Maintenance of Genomic Stability in Mouse Embryonic Stem Cells: Relevance in Aging and Disease


Abstract
:1. Introduction
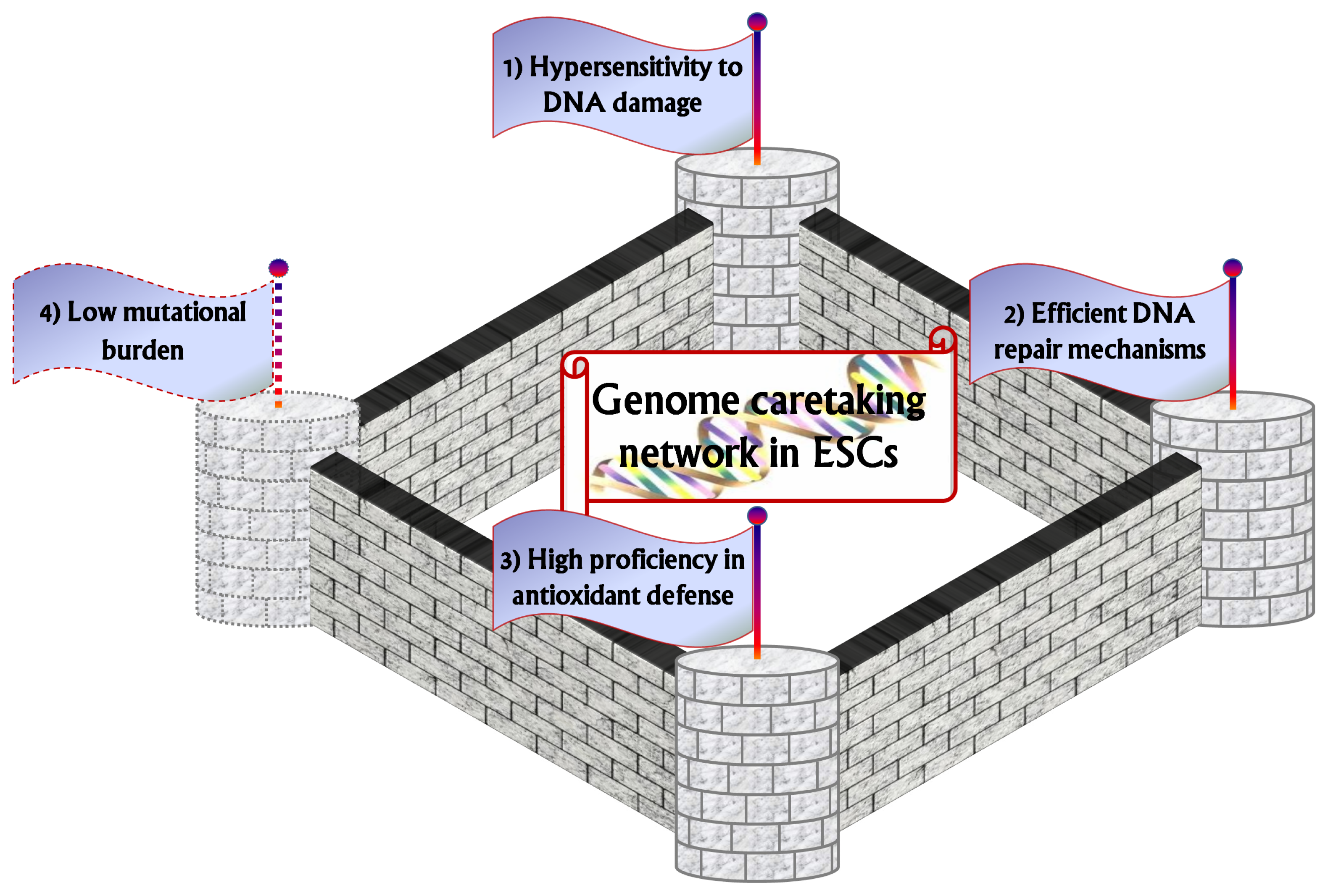
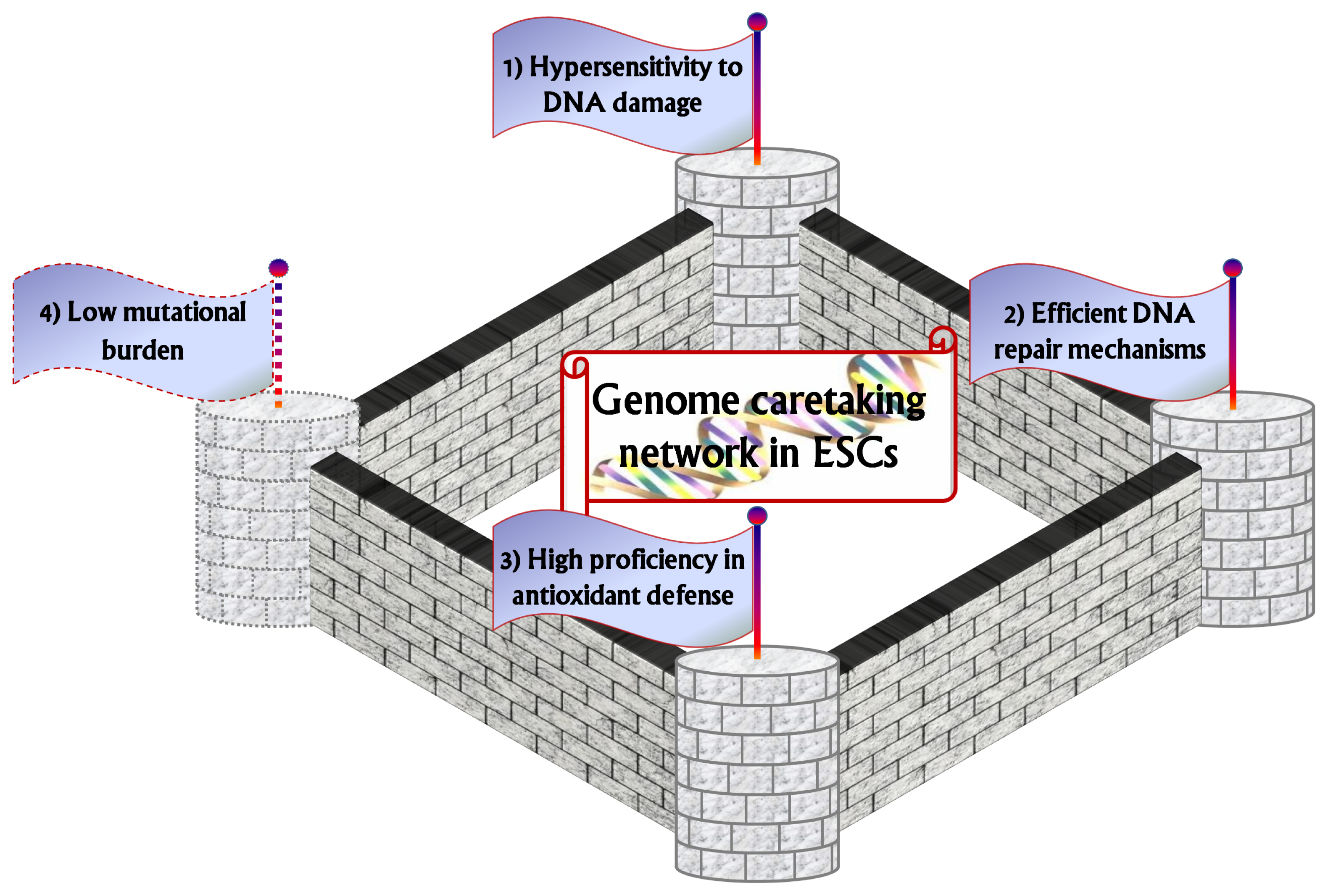
2. Hypersensitivity to DNA Damage: The First Watchtower
3. Efficient DNA Repair Mechanisms as the Second Watchtower
3.1. Base Excision Repair
3.2. Double Strand Break Repair
3.3. Mismatch Repair
3.4. Nucleotide Excision Repair
4. The Third Watchtower: High Proficiency in Antioxidant Defense
5. Low Mutational Burden as a Putative Fourth Watchtower?
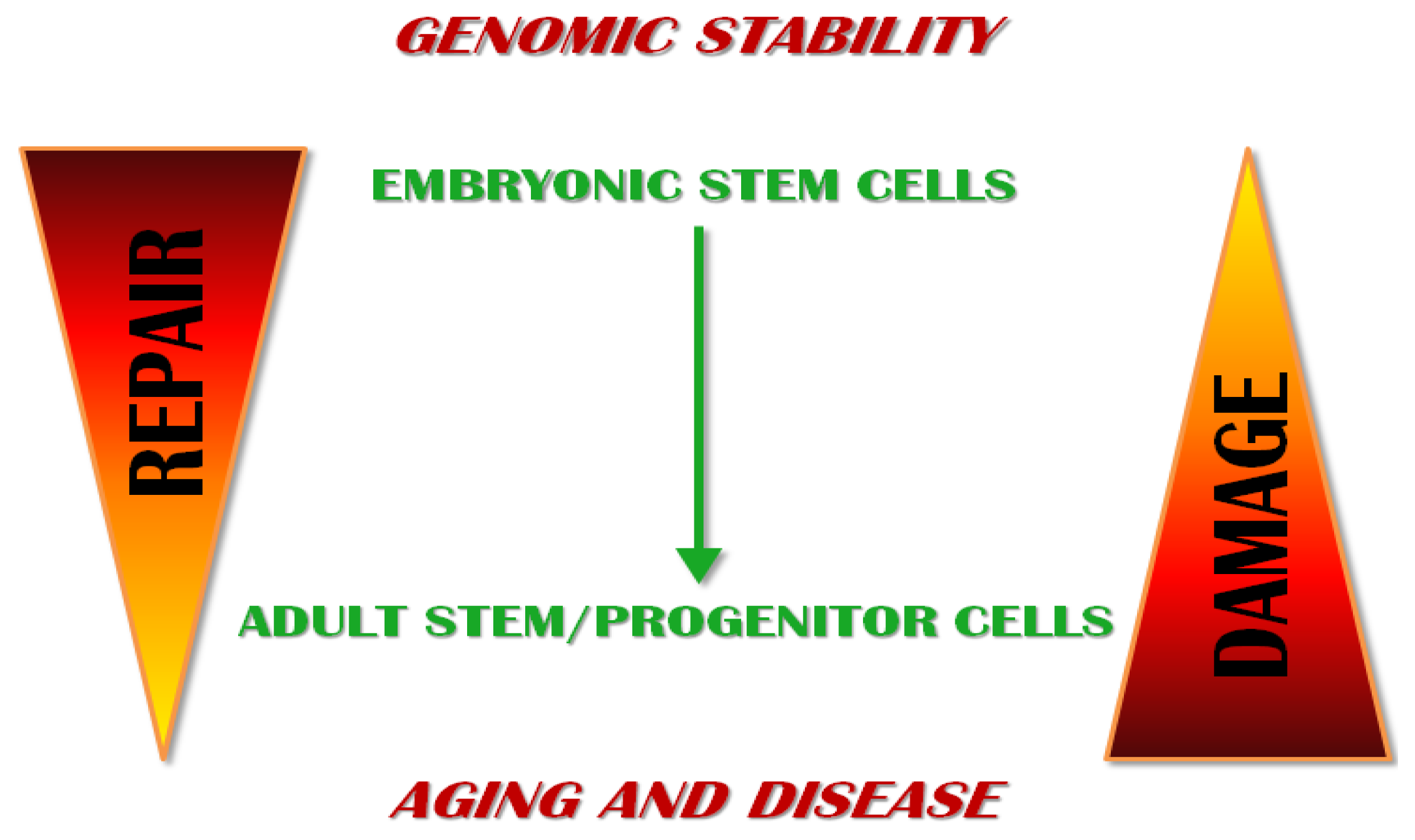
6. Relevance in Aging and Disease
6.1. Aging and ESCs
6.2. Aging and Adult Stem/Progenitor Cells
6.3. Disease and ESCs
6.4. Disease and Adult Stem/Progenitor Cells
7. Conclusions
Acknowledgments
Conflict of Interest
References
- Ben-David, U.; Kopper, O.; Benvenisty, N. Expanding the boundaries of embryonic stem cells. Cell Stem Cell 2012, 10, 666–677. [Google Scholar]
- Sancar, A.; Lindsey-Boltz, L.A.; Ünsal-Kaçmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Ann. Rev. Biochem 2004, 73, 39–85. [Google Scholar]
- Tichy, E.D. Mechanisms maintaining genomic integrity in embryonic stem cells and induced pluripotent stem cells. Exp. Biol. Med 2011, 236, 987–996. [Google Scholar]
- Tichy, E.D.; Stambrook, P.J. DNA repair in murine embryonic stem cells and differentiated cells. Exp. Cell Res 2008, 314, 1929–1936. [Google Scholar]
- Savatier, P.; Huang, S.; Szekely, L.; Wiman, K.G.; Samarut, J. Contrasting patterns of retinoblastoma protein expression in mouse embryonic stem cells and embryonic fibroblasts. Oncogene 1994, 9, 809–818. [Google Scholar]
- Aladjem, M.I.; Spike, B.T.; Rodewald, L.W.; Hope, T.J.; Klemm, M.; Jaenisch, R.; Wahl, G.M. ES cells do not activate p53-dependent stress responses and undergo p53-independent apoptosis in response to DNA damage. Curr. Biol 1998, 8, 145–155. [Google Scholar]
- Van Sloun, P.P.H.; Jansen, J.G.; Weeda, G.; Mullenders, L.H.F.; van Zeeland, A.A.; Lohman, P.H.M.; Vrieling, H. The role of nucleotide excision repair in protecting embryonic stem cells from genotoxic effects of UV-induced DNA damage. Nucleic Acids Res 1999, 27, 3276–3282. [Google Scholar]
- Roos, W.; Christmann, M.; Fraser, S.; Kaina, B. Mouse embryonic stem cells are hypersensitive to apoptosis triggered by the DNA damage O6-methylguanine due to high E2F1 regulated mismatch repair. Cell Death Differ 2007, 14, 1422–1432. [Google Scholar]
- Neely, K.E.; Piwnica-Worms, H. Cdc25A regulation: To destroy or not to destroy, is that the only question? Cell Cycle 2003, 2, 453–455. [Google Scholar]
- Hong, Y.; Cervantes, R.; Tichy, E.; Tischfield, J.; Stambrook, P. Protecting genomic integrity in somatic cells and embryonic stem cells. Mutat. Res. Fundam. Mol. Mech. Mutagenesis 2007, 614, 48–55. [Google Scholar]
- Prost, S.; Bellamy, C.O.C.; Clarke, A.R.; Wyllie, A.H.; Harrison, D.J. p53-independent DNA repair and cell cycle arrest in embryonic stem cells. FEBS Lett 1998, 425, 499–504. [Google Scholar]
- Hong, Y.; Stambrook, P.J. Restoration of an absent G1 arrest and protection from apoptosis in embryonic stem cells after ionizing radiation. Proc. Natl. Acad. Sci. USA 2004, 101, 14443–14448. [Google Scholar]
