Selection of Reliable Reference Genes for Gene Expression Studies in the Biofuel Plant Jatropha curcas Using Real-Time Quantitative PCR


Abstract
:1. Introduction
2. Results and Discussion
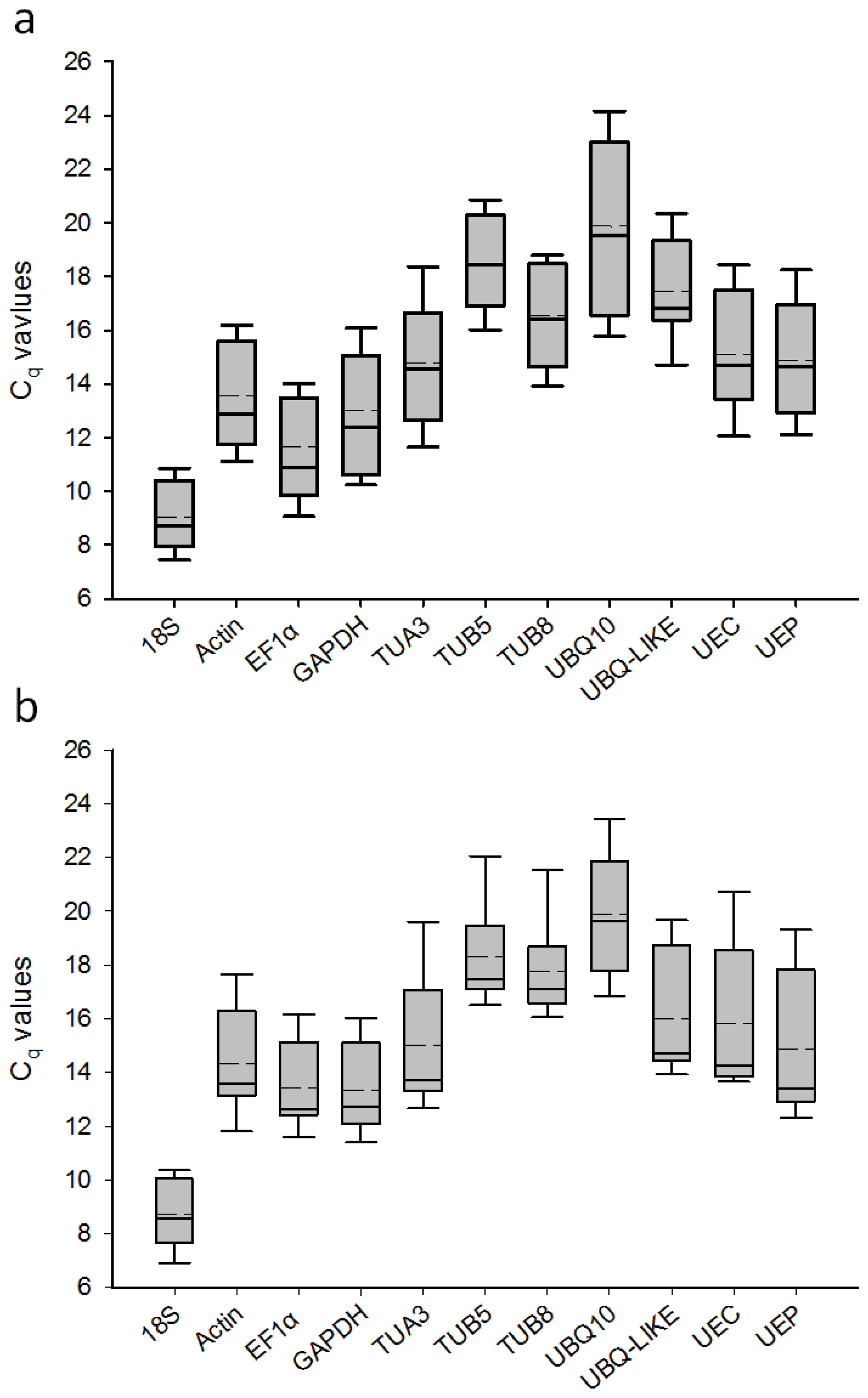
2.1. PCR Amplification Specificity and PCR Efficiency
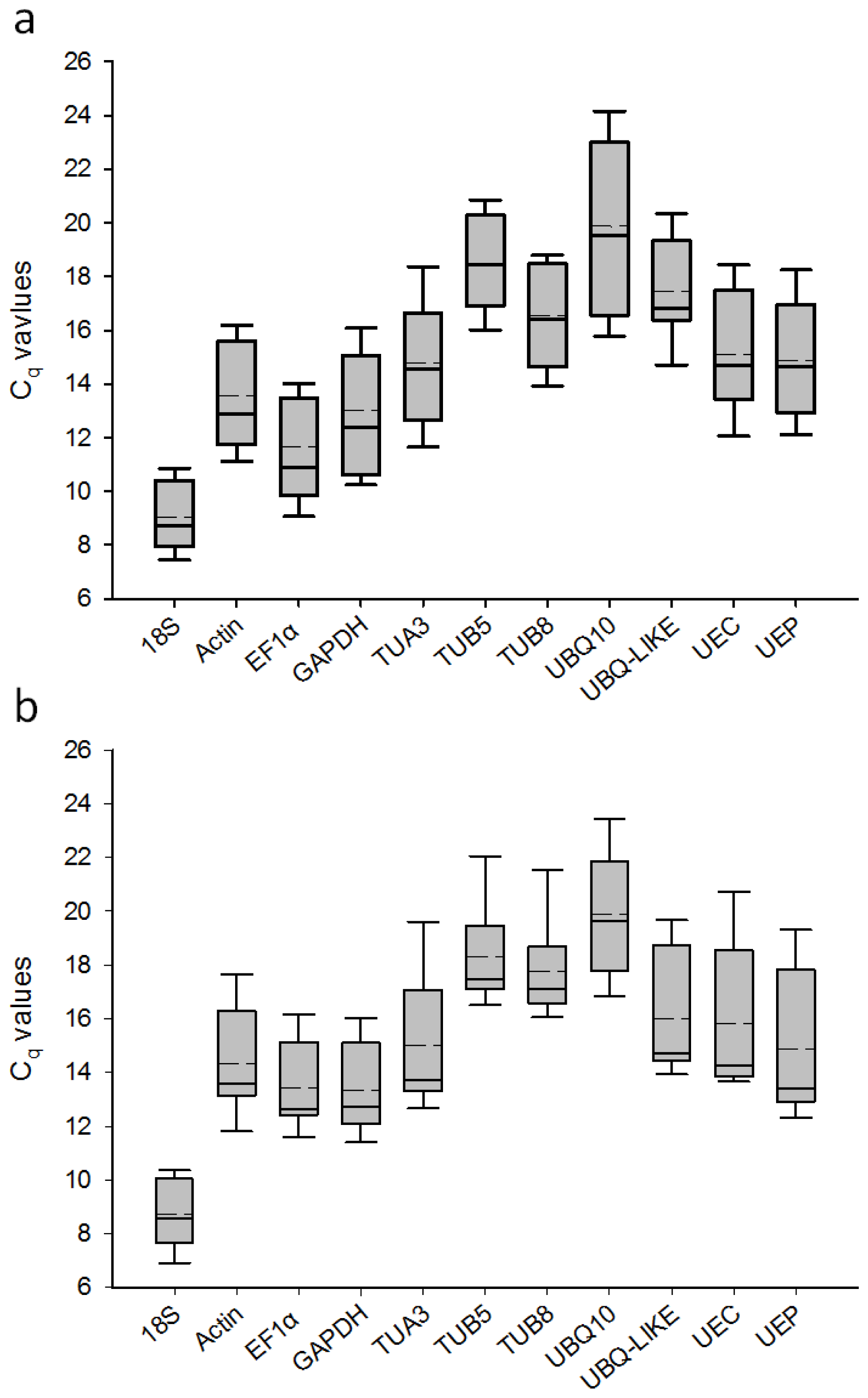
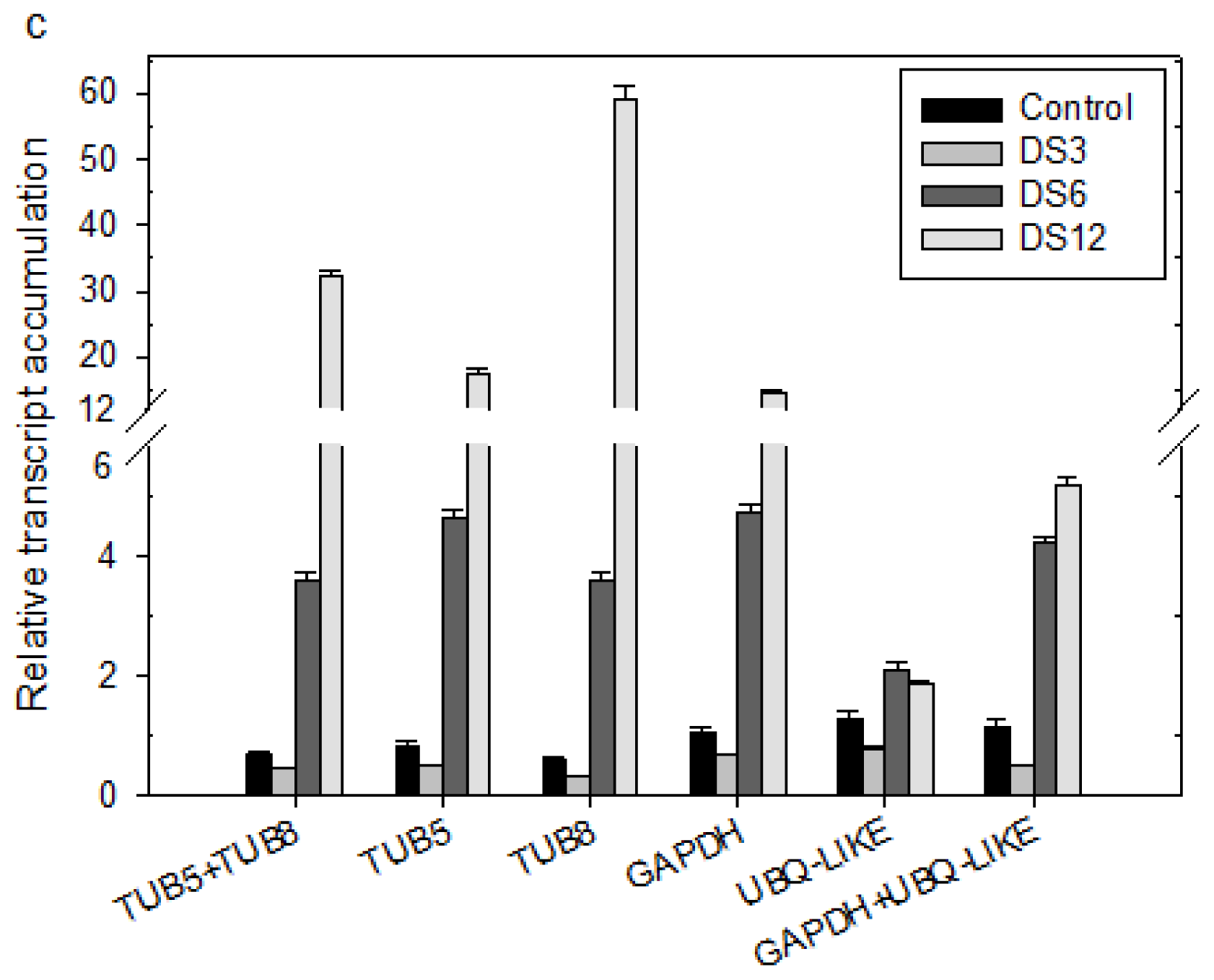
2.2. Transcript Accumulation of Candidate Reference Genes
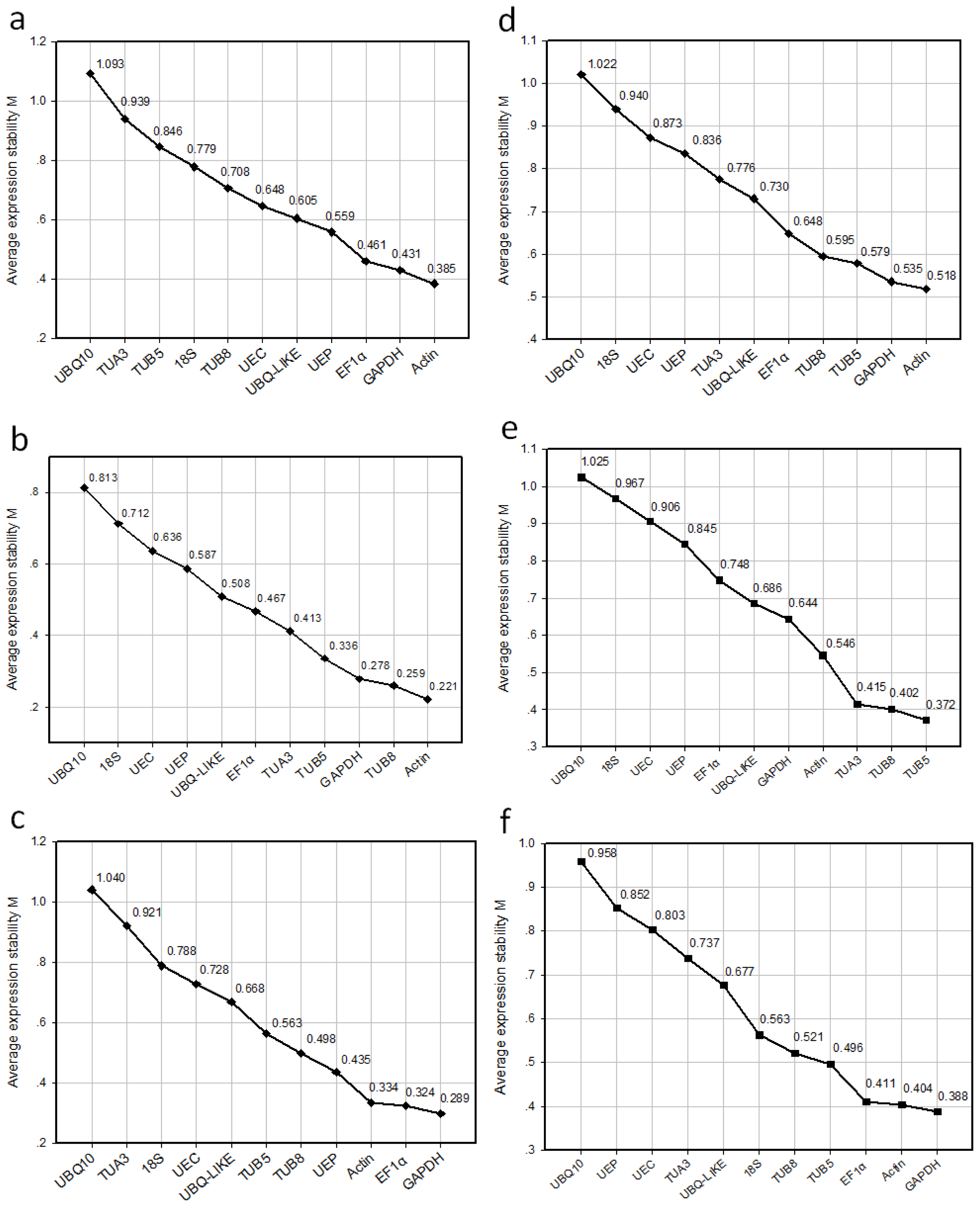
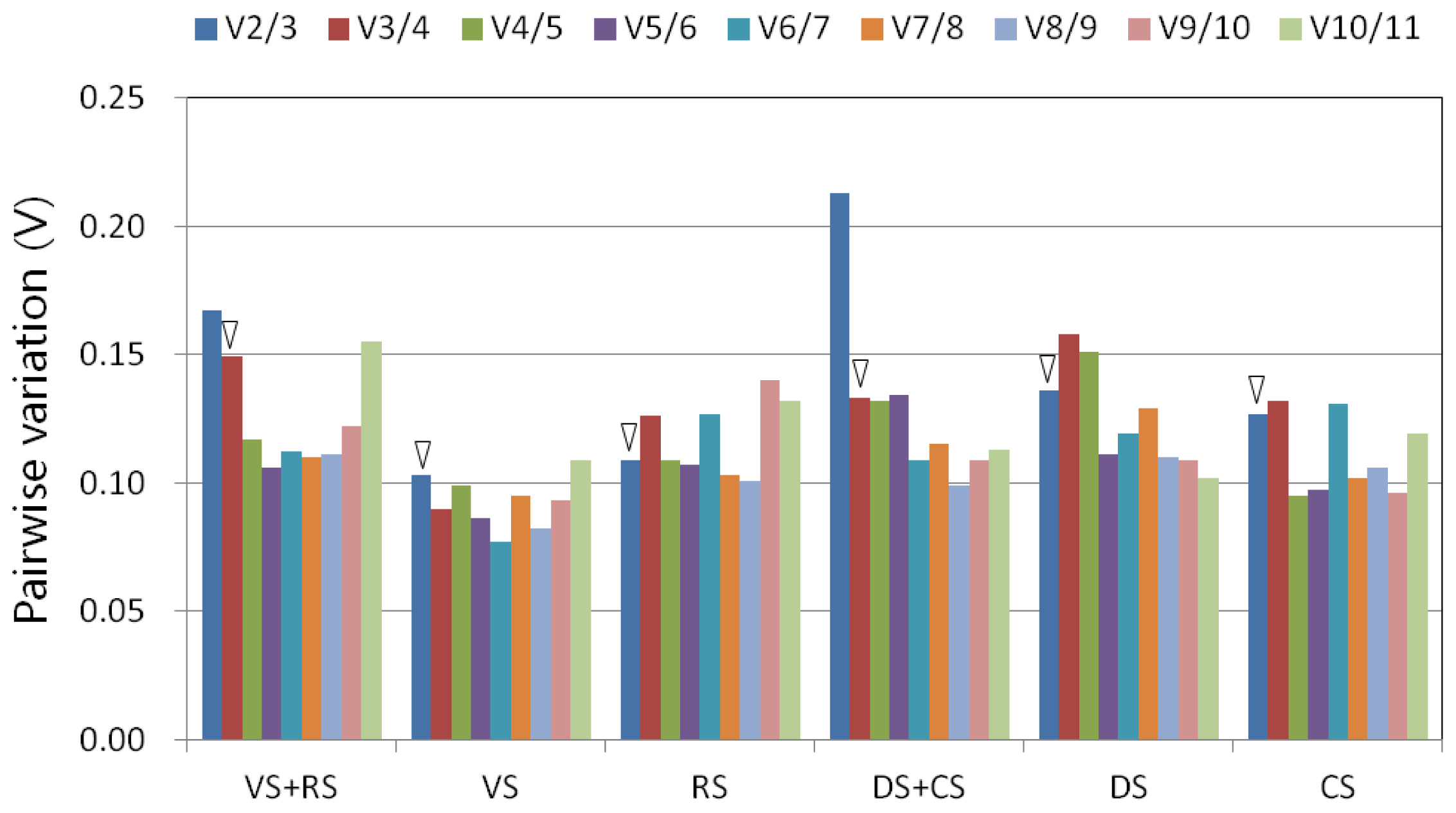
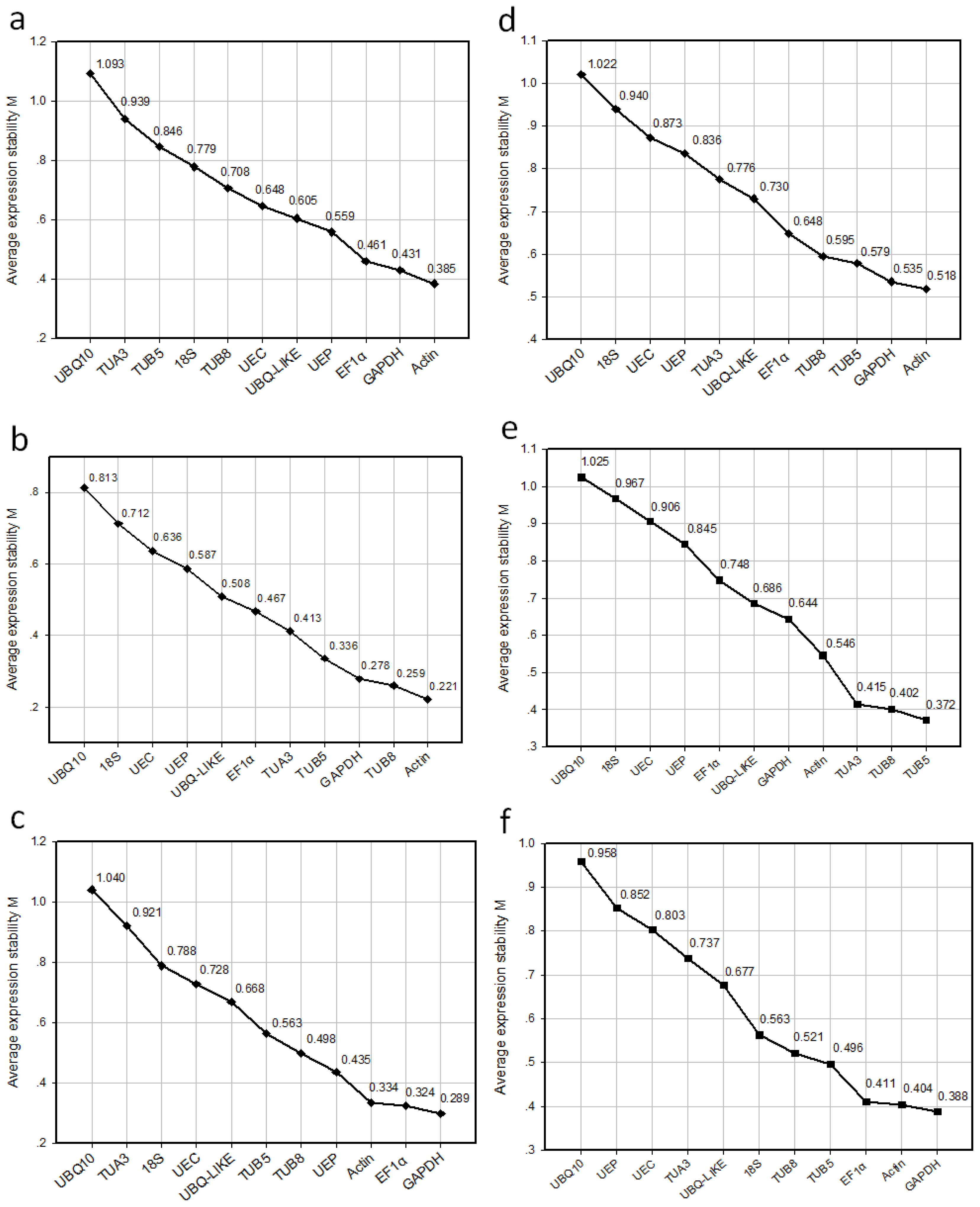
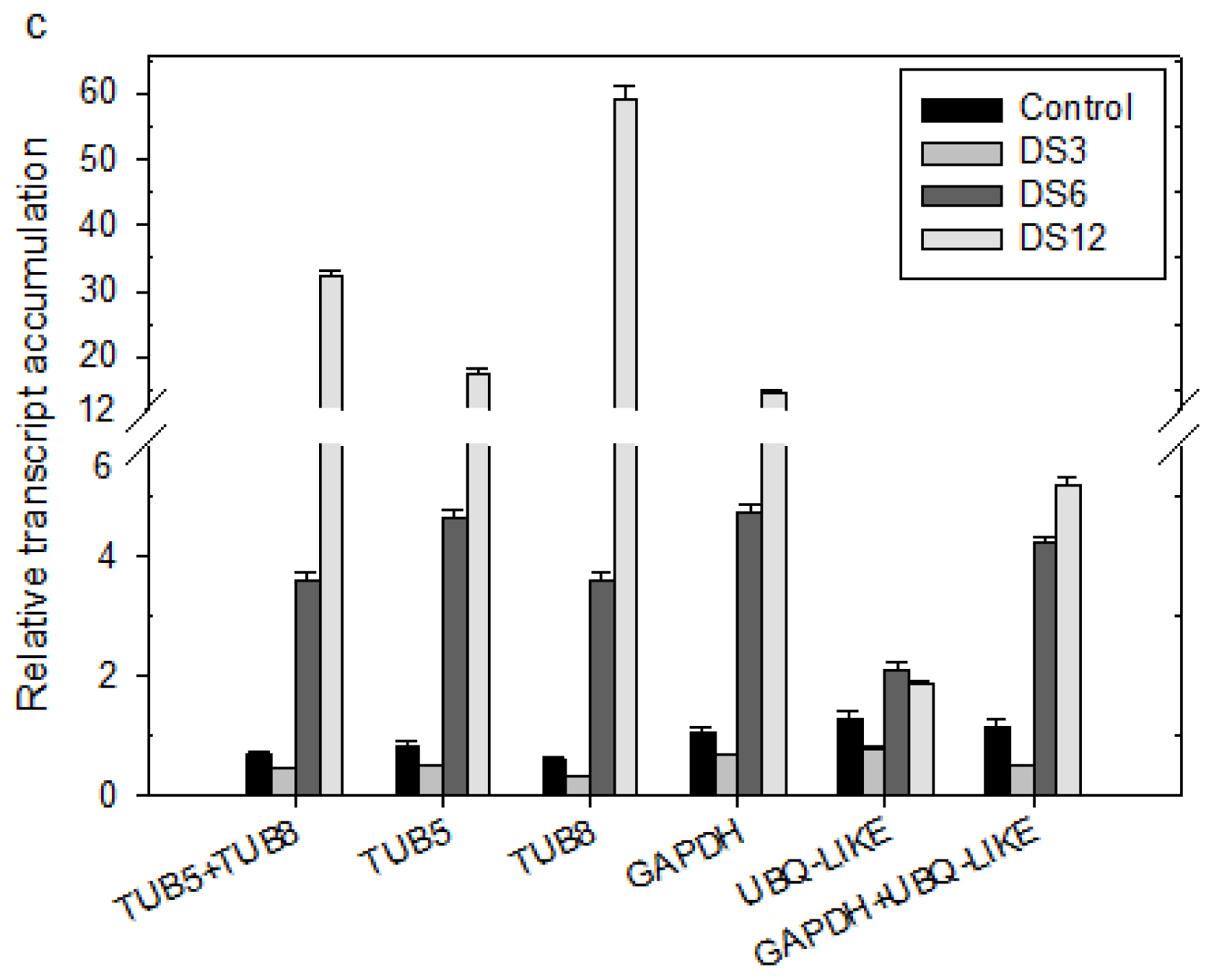
2.3. Ranking of Candidate Reference Genes and Determination of Optimal Reference Genes
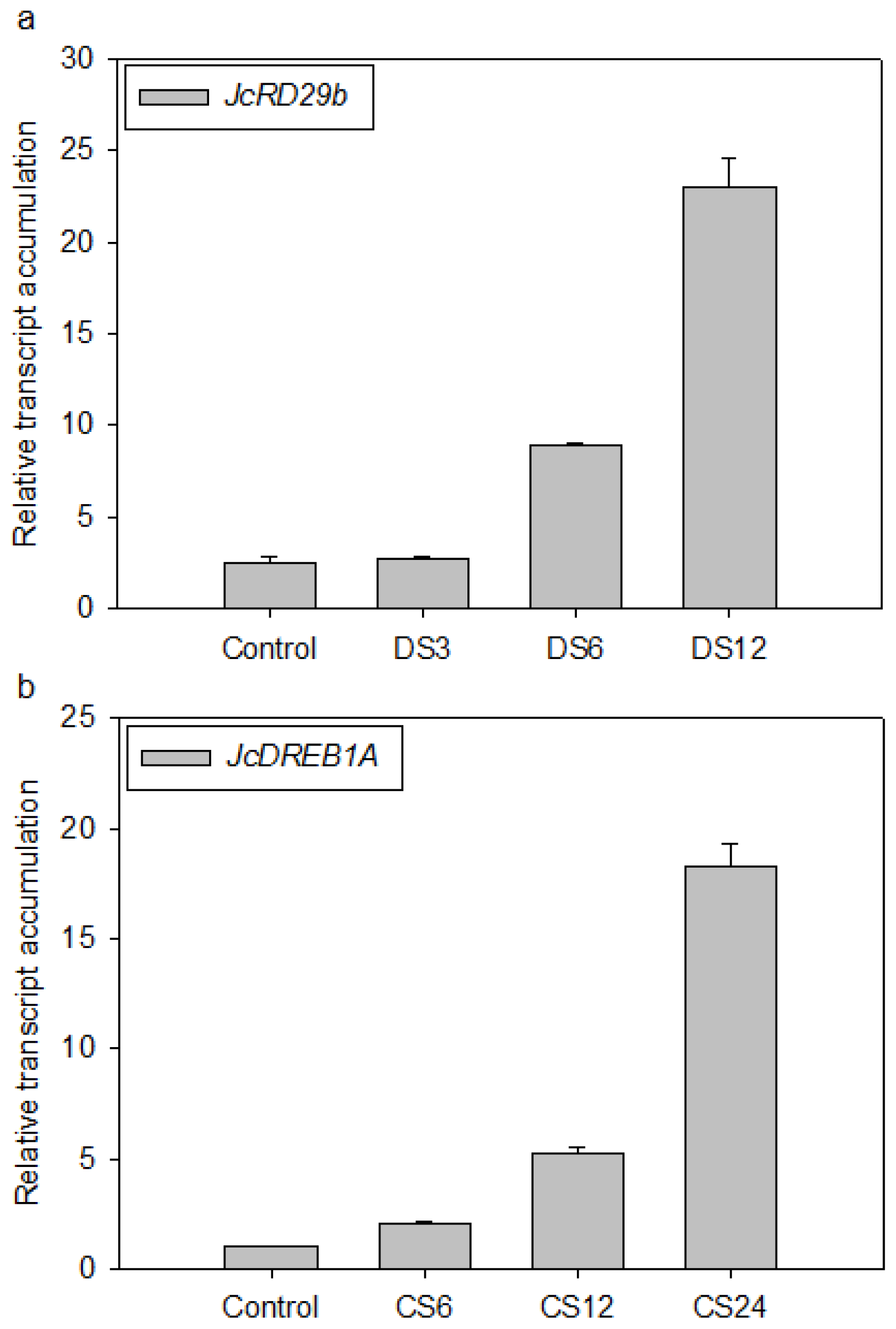
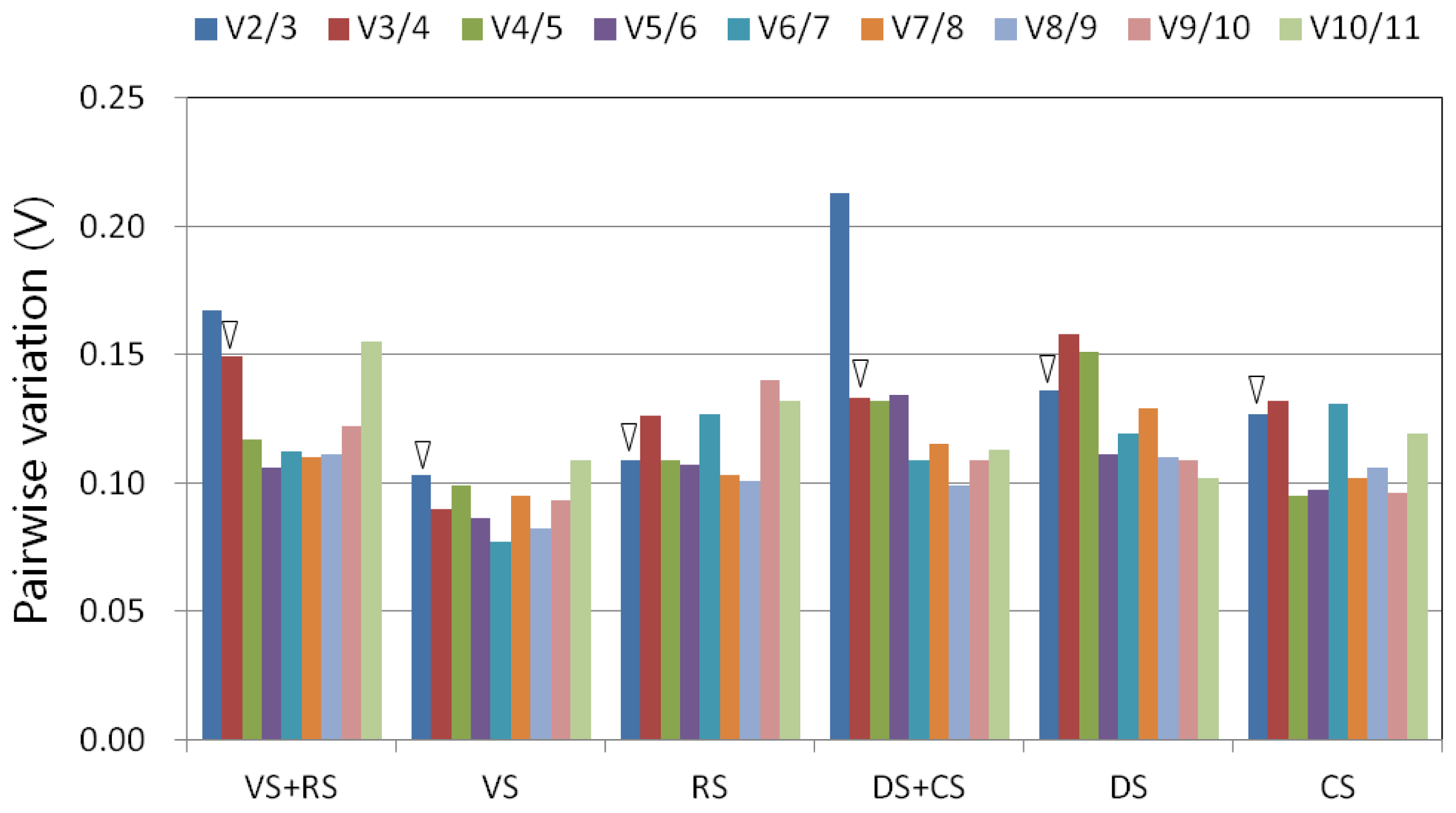
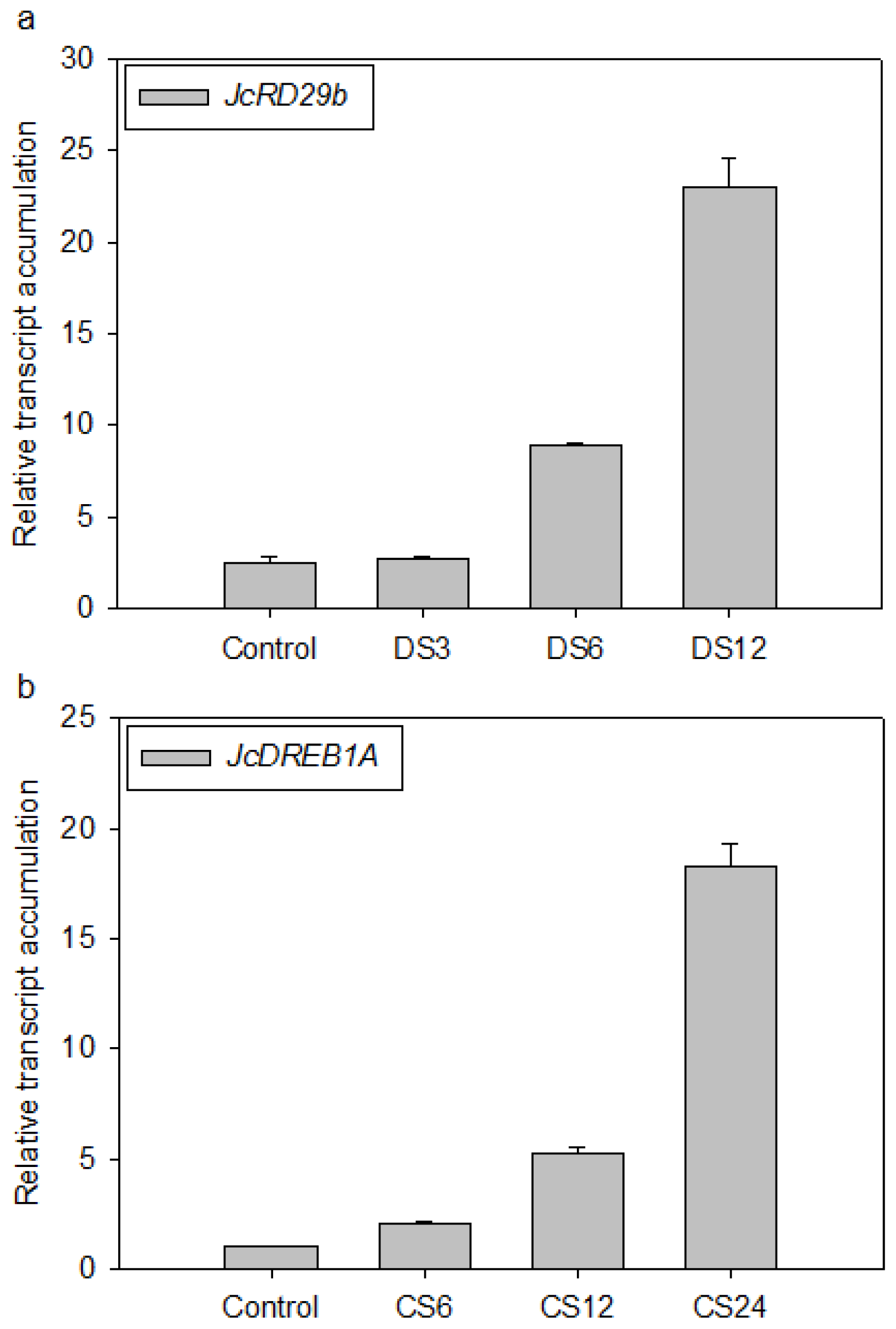
2.4. Validation of the Selected Reference Genes in Leaf Samples Treated with Desiccation or Cold Stress
3. Experimental Section
3.1. Plant Materials and Stress Treatments
3.2. Candidate Reference Genes from Jatropha
3.3. Desiccation Stress- and Cold Stress-Responsive Genes from Jatropha
3.4. RNA Isolation and Purification and cDNA Synthesis
3.5. Primer Design and RT-qPCR Analysis
3.6. Data Analysis
4. Conclusions
Supplementary Information
ijms-14-24338-s002.pdfAcknowledgments
Conflicts of Interest
References
- Carels, N. Jatropha curcas: A review. Adv. Bot. Res 2009, 50, 39–86. [Google Scholar]
- Fairless, D. Biofuel: The little shrub that could—Maybe. Nature 2007, 449, 652–655. [Google Scholar]
- Francis, G.; Edinger, R.; Becker, K. A concept for simultaneous wasteland reclamation, fuel production, and socio-economic development in degraded areas in India: Need, potential and perspectives of Jatropha plantations. Nat. Resour. Forum 2005, 29, 12–24. [Google Scholar]
