.8% | 1 | 1.8% | 3 | 1.5% | 3 | 1.6% | 0 |
| Energy | 3 | 1.5% | 2 | 1.4% | 1 | 1.8% | 2 | 1.0% | 2 | 1.0% | 0 |
| DNA or RNA binding | 0 | 0.0% | 0 | 0.0% | 0 | 0.0% | 2 | 1.0% | 2 | 1.0% | 0 |
| Cell wall | 5 | 2.5% | 5 | 3.5% | 0 | 0.0% | 2 | 1.0% | 2 | 1.0% | 0 |
| Small molecular | 2 | 1.0% | 2 | 1.4% | 0 | 0.0% | 1 | 0.5% | 1 | 0.5% | 0 |
| Cytoskeleton | 5 | 2.5% | 5 | 3.5% | 0 | 0.0% | 0 | 0.0% | 0 | 0.0% | 0 |
| Total | 200 | - | 144 | - | 56 | - | 200 | - | 191 | - | 9 |
| Percentagef | - | - | 72.0% | 28.0% | - | - | 95.5% | 4.5% |
| Gene ID | Annotation | Fold change by DGE a | Fold change by Q-PCR |
|---|---|---|---|
| Bra038700 | polygalacturonase inhibitory protein | 3.2 | 9.8 ± 1.4 |
| Bra021558 | nine-cis-epoxycarotenoid dioxygenase3 | 3.1 | 4.9 ± 1.1 |
| Bra003701 | AP2 domain containing protein RAP2.5 | 2.5 | 7.4 ± 1.5 |
| Bra014080 | hydrolase | 2.4 | 24.9 ± 1.1 |
| Bra007609 | glycoside hydrolase family 28 protein | 2.3 | 12.2 ± 1.3 |
| Bra016729 | glyceraldehyde-3-phosphate dehydrogenase 1 | 4.1 | 6.4 ± 1.3 |
| Bra022115 | transcription factor | 2.1 | 8.9 ± 1.2 |
| Bra004778 | Stearoyl-acyl carrier protein desaturase | 17.0 | 73.0 ± 1.4 |
| Bra012551 | abscisic acid 8′-hydroxylase/oxygen binding | 0.3 | 0.24 ± 0.01 |
| Bra019528 | betaine aldehyde dehydrogenase | 0.3 | 0.3 ± 0.01 |
| Bra015693 | alcohol dehydrogenase | 16.2 | 12.1 ± 1.2 |
| Bra030945 | phosphoenolpyruvate carboxylase | 0.3 | 0.2 ± 0.03 |
Supplementary Files
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zou, X.; Tan, X.; Hu, C.; Zeng, L.; Lu, G.; Fu, G.; Cheng, Y.; Zhang, X. The Transcriptome of Brassica napus L. Roots under Waterlogging at the Seedling Stage. Int. J. Mol. Sci. 2013, 14, 2637-2651. https://doi.org/10.3390/ijms14022637
Zou X, Tan X, Hu C, Zeng L, Lu G, Fu G, Cheng Y, Zhang X. The Transcriptome of Brassica napus L. Roots under Waterlogging at the Seedling Stage. International Journal of Molecular Sciences. 2013; 14(2):2637-2651. https://doi.org/10.3390/ijms14022637
Chicago/Turabian StyleZou, Xiling, Xiaoyu Tan, Chengwei Hu, Liu Zeng, Guangyuan Lu, Guiping Fu, Yong Cheng, and Xuekun Zhang. 2013. "The Transcriptome of Brassica napus L. Roots under Waterlogging at the Seedling Stage" International Journal of Molecular Sciences 14, no. 2: 2637-2651. https://doi.org/10.3390/ijms14022637