- Cohen, H.Y.; Lavu, S.; Bitterman, K.J.; Hekking, B.; Imahiyerobo, T.A.; Miller, C.; Frye, R.; Ploegh, H.; Kessler, B.M.; Sinclair, D.A. Acetylation of the C terminus of Ku70 by CBP and PCAF controls Bax-mediated apoptosis. Mol. Cell 2004, 13, 627–638. [Google Scholar]
- Subramanian, C.; Opipari, A.W.; Bian, X.; Castle, V.P.; Kwok, R.P.S. Ku70 acetylation mediates neuroblastoma cell death induced by histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 2005, 102, 4842–4847. [Google Scholar]
- Dumitru, R.; Gama, V.; Fagan, B.M.; Bower, J.J.; Swahari, V.; Pevny, L.H.; Deshmukh, M. Human embryonic stem cells have constitutively active bax at the golgi and are primed to undergo rapid apoptosis. Mol. Cell 2012, 46, 573–583. [Google Scholar]
- Lin, T.; Chao, C.; Saito, S.; Mazur, S.J.; Murphy, M.E.; Appella, E.; Xu, Y. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat. Cell Biol 2004, 7, 165–171. [Google Scholar]
- Qin, H.; Yu, T.; Qing, T.; Liu, Y.; Zhao, Y.; Cai, J.; Li, J.; Song, Z.; Qu, X.; Zhou, P. Regulation of apoptosis and differentiation by p53 in human embryonic stem cells. J. Biol. Chem 2007, 282, 5842–5852. [Google Scholar]
- Tichy, E.D.; Liang, L.; Deng, L.; Tischfield, J.; Schwemberger, S.; Babcock, G.; Stambrook, P.J. Mismatch and base excision repair proficiency in murine embryonic stem cells. DNA Repair 2011, 10, 445–451. [Google Scholar]
- Maynard, S.; Swistowska, A.M.; Lee, J.W.; Liu, Y.; Liu, S.T.; Da Cruz, A.B.; Rao, M.; de Souza-Pinto, N.C.; Zeng, X.; Bohr, V.A. Human embryonic stem cells have enhanced repair of multiple forms of DNA damage. Stem Cells 2008, 26, 2266–2274. [Google Scholar]
- Banuelos, C.; Banath, J.; MacPhail, S.; Zhao, J.; Eaves, C.; O’Connor, M.; Lansdorp, P.; Olive, P. Mouse but not human embryonic stem cells are deficient in rejoining of ionizing radiation-induced DNA double-strand breaks. DNA Repair 2008, 7, 1471–1483. [Google Scholar]
- Tichy, E.D.; Pillai, R.; Deng, L.; Liang, L.; Tischfield, J.; Schwemberger, S.J.; Babcock, G.F.; Stambrook, P.J. Mouse embryonic stem cells, but not somatic cells, predominantly use homologous recombination to repair double-strand DNA breaks. Stem Cells Dev 2010, 19, 1699–1711. [Google Scholar]
- Adams, B.R.; Golding, S.E.; Rao, R.R.; Valerie, K. Dynamic dependence on ATR and ATM for double-strand break repair in human embryonic stem cells and neural descendants. PLoS One 2010, 5, e10001. [Google Scholar]
- De Waard, H.; Sonneveld, E.; de Wit, J.; Esveldt-van Lange, R.; Hoeijmakers, J.; Vrieling, H.; van Der Horst, G. Cell-type-specific consequences of nucleotide excision repair deficiencies: Embryonic stem cells versus fibroblasts. DNA Repair 2008, 7, 1659–1669. [Google Scholar]
- Saretzki, G.; Armstrong, L.; Leake, A.; Lako, M.; von Zglinicki, T. Stress defense in murine embryonic stem cells is superior to that of various differentiated murine cells. Stem Cells 2004, 22, 962–971. [Google Scholar]
- Ito, K.; Hirao, A.; Arai, F.; Takubo, K.; Matsuoka, S.; Miyamoto, K.; Ohmura, M.; Naka, K.; Hosokawa, K.; Ikeda, Y. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat. Med 2006, 12, 446–451. [Google Scholar]