- Jones, N.; Miller, J.H. Jatropha curcas: A Multipurpose Species for Problematic Sites; World Bank: Washington, DC, USA, 1992; pp. 1–12. [Google Scholar]
- Kumar, A.; Sharma, S. An evaluation of multipurpose oil seed crop for industrial uses (Jatropha curcas L.): A review. Ind. Crops Prod 2008, 28, 1–10. [Google Scholar]
- Makkar, H.P.; Becker, K. Jatropha curcas, a promising crop for the generation of biodiesel and value–added coproducts. Eur. J. Lipid Sci. Technol 2009, 111, 773–787. [Google Scholar]
- Abdulla, R.; Chan, E.S.; Ravindra, P. Biodiesel production from Jatropha curcas: A critical review. Crit. Rev. Biotechnol 2011, 31, 53–64. [Google Scholar]
- Bonnet, S.; Gheewala, S. Potential of Jatropha as an Energy Crop. In Jatropha, Challenges for a New Energy Crop; Carels, N., Sujatha, M., Bahadur, B., Eds.; Springer: New York, NY, USA, 2012; Volume 1, pp. 571–582. [Google Scholar]
- Li, L.; Coppola, E.; Rine, J.; Miller, J.L.; Walker, D. Catalytic hydrothermal conversion of triglycerides to non-ester biofuels. Energy Fuels 2010, 24, 1305–1315. [Google Scholar]
- Chikara, J.; Prakash, A.; Mastan, S.G.; Ghosh, A. Genetic Improvement in Jatropha curcas through Selection and Breeding. In Jatropha, Challenges for a New Energy Crop; Bahadur, B., Sujatha, M., Carels, N., Eds.; Springer: New York, NY, USA, 2013; Volume 2, pp. 119–133. [Google Scholar]
- Divakara, B.N.; Upadhyaya, H.D.; Wani, S.P.; Gowda, C.L.L. Biology and genetic improvement of Jatropha curcas L.: A review. Appl. Energy 2010, 87, 732–742. [Google Scholar]
- Pan, B.-Z.; Xu, Z.-F. Benzyladenine treatment significantly increases the seed yield of the biofuel plant Jatropha curcas. J. Plant Growth Regul 2011, 30, 166–174. [Google Scholar]