- Kim, W.Y.; Sharpless, N.E. The regulation of INK4/ARF in cancer and aging. Cell 2006, 127, 265–275. [Google Scholar]
- Molofsky, A.V.; Slutsky, S.G.; Joseph, N.M.; He, S.; Pardal, R.; Krishnamurthy, J.; Sharpless, N.E.; Morrison, S.J. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature 2006, 443, 448–452. [Google Scholar]
- Yang, D.G.; Liu, L.; Zheng, X.Y. Cyclin-dependent kinase inhibitor p16(INK4a) and telomerase may co-modulate endothelial progenitor cells senescence. Ageing Res. Rev 2008, 7, 137–146. [Google Scholar]
- Krishnamurthy, J.; Torrice, C.; Ramsey, M.R.; Kovalev, G.I.; Al-Regaiey, K.; Su, L.; Sharpless, N.E. Ink4a/Arf expression is a biomarker of aging. J. Clin. Invest 2004, 114, 1299–1307. [Google Scholar]
- Zindy, F.; Quelle, D.E.; Roussel, M.F.; Sherr, C.J. Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene 1997, 15, 203–211. [Google Scholar]
- Rossi, D.J.; Jamieson, C.H.M.; Weissman, I.L. Stems cells and the pathways to aging and cancer. Cell 2008, 132, 681–696. [Google Scholar]
- Loh, Y.H.; Wu, Q.; Chew, J.L.; Vega, V.B.; Zhang, W.; Chen, X.; Bourque, G.; George, J.; Leong, B.; Liu, J. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat. Genet 2006, 38, 431–440. [Google Scholar]
- Mitsui, K.; Tokuzawa, Y.; Itoh, H.; Segawa, K.; Murakami, M.; Takahashi, K.; Maruyama, M.; Maeda, M.; Yamanaka, S. The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell 2003, 113, 631–642. [Google Scholar]
- Chambers, I.; Colby, D.; Robertson, M.; Nichols, J.; Lee, S.; Tweedie, S.; Smith, A. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell 2003, 113, 643–655. [Google Scholar]
- Nichols, J.; Zevnik, B.; Anastassiadis, K.; Niwa, H.; Klewe-Nebenius, D.; Chambers, I.; Schöler, H.; Smith, A. Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell 1998, 95, 379–391. [Google Scholar]
- Wilson, D.M.; Bohr, V.A. The mechanics of base excision repair and its relationship to aging and disease. DNA Repair 2007, 6, 544–559. [Google Scholar]
- Starcevic, D.; Dalal, S.; Sweasy, J.B. Is there a link between DNA polymerase beta and cancer? Cell Cycle 2004, 3, 996–999. [Google Scholar]
- Al-Tassan, N.; Chmiel, N.H.; Maynard, J.; Fleming, N.; Livingston, A.L.; Williams, G.T.; Hodges, A.K.; Davies, D.R.; David, S.S.; Sampson, J.R. Inherited variants of MYH associated with somatic G: C→ T: A mutations in colorectal tumors. Nat. Genet 2002, 30, 227–232. [Google Scholar]
- Fortini, P.; Pascucci, B.; Parlanti, E.; Sobol, R.W.; Wilson, S.H.; Dogliotti, E. Different DNA polymerases are involved in the short-and long-patch base excision repair in mammalian cells. Biochemistry 1998, 37, 3575–3580. [Google Scholar]
- Khanna, K.K.; Jackson, S.P. DNA double-strand breaks: Signaling, repair and the cancer connection. Nat. Genet 2001, 27, 247–254. [Google Scholar]
- Rolig, R.L.; McKinnon, P.J. Linking DNA damage and neurodegeneration. Trends Neurosci 2000, 23, 417–424. [Google Scholar]
- Johnson, R.; Jasin, M. Double-strand-break-induced homologous recombination in mammalian cells. Biochem. So. Trans 2001, 29, 196–201. [Google Scholar]
- Tsukamoto, Y.; Ikeda, H. Double-strand break repair mediated by DNA end-joining. Genes Cells 2003, 3, 135–144. [Google Scholar]
- Rassool, F.V.; Tomkinson, A.E. Targeting abnormal DNA double strand break repair in cancer. Cell. Mol. Life Sci 2010, 67, 3699–3710. [Google Scholar]
- Savatier, P.; Lapillonne, H.; Van Grunsven, L.; Rudkin, B.; Samarut, J. Withdrawal of differentiation inhibitory activity/leukemia inhibitory factor up-regulates D-type cyclins and cyclin-dependent kinase inhibitors in mouse embryonic stem cells. Oncogene 1996, 12, 309–322. [Google Scholar]
- Nagaria, P.; Robert, C.; Rassool, F. DNA double-strand break response in stem cells: Mechanisms to maintain genomic integrity. Biochim. Biophys. Acta (BBA) 2012, 1830, 2345–2353. [Google Scholar]
- Serrano, L.; Liang, L.; Chang, Y.; Deng, L.; Maulion, C.; Nguyen, S.; Tischfield, J.A. Homologous recombination conserves DNA sequence integrity throughout the cell cycle in embryonic stem cells. Stem Cells Dev 2011, 20, 363–374. [Google Scholar]
- Fung, H.; Weinstock, D.M. Repair at single targeted DNA double-strand breaks in pluripotent and differentiated human cells. PLoS One 2011, 6, e20514. [Google Scholar]
- Banth, J.P.; Bauelos, C.A.; Klokov, D.; MacPhail, S.M.; Lansdorp, P.M.; Olive, P.L. Explanation for excessive DNA single-strand breaks and endogenous repair foci in pluripotent mouse embryonic stem cells. Exp. Cell Res 2009, 315, 1505–1520. [Google Scholar]
- Turinetto, V.; Orlando, L.; Sanchez Ripoll, Y.; Kumpfmueller, B.; Storm, M.; Porcedda, P.; Minieri, V.; Saviozzi, S.; Accomasso, L.; Cibrario Rocchietti, E.; et al. High basal γH2AX levels sustain self-renewal of mouse embryonic and induced pluripotent stem cells. Stem Cells 2012, 30, 1414–1423. [Google Scholar]
- Andng, M.; Hjerling Leffler, J.; Moliner, A.; Lundgren, T.K.; Castelo-Branco, G.; Nanou, E.; Pozas, E.; Bryja, V.; Halliez, S.; Nishimaru, H.; et al. Histone H2AX-dependent GABA(A) receptor regulation of stem cell proliferation. Nature 2008, 451, 460–464. [Google Scholar]
- Kunkel, T.A.; Erie, D.A. Dna mismatch repair. Ann. Rev. Biochem 2005, 74, 681–710. [Google Scholar]
- Modrich, P. Mechanisms in eukaryotic mismatch repair. J. Biol. Chem 2006, 281, 30305–30309. [Google Scholar]
- Fishel, R.; Lescoe, M.K.; Rao, M.; Copeland, N.G.; Jenkins, N.A.; Garber, J.; Kane, M.; Kolodner, R. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 1993, 75, 1027–1038. [Google Scholar]
- Leach, F.S.; Nicolaides, N.C.; Papadopoulos, N.; Liu, B.; Jen, J.; Parsons, R.; Peltomäki, P.; Sistonen, P.; Aaltonen, L.A.; Nyström-Lahti, M. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell 1993, 75, 1215–1225. [Google Scholar]
- Borgdorff, V.; Pauw, B.; van Hees-Stuivenberg, S.; de Wind, N. DNA mismatch repair mediates protection from mutagenesis induced by short-wave ultraviolet light. DNA Repair 2006, 5, 1364–1372. [Google Scholar]
- Zhou, B.; Huang, C.; Yang, J.; Lu, J.; Dong, Q.; Sun, L.-Z. Preparation of heteroduplex enhanced green fluorescent protein plasmid for in vivo mismatch repair activity assay. Anal. Biochem 2009, 388, 167–169. [Google Scholar]