- Sanderson, K. Wonder weed plans fail to flourish. Nature 2009, 461, 328–329. [Google Scholar]
- Carels, N. Towards the Domestication of Jatropha: The Integration of Sciences. In Jatropha, Challenges for a New Energy Crop; Bahadur, B., Sujatha, M., Carels, N., Eds.; Springer: New York, NY, USA, 2013; Volume 2, pp. 263–299. [Google Scholar]
- Gressel, J. Transgenics are imperative for biofuel crops. Plant Sci 2008, 174, 246–263. [Google Scholar]
- Sujatha, M.; Reddy, T.P.; Mahasi, M.J. Role of biotechnological interventions in the improvement of castor (Ricinus communis L.) and Jatropha curcas L. Biotechnol. Adv 2008, 26, 424–435. [Google Scholar]
- Hirakawa, H.; Tsuchimoto, S.; Sakai, H.; Nakayama, S.; Fujishiro, T.; Kishida, Y.; Kohara, M.; Watanabe, A.; Yamada, M.; Aizu, T.; et al. Upgraded genomic information of Jatropha curcas L. Plant Biotechnol 2012, 29, 123–130. [Google Scholar]
- Sato, S.; Hirakawa, H.; Isobe, S.; Fukai, E.; Watanabe, A.; Kato, M.; Kawashima, K.; Minami, C.; Muraki, A.; Nakazaki, N.; et al. Sequence analysis of the genome of an oil-bearing tree, Jatropha curcas L. DNA Res 2011, 18, 65–76. [Google Scholar]
- Kajikawa, M.; Morikawa, K.; Inoue, M.; Widyastuti, U.; Suharsono, S.; Yokota, A.; Akashi, K. Establishment of bispyribac selection protocols for Agrobacterium tumefaciens- and Agrobacterium rhizogenes-mediated transformation of the oil seed plant Jatropha curcas L. Plant Biotechnol 2012, 29, 145–153. [Google Scholar]
- Kumar, N.; Reddy, M.; Sujatha, M. Genetic transformation of Jatropha curcas: Current status and future prospects. In Jatropha, Challenges for a New Energy Crop; Bahadur, B., Sujatha, M., Carels, N., Eds.; Springer: New York, NY, USA, 2013; Volume 2, pp. 535–546. [Google Scholar]
- Li, M.R.; Li, H.Q.; Jiang, H.W.; Pan, X.P.; Wu, G.J. Establishment of an Agrobacteriuim-mediated cotyledon disc transformation method for Jatropha curcas. Plant Cell Tiss. Organ Cult 2008, 92, 173–181. [Google Scholar]
- Pan, J.L.; Fu, Q.T.; Xu, Z.F. Agrobacterium tumefaciens-mediated transformation of biofuel plant Jatropha curcas using kanamycin selection. Afr. J. Biotechnol 2010, 9, 6477–6481. [Google Scholar]
- Bustin, S.A. Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): Trends and problems. J. Mol. Endocrinol 2002, 29, 23–39. [Google Scholar]
- Phillips, M.A.; D’Auria, J.C.; Luck, K.; Gershenzon, J. Evaluation of candidate reference genes for real-time quantitative PCR of plant samples using purified cDNA as template. Plant Mol. Biol. Report 2009, 27, 407–416. [Google Scholar]
- Guénin, S.; Mauriat, M.; Pelloux, J.; Van Wuytswinkel, O.; Bellini, C.; Gutierrez, L. Normalization of qRT-PCR data: The necessity of adopting a systematic, experimental conditions-specific, validation of references. J. Exp. Bot 2009, 60, 487–493. [Google Scholar]
- Gutierrez, L.; Mauriat, M.; Guénin, S.; Pelloux, J.; Lefebvre, J.F.; Louvet, R.; Rusterucci, C.; Moritz, T.; Guerineau, F.; Bellini, C. The lack of a systematic validation of reference genes: A serious pitfall undervalued in reverse transcription–polymerase chain reaction (RT–PCR) analysis in plants. Plant Biotechnol. J 2008, 6, 609–618. [Google Scholar]
- Huggett, J.; Dheda, K.; Bustin, S.; Zumla, A. Real-time RT-PCR normalisation; Strategies and considerations. Genes Immun 2005, 6, 279–284. [Google Scholar]