- Andressoo, J.O.; Hoeijmakers, J.H.J.; Mitchell, J.R. Nucleotide excision repair disorders and the balance between cancer and aging. Cell Cycle 2006, 5, 2886–2888. [Google Scholar]
- Costa, R.; Chiganças, V.; da Silva Galhardo, R.; Carvalho, H.; Menck, C.F.M. The eukaryotic nucleotide excision repair pathway. Biochimie 2003, 85, 1083–1099. [Google Scholar]
- Pedersen, R.; Cleaver, J. Repair of UV damage to DNA of implantation-stage mouse embryos in vitro. Exp. Cell Res 1975, 95, 247–253. [Google Scholar]
- Cho, Y.M.; Kwon, S.; Pak, Y.K.; Seol, H.W.; Choi, Y.M.; Park, D.J.; Park, K.S.; Lee, H.K. Dynamic changes in mitochondrial biogenesis and antioxidant enzymes during the spontaneous differentiation of human embryonic stem cells. Biochem. Biophys. Res. Commun 2006, 348, 1472–1478. [Google Scholar]
- Schmelter, M.; Ateghang, B.; Helmig, S.; Wartenberg, M.; Sauer, H. Embryonic stem cells utilize reactive oxygen species as transducers of mechanical strain-induced cardiovascular differentiation. FASEB J 2006, 20, 1182–1184. [Google Scholar]
- Li, T.S.; Marbán, E. Physiological levels of reactive oxygen species are required to maintain genomic stability in stem cells. Stem Cells 2010, 28, 1178–1185. [Google Scholar]
- Cervantes, R.B.; Stringer, J.R.; Shao, C.; Tischfield, J.A.; Stambrook, P.J. Embryonic stem cells and somatic cells differ in mutation frequency and type. Proc. Natl. Acad. Sci. USA 2002, 99, 3586–3590. [Google Scholar]
- Fischer, J.M.; Stringer, J.R. Visualizing loss of heterozygosity in living mouse cells and tissues. Mutat. Res. Fundam. Mol. Mech. Mutagenesis 2008, 645, 1–8. [Google Scholar]
- Larson, J.S.; Yin, M.; Fischer, J.M.; Stringer, S.L.; Stringer, J.R. Expression and loss of alleles in cultured mouse embryonic fibroblasts and stem cells carrying allelic fluorescent protein genes. BMC Mol. Biol 2006, 7, 36. [Google Scholar]
- Donahue, S.L.; Lin, Q.; Cao, S.; Ruley, H.E. Carcinogens induce genome-wide loss of heterozygosity in normal stem cells without persistent chromosomal instability. Proc. Natl. Acad. Sci. USA 2006, 103, 11642–11646. [Google Scholar]
- Mimeault, M.; Batra, S.K. Recent insights into the molecular mechanisms involved in aging and the malignant transformation of adult stem/progenitor cells and their therapeutic implications. Ageing Res. Rev 2009, 8, 94. [Google Scholar]
- Smith, J.R.; Pereira-Smith, O.M. Replicative senescence: Implications for in vivo aging and tumor suppression. Science 1996, 273, 63–67. [Google Scholar]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar]
- Hathcock, K.S.; Jeffrey Chiang, Y.; Hodes, R.J. In vivo regulation of telomerase activity and telomere length. Immunol. Rev 2005, 205, 104–113. [Google Scholar]
- Miura, T.; Mattson, M.P.; Rao, M.S. Cellular lifespan and senescence signaling in embryonic stem cells. Aging Cell 2004, 3, 333–343. [Google Scholar]
- Szibor, M.; Holtz, J. Mitochondrial ageing. Basic. Res. Cardiol 2003, 98, 210–218. [Google Scholar]
- Bertram, M.J.; Berube, N.G.; Hang-Swanson, X.; Ran, Q.; Leung, J.K.; Bryce, S.; Spurgers, K.; Bick, R.J.; Baldini, A.; Ning, Y.; et al. Identification of a gene that reverses the immortal phenotype of a subset of cells and is a member of a novel family of transcription factor-like genes. Mol. Cell. Biol. 1999, 19, 1479–1485. [Google Scholar]