- Bustin, S.A. Why the need for qPCR publication guidelines?—The case for MIQE. Methods 2010, 50, 217–226. [Google Scholar]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem 2009, 55, 611–622. [Google Scholar]
- Czechowski, T.; Stitt, M.; Altmann, T.; Udvardi, M.K.; Scheible, W.-R. Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol 2005, 139, 5–17. [Google Scholar]
- Dekkers, B.J.W.; Willems, L.; Bassel, G.W.; van Bolderen-Veldkamp, R.P.; Ligterink, W.; Hilhorst, H.W.M.; Bentsink, L. Identification of reference genes for RT–qPCR expression analysis in Arabidopsis and tomato seeds. Plant Cell Physiol 2012, 53, 28–37. [Google Scholar]
- Hong, S.M.; Bahn, S.C.; Lyu, A.; Jung, H.S.; Ahn, J.H. Identification and testing of superior reference genes for a starting pool of transcript normalization in Arabidopsis. Plant Cell Physiol 2010, 51, 1694–1706. [Google Scholar]
- Remans, T.; Smeets, K.; Opdenakker, K.; Mathijsen, D.; Vangronsveld, J.; Cuypers, A. Normalisation of real-time RT-PCR gene expression measurements in Arabidopsis thaliana exposed to increased metal concentrations. Planta 2008, 227, 1343–1349. [Google Scholar]
- Jain, M.; Nijhawan, A.; Tyagi, A.K.; Khurana, J.P. Validation of housekeeping genes as internal control for studying gene expression in rice by quantitative real-time PCR. Biochem. Biophys. Res. Commun 2006, 345, 646–651. [Google Scholar]
- Li, Q.-F.; Sun, S.M.; Yuan, D.-Y.; Yu, H.-X.; Gu, M.-H.; Liu, Q.-Q. Validation of candidate reference genes for the accurate normalization of real-time quantitative RT-PCR data in rice during seed development. Plant Mol. Biol. Report 2010, 28, 49–57. [Google Scholar]