- Krtolica, A. Stem cell: Balancing aging and cancer. Int. J. Biochem. Cell Biol 2005, 37, 935–941. [Google Scholar]
- Chambers, S.; Goodell, M. Hematopoietic stem cell aging: Wrinkles in stem cell potential. Stem Cell Rev. Rep 2007, 3, 201–211. [Google Scholar]
- Fraga, M.F.; Esteller, M. Epigenetics and aging: The targets and the marks. Trends Genet 2007, 23, 413–418. [Google Scholar]
- Gopinath, S.D.; Rando, T.A. Stem cell review series: Aging of the skeletal muscle stem cell niche. Aging Cell 2008, 7, 590–598. [Google Scholar]
- Sharpless, N.E.; DePinho, R.A. How stem cells age and why this makes us grow old. Nat. Rev. Mol. Cell Biol 2007, 8, 703–713. [Google Scholar]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar]
- Maslov, A.Y.; Barone, T.A.; Plunkett, R.J.; Pruitt, S.C. Neural stem cell detection, characterization and age-related changes in the subventricular zone of mice. J. Neurosci 2004, 24, 1726–1733. [Google Scholar]
- Janzen, V.; Forkert, R.; Fleming, H.E.; Saito, Y.; Waring, M.T.; Dombkowski, D.M.; Cheng, T.; DePinho, R.A.; Sharpless, N.E.; Scadden, D.T. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature 2006, 443, 421–426. [Google Scholar]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A., Jr; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumors. Nature 1992, 356, 215. [Google Scholar]
- Tyner, S.D.; Venkatachalam, S.; Choi, J.; Jones, S.; Ghebranious, N.; Igelmann, H.; Lu, X.; Soron, G.; Cooper, B.; Brayton, C. p53 mutant mice that display early ageing-associated phenotypes. Nature 2002, 415, 45–53. [Google Scholar]
- Matheu, A.; Maraver, A.; Klatt, P.; Flores, I.; Garcia-Cao, I.; Borras, C.; Flores, J.M.; Viña, J.; Blasco, M.A.; Serrano, M. Delayed ageing through damage protection by the Arf/p53 pathway. Nature 2007, 448, 375–379. [Google Scholar]
- Mendrysa, S.M.; O’Leary, K.A.; McElwee, M.K.; Michalowski, J.; Eisenman, R.N.; Powell, D.A.; Perry, M.E. Tumor suppression and normal aging in mice with constitutively high p53 activity. Genes Dev 2006, 20, 16–21. [Google Scholar]
- Dumble, M.; Moore, L.; Chambers, S.M.; Geiger, H.; van Zant, G.; Goodell, M.A.; Donehower, L.A. The impact of altered p53 dosage on hematopoietic stem cell dynamics during aging. Blood 2007, 109, 1736–1742. [Google Scholar]
- Lowe, S.W.; Cepero, E.; Evan, G. Intrinsic tumor suppression. Nature 2004, 432, 307–315. [Google Scholar]
- Ungewitter, E.; Scrable, H. Antagonistic pleiotropy and p53. Mech. Ageing Dev 2009, 130, 10–17. [Google Scholar]
- Collado, M.; Gil, J.; Efeyan, A.; Guerra, C.; Schuhmacher, A.J.; Barradas, M.; Benguría, A.; Zaballos, A.; Flores, J.M.; Barbacid, M. Tumour biology: Senescence in premalignant tumors. Nature 2005, 436, 642. [Google Scholar]
- Dansen, T.B.; Burgering, B.M.T. Unravelling the tumor-suppressive functions of FOXO proteins. Trends Cell Biol 2008, 18, 421–429. [Google Scholar]
- Shay, J.; Wright, W. Hallmarks of telomeres in ageing research. J. Pathol 2007, 211, 114–123. [Google Scholar]
- Hendricks, C.A.; Engelward, B.P. “Recombomice”: The past, present and future of recombination-detection in mice”. DNA Repair (Amst) 2004, 3, 1255–1261. [Google Scholar]
- Bielas, J.H.; Venkatesan, R.N.; Loeb, L.A. LOH-proficient embryonic stem cells: A model of cancer progenitor cells? Trends Genet 2007, 23, 154–157. [Google Scholar]
- Lin, Q.; Donahue, S.L.; Ruley, H.E. Genome maintenance and mutagenesis in embryonic stem cells. Cell Cycle 2006, 5, 2710–2714. [Google Scholar]
- Wicha, M.S.; Liu, S.; Dontu, G. Cancer stem cells: An old idea—A paradigm shift. Cancer Res 2006, 66, 1883–1890. [Google Scholar]
- Cairns, J. Somatic stem cells and the kinetics of mutagenesis and carcinogenesis. Proc. Natl. Acad. Sci. USA 2002, 99, 10567–10570. [Google Scholar]
- Armakolas, A.; Klar, A.J. Cell type regulates selective segregation of mouse chromosome 7 DNA strands in mitosis. Science 2006, 311, 1146–1149. [Google Scholar]
- Damelin, M.; Sun, Y.E.; Sodja, V.B.; Bestor, T.H. Decatenation checkpoint deficiency in stem and progenitor cells. Cancer Cell 2005, 8, 479–484. [Google Scholar]
- Valent, P.; Bonnet, D.; de Maria, R.; Lapidot, T.; Copland, M.; Melo, J.V.; Chomienne, C.; Ishikawa, F.; Schuringa, J.J.; Stassi, G. Cancer stem cell definitions and terminology: The devil is in the details. Nat. Rev. Cancer 2012, 12, 767–775. [Google Scholar]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells: Current status and evolving complexities. Cell Stem Cell 2012, 10, 717–728. [Google Scholar]
- Hope, K.J.; Jin, L.; Dick, J.E. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat. Immunol 2004, 5, 738–743. [Google Scholar]
- Jamieson, C.H.M.; Ailles, L.E.; Dylla, S.J.; Muijtjens, M.; Jones, C.; Zehnder, J.L.; Gotlib, J.; Li, K.; Manz, M.G.; Keating, A. Granulocyte–macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. New Engl. J. Med 2004, 351, 657–667. [Google Scholar]
- Kim, C.F.B.; Jackson, E.L.; Woolfenden, A.E.; Lawrence, S.; Babar, I.; Vogel, S.; Crowley, D.; Bronson, R.T.; Jacks, T. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 2005, 121, 823–835. [Google Scholar]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumor initiating cells. Nature 2004, 432, 396–401. [Google Scholar]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.; Liu, S. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; de Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2006, 445, 111–115. [Google Scholar]
- Schatton, T.; Murphy, G.F.; Frank, N.Y.; Yamaura, K.; Waaga-Gasser, A.M.; Gasser, M.; Zhan, Q.; Jordan, S.; Duncan, L.M.; Weishaupt, C. Identification of cells initiating human melanomas. Nature 2008, 451, 345–349. [Google Scholar]
- Yang, Z.; Ho, D.; Ng, M.; Lau, C.; Yu, W.; Ngai, P.; Chu, P.W.K.; Lam, C.; Poon, R.T.P.; Fan, S. Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell 2008, 13, 153–166. [Google Scholar]
- Miura, M.; Miura, Y.; Padilla-Nash, H.M.; Molinolo, A.A.; Fu, B.; Patel, V.; Seo, B.M.; Sonoyama, W.; Zheng, J.J.; Baker, C.C. Accumulated chromosomal instability in murine bone marrow mesenchymal stem cells leads to malignant transformation. Stem Cells 2005, 24, 1095–1103. [Google Scholar]