- Qi, J.; Yu, S.; Zhang, F.; Shen, X.; Zhao, X.; Yu, Y.; Zhang, D. Reference gene selection for real-time quantitative polymerase chain reaction of mRNA transcript levels in Chinese cabbage (Brassica rapa L. ssp. pekinensis). Plant Mol. Biol. Report 2010, 28, 597–604. [Google Scholar]
- Schmittgen, T.D.; Zakrajsek, B.A. Effect of experimental treatment on housekeeping gene expression: Validation by real-time, quantitative RT-PCR. J. Biochem. Biophys. Methods 2000, 46, 69–81. [Google Scholar]
- Vandesompele, J.; de Preter, K.; Pattyn, F.; Poppe, B.; van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 2002, 3, research0034. [Google Scholar]
- Andersen, C.L.; Jensen, J.L.; Ørntoft, T.F. Normalization of real-time quantitative reverse transcription-PCR data: A model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res 2004, 64, 5245–5250. [Google Scholar]
- Hellemans, J.; Mortier, G.; de Paepe, A.; Speleman, F.; Vandesompele, J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol 2007, 8, R19. [Google Scholar]
- Biogazelle. http://www.biogazelle.com/qbaseplus (accessed on 18 May 2012).
- Multid Analyses. http://www.multid.se/ (accessed on 18 May 2012).
- Liu, B.; Wang, W.; Gao, J.; Chen, F.; Wang, S.; Xu, Y.; Tang, L.; Jia, Y. Molecular cloning and characterization of a jasmonate biosynthetic pathway gene for allene oxide cyclase from Jatropha curcas. Acta Physiol. Plant 2010, 32, 531–539. [Google Scholar]
- Zhang, F.-L.; Niu, B.; Wang, Y.-C.; Chen, F.; Wang, S.-H.; Xu, Y.; Jiang, L.-D.; Gao, S.; Wu, J.; Tang, L. A novel betaine aldehyde dehydrogenase gene from Jatropha curcas, encoding an enzyme implicated in adaptation to environmental stress. Plant Sci 2008, 174, 510–518. [Google Scholar]
- Li, J.; Li, M.-R.; Wu, P.-Z.; Tian, C.-E.; Jiang, H.-W.; Wu, G.-J. Molecular cloning and expression analysis of a gene encoding a putative β-ketoacyl-acyl carrier protein (ACP) synthase III (KAS III) from Jatropha curcas. Tree Physiol 2008, 28, 921–927. [Google Scholar]
- Omar, S.A.; Fu, Q.-T.; Chen, M.-S.; Wang, G.-J.; Song, S.-Q.; Elsheery, N.I.; Xu, Z.F. Identification and expression analysis of two small heat shock protein cDNAs from developing seeds of biodiesel feedstock plant Jatropha curcas. Plant Sci 2011, 181, 632–637. [Google Scholar]
- Xu, R.; Wang, R.; Liu, A. Expression profiles of genes involved in fatty acid and triacylglycerol synthesis in developing seeds of Jatropha (Jatropha curcas L.). Biomass Bioenergy 2011, 35, 1683–1692. [Google Scholar]
- Yang, J.; Yang, M.F.; Wang, D.; Chen, F.; Shen, S.H. JcDof1, a Dof transcription factor gene, is associated with the light-mediated circadian clock in Jatropha curcas. Physiol. Plant 2010, 139, 324–334. [Google Scholar]
- Exposito-Rodriguez, M.; Borges, A.; Borges-Perez, A.; Perez, J. Selection of internal control genes for quantitative real-time RT-PCR studies during tomato development process. BMC Plant Biol 2008, 8, 131. [Google Scholar]
- Liu, Q.; Kasuga, M.; Sakuma, Y.; Abe, H.; Miura, S.; Yamaguchi-Shinozaki, K.; Shinozaki, K. Two transcription factors, DREB1 and DREB2, with an EREBP/AP2 DNA binding domain separate two cellular signal transduction pathways in drought-and low-temperature-responsive gene expression, respectively, in Arabidopsis. Plant Cell 1998, 10, 1391–1406. [Google Scholar]
- Jatropha Genome Database (Contents Release 4.5). http://www.kazusa.or.jp/jatropha (accessed on 20 January 2011).
- National Center for Biotechnology Information. http://www.ncbi.nlm.nih.gov/ (accessed on 8 February 2011).