- Shiras, A.; Chettiar, S.T.; Shepal, V.; Rajendran, G.; Prasad, G.R.; Shastry, P. Spontaneous transformation of human adult nontumorigenic stem cells to cancer stem cells is driven by genomic instability in a human model of glioblastoma. Stem Cells 2007, 25, 1478–1489. [Google Scholar]
- Brabletz, T.; Jung, A.; Spaderna, S.; Hlubek, F.; Kirchner, T. Migrating cancer stem cells—An integrated concept of malignant tumor progression. Nat. Rev. Cancer 2005, 5, 744–749. [Google Scholar]
- Mimeault, M.; Hauke, R.; Batra, S.K. Recent advances on the molecular mechanisms involved in the drug resistance of cancer cells and novel targeting therapies. Clin. Pharmacol. Ther 2008, 83, 673–691. [Google Scholar]
- Rich, J. Cancer stem cells in radiation resistance. Cancer Res 2007, 67, 8980–8984. [Google Scholar]


{kind=link}
{kind=link}
| ESCs hypersensitivity to DNA damage | |||
|---|---|---|---|
| Mechanism | Pathway involved | Species | References |
| G1 arrest impairment | Chk2 centrosome sequestration; p53 cytoplasm sequestration. | Mouse | [4,10] |
| Rapid apoptotic response following DNA damage | Constitutively activated form of BAX | Mouse, Human | [13–15] |
| Differentiation | p53 mediated repression of Nanog and Oct4 promoter | Mouse, Human | [16,17] |
| ESCs DNA repair mechanisms | |||
| Mechanism | Pathway involved | Species | References |
| High efficiency in Base Excision Repair | BER pathway proteins over expressed in ESCs | Mouse, Human | [18,19] |
| Preferential repair of DSBs through HR rather than NHEJ | High level of HR pathway proteins, such as RAD51, RAD52, RAD 54 | Mouse | [20,21] |
| ATR dependent HR | Human | [22] | |
| High efficiency in Mismatch Repair | High basal level of Msh2 and Msh6 | Mouse | [18] |
| Fine regulation of Nucleotide Excision Repair | Shutting down of NER activity when high amount of DNA damage occurs | Mouse | [7,23] |
| ESCs high proficiency in antioxidant defense | |||
| Mechanism | Pathway involved | Species | References |
| Ability to proliferate in hyperoxic conditions | High level of ROS inactivating enzymes | Mouse | [24] |
| Adult/progenitor stem cells aging and disease | |||
| Mechanism | Pathway involved | Species | References |
| Age associated pathophysiological changes | Upregulation of tumor suppressor gene products like p16INK4A and p19ARF Misregulation of p53 levels | Mouse, Human Mouse, Human | [25–30] [31] |
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Giachino, C.; Orlando, L.; Turinetto, V. Maintenance of Genomic Stability in Mouse Embryonic Stem Cells: Relevance in Aging and Disease. Int. J. Mol. Sci. 2013, 14, 2617-2636. https://doi.org/10.3390/ijms14022617
Giachino C, Orlando L, Turinetto V. Maintenance of Genomic Stability in Mouse Embryonic Stem Cells: Relevance in Aging and Disease. International Journal of Molecular Sciences. 2013; 14(2):2617-2636. https://doi.org/10.3390/ijms14022617
Chicago/Turabian StyleGiachino, Claudia, Luca Orlando, and Valentina Turinetto. 2013. "Maintenance of Genomic Stability in Mouse Embryonic Stem Cells: Relevance in Aging and Disease" International Journal of Molecular Sciences 14, no. 2: 2617-2636. https://doi.org/10.3390/ijms14022617