- Yamaguchi-Shinozaki, K.; Shinozaki, K. Characterization of the expression of a desiccation-responsive rd29 gene of Arabidopsis thaliana and analysis of its promoter in transgenic plants. Mol. Gen. Genet 1993, 236, 331–340. [Google Scholar]
- Ding, L.W.; Sun, Q.Y.; Wang, Z.Y.; Sun, Y.B.; Xu, Z.F. Using silica particles to isolate total RNA from plant tissues recalcitrant to extraction in guanidine thiocyanate. Anal. Biochem 2008, 374, 426–428. [Google Scholar]
- Ramakers, C.; Ruijter, J.M.; Deprez, R.H.L.; Moorman, A.F. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci. Lett 2003, 339, 62–66. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene symbol | Slope | E (%) | R2 | Ta (°C) | Tm (°C) |
|---|---|---|---|---|---|
| 18S | −3.21 | 105% | 0.999 | 58 | 85.57 |
| Actin | −3.299 | 101% | 0.995 | 58 | 83.90 |
| EF1α | −3.158 | 107% | 0.999 | 59 | 82.45 |
| GAPDH | −3.173 | 107% | 0.998 | 58 | 85.72 |
| TUA3 | −3.353 | 99% | 0.999 | 58 | 86.46 |
| TUB5 | −3.462 | 94% | 0.999 | 58 | 84.05 |
| TUB8 | −3.285 | 102% | 0.998 | 58 | 82.58 |
| UBQ10 | −3.129 | 109% | 0.998 | 59 | 79.79 |
| UBQ-LIKE | −3.595 | 90% | 0.996 | 59 | 84.38 |
| UEC | −3.149 | 107% | 0.999 | 58 | 85.79 |
| UEP | −3.23 | 104% | 0.999 | 58 | 86.24 |
| Ranking | VS + RS | VS | RS | DS + CS | DS | CS |
|---|---|---|---|---|---|---|
| Gene (M value) | Gene (M value) | Gene (M value) | Gene (M value) | Gene (M value) | Gene (M value) | |
| 1 | Actin (0.112) | Actin (0.112) | EF1α (0.020) | Actin (0.139) | Actin (0.116) | Actin (0.133) |
| 2 | EF1α (0.194) | GAPDH (0.185) | UEP (0.110) | GAPDH (0.256) | UBQ-LIKE (0.268) | GAPDH (0.136) |
| 3 | GAPDH (0.230) | TUB8 (0.187) | GAPDH (0.200) | UBQ-LIKE (0.349) | GAPDH (0.309) | EF1α (0.279) |
| 4 | UEP (0.287) | UBQ-LIKE (0.229) | Actin (0.201) | TUB5 (0.419) | TUB5 (0.432) | UBQ-LIKE (0.387) |
| 5 | UBQ-LIKE (0.406) | EF1α (0.289) | TUB8 (0.429) | EF1α (0.434) | TUB8 (0.476) | TUB5 (0.436) |
| 6 | TUB8 (0.424) | TUA3 (0.364) | UEC (0.459) | TUA3 (0.467) | EF1α (0.489) | TUA3 (0.470) |
| 7 | UEC (0.486) | UEP (0.394) | UBQ-LIKE (0.509) | TUB8 (0.497) | TUA3 (0.549) | TUB8 (0.475) |
| 8 | 18S (0.646) | TUB5 (0.416) | TUB5 (0.597) | UEP (0.593) | UEP (0.586) | UEC (0.592) |
| 9 | TUB5 (0.678) | UEC (0.459) | 18S (0.654) | UEC (0.664) | UBQ10 (0.662) | UEP (0.631) |
| 10 | TUA3 (0.886) | 18S (0.692) | TUA3 (0.988) | 18S (0.763) | UEC (0.686) | 18S (0.634) |
| 11 | UBQ10 (1.185) | UBQ10 (0.807) | UBQ10 (1.030) | UBQ10 (0.848) | 18S (0.757) | UBQ10 (0.897) |
| Gene ID | Gene symbol | Primer sequences | Amplicon length |
|---|---|---|---|
| Jcr4S06558.10 a | Actin | F: 5′-CTCCTCTCAACCCCAAAGCCAA-3′ R: 5′-CACCAGAATCCAGCACGATACCA-3′ | 147 bp |
| Jcr4U29393.10 a | GAPDH | F: 5′-TGAAGGACTGGAGAGGTGGAAGAGC-3′ R: 5′-ATCAACAGTTGGAACACGGAAAGCC-3′ | 140 bp |
| Jcr4S00045.200 a | UBQ10 | F: 5′-AAAGCAGTTGGAGGATGGAAGGAC-3′ R: 5′-GCGAAGCCTGAGAACAAGGTGAAG-3′ | 82 bp |
| Jcr4S10519.50 a | UBQ-LIKE | F: 5′-GGTGAGAGTGAAGTGTAATGATGACGAC-3′ R: 5′-CCTCAGAGTTATATGGTCCTTGTAAATGG-3′ | 136 bp |
| Jcr4508473.50 a | UEP | F: 5′-AATCCCTCCAGACCAGCAGCGACT-3′ R: 5′-GCTCTTGTAGAACTGAAGCACGGC-3′ | 220 bp |
| Jcr4S00542.10 a | EF1α | F: 5′-AAGATGATTCCCACCAAGCCCA-3′ R: 5′-CACAGCAAAACGACCCAGAGGA-3′ | 72 bp |
| GW880075.1 b | TUA3 | F: 5′-TTCAATCAGCGAAAATGAGAGAGTG-3′ R: 5′-TCACTGAAAAAGGTGTTGAAGGCA-3′ | 178 bp |
| GW877086.1 b | TUB8 | F: 5′-GCAGGGAATAACTGGGCTAAAGGT-3′ R: 5′-CTCCACCCAACGAATGACAAACTT-3′ | 136 bp |
| EZ114400.1 b | UEC | F: 5′-GTCCCTGATTTTGAGATGGCGTC-3′ R: 5′-CAATATGTCAAGACAAATGCTCCCG-3′ | 284 bp |
| GW878948.1 b | TUB5 | F: 5′-TATGTTCCCAGGGCGGTTCTAATG-3′ R: 5′-GGACTGCCCAAAGACAAAGTTATCG-3′ | 111 bp |
| AY823528.1 b | 18S | F: 5′-CTCAACCATAAACGATGCCGACC-3′ R: 5′-TTCAGCCTTGCGACCATACTCCC-3′ | 117 bp |
| Jcr4S01474.40 a | JcRD29b | F: 5′-AATCTCCGCAAAGAATGTTGTAGC-3′ R: 5′-CTCCCTGTCTCAGCAACTTTCTCATA-3′ | 180 bp |
| Jcr4S27135.10 a | JcDREB1A | F: 5′-CGGATGGACTTTTAGGGGATGAAT-3′ R: 5′-CACTGAGGTGGAGGCAACAACA-3′ | 160 bp |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, L.; He, L.-L.; Fu, Q.-T.; Xu, Z.-F. Selection of Reliable Reference Genes for Gene Expression Studies in the Biofuel Plant Jatropha curcas Using Real-Time Quantitative PCR. Int. J. Mol. Sci. 2013, 14, 24338-24354. https://doi.org/10.3390/ijms141224338
Zhang L, He L-L, Fu Q-T, Xu Z-F. Selection of Reliable Reference Genes for Gene Expression Studies in the Biofuel Plant Jatropha curcas Using Real-Time Quantitative PCR. International Journal of Molecular Sciences. 2013; 14(12):24338-24354. https://doi.org/10.3390/ijms141224338
Chicago/Turabian StyleZhang, Lu, Liang-Liang He, Qian-Tang Fu, and Zeng-Fu Xu. 2013. "Selection of Reliable Reference Genes for Gene Expression Studies in the Biofuel Plant Jatropha curcas Using Real-Time Quantitative PCR" International Journal of Molecular Sciences 14, no. 12: 24338-24354. https://doi.org/10.3390/ijms141224338