The Dual Role of Smad7 in the Control of Cancer Growth and Metastasis

 ,
,
Abstract
:1. Introduction
2. Expression and Role of Smad7 in Cancer
2.1. Colorectal Cancer
2.2. Pancreatic Cancer
2.3. Gastric Cancer
2.4. Skin Cancer
2.5. Breast Cancer
2.6. Liver Cancer
2.7. Prostate Cancer
3. Conclusions
Acknowledgments
Conflicts of Interest
References
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar]
- Liotta, L.A.; Stetler-Stevenson, W.G. Tumor invasion and metastasis: An imbalance of positive and negative regulation. Cancer Res 1991, 51, 5054s–5059s. [Google Scholar]
- Oft, M.; Akhurst, R.J.; Balmain, A. Metastasis is driven by sequential elevation of H-ras and Smad2 levels. Nat. Cell Biol 2002, 4, 487–494. [Google Scholar]
- Katsuno, Y.; Lamouille, S.; Derynck, R. Tgf-beta signaling and epithelial-mesenchymal transition in cancer progression. Curr. Opin. Oncol 2013, 25, 76–84. [Google Scholar]
- Piek, E.; Heldin, C.H.; Ten Dijke, P. Specificity, diversity, and regulation in TGF-β superfamily signaling. FASEB J 1999, 13, 2105–2124. [Google Scholar]
- Massague, J.; Seoane, J.; Wotton, D. Smad transcription factors. Genes Dev 2005, 19, 2783–2810. [Google Scholar]
- Kretzschmar, M.; Doody, J.; Timokhina, I.; Massague, J. A mechanism of repression of TGFβ/Smad signaling by oncogenic ras. Genes Dev 1999, 13, 804–816. [Google Scholar]
- Luo, K.; Stroschein, S.L.; Wang, W.; Chen, D.; Martens, E.; Zhou, S.; Zhou, Q. The Ski oncoprotein interacts with the Smad proteins to repress tgfbeta signaling. Genes Dev 1999, 13, 2196–2206. [Google Scholar]
- Stroschein, S.L.; Wang, W.; Zhou, S.; Zhou, Q.; Luo, K. Negative feedback regulation of TGF-β signaling by the snon oncoprotein. Science 1999, 286, 771–774. [Google Scholar]
- Deheuninck, J.; Luo, K. Ski and snon, potent negative regulators of tgf-beta signaling. Cell Res 2009, 19, 47–57. [Google Scholar]
- Briones-Orta, M.A.; Tecalco-Cruz, A.C.; Sosa-Garrocho, M.; Caligaris, C.; Macias-Silva, M. Inhibitory Smad7: Emerging roles in health and disease. Curr. Mol. Pharmacol 2011, 4, 141–153. [Google Scholar]
- Yan, X.; Chen, Y.G. Smad7: Not only a regulator, but also a cross-talk mediator of TGF-β signalling. Biochem. J 2011, 434, 1–10. [Google Scholar]
- Nakao, A.; Afrakhte, M.; Moren, A.; Nakayama, T.; Christian, J.L.; Heuchel, R.; Itoh, S.; Kawabata, M.; Heldin, N.E.; Heldin, C.H.; et al. Identification of Smad7, a tgfbeta-inducible antagonist of tgf-beta signalling. Nature 1997, 389, 631–635. [Google Scholar]
- Hayashi, H.; Abdollah, S.; Qiu, Y.; Cai, J.; Xu, Y.Y.; Grinnell, B.W.; Richardson, M.A.; Topper, J.N.; Gimbrone, M.A., Jr.; Wrana, J.L.; et al. The Mad-Related protein Smad7 associates with the tgfbeta receptor and functions as an antagonist of tgfbeta signaling. Cell 1997, 89, 1165–1173. [Google Scholar]
- Ebisawa, T.; Fukuchi, M.; Murakami, G.; Chiba, T.; Tanaka, K.; Imamura, T.; Miyazono, K. Smurf1 interacts with transforming growth factor-beta type i receptor through Smad7 and induces receptor degradation. J. Biol. Chem 2001, 276, 12477–12480. [Google Scholar]
- Kavsak, P.; Rasmussen, R.K.; Causing, C.G.; Bonni, S.; Zhu, H.; Thomsen, G.H.; Wrana, J.L. Smad7 binds to smurf2 to form an e3 ubiquitin ligase that targets the tgf beta receptor for degradation. Mol. Cell 2000, 6, 1365–1375. [Google Scholar]
- Kuratomi, G.; Komuro, A.; Goto, K.; Shinozaki, M.; Miyazawa, K.; Miyazono, K.; Imamura, T. Nedd4-2 (neural precursor cell expressed, developmentally down-regulated 4-2) negatively regulates tgf-beta (transforming growth factor-beta) signalling by inducing ubiquitin-mediated degradation of Smad2 and tgf-beta type i receptor. Biochem. J 2005, 386, 461–470. [Google Scholar]
- Shi, W.; Sun, C.; He, B.; Xiong, W.; Shi, X.; Yao, D.; Cao, X. Gadd34-pp1c recruited by Smad7 dephosphorylates tgfbeta type i receptor. J. Cell Biol 2004, 164, 291–300. [Google Scholar]
- Zhang, S.; Fei, T.; Zhang, L.; Zhang, R.; Chen, F.; Ning, Y.; Han, Y.; Feng, X.H.; Meng, A.; Chen, Y.G. Smad7 antagonizes transforming growth factor beta signaling in the nucleus by interfering with functional Smad-DNA complex formation. Mol. Cell Biol 2007, 27, 4488–4499. [Google Scholar]
- Hong, S.; Lee, C.; Kim, S.J. Smad7 sensitizes tumor necrosis factor induced apoptosis through the inhibition of antiapoptotic gene expression by suppressing activation of the nuclear factor-κb pathway. Cancer Res 2007, 67, 9577–9583. [Google Scholar]
- Lee, Y.S.; Kim, J.H.; Kim, S.T.; Kwon, J.Y.; Hong, S.; Kim, S.J.; Park, S.H. Smad7 and Smad6 bind to discrete regions of pellino-1 via their mh2 domains to mediate TGF-β1-induced negative regulation of IL-1R/TLR signaling. Biochem. Biophys. Res. Commun 2010, 393, 836–843. [Google Scholar]
- Hoffmann, A.; Preobrazhenska, O.; Wodarczyk, C.; Medler, Y.; Winkel, A.; Shahab, S.; Huylebroeck, D.; Gross, G.; Verschueren, K. Transforming growth factor-β-activated kinase-1 (TAK1), a MAP3K, interacts with Smad proteins and interferes with osteogenesis in murine mesenchymal progenitors. J. Biol. Chem 2005, 280, 27271–27283. [Google Scholar]
- Hong, S.; Lim, S.; Li, A.G.; Lee, C.; Lee, Y.S.; Lee, E.K.; Park, S.H.; Wang, X.J.; Kim, S.J. Smad7 binds to the adaptors TAB2 and TAB3 to block recruitment of the kinase TAK1 to the adaptor TRAF2. Nat. Immunol 2007, 8, 504–513. [Google Scholar]
- Han, G.; Li, A.G.; Liang, Y.Y.; Owens, P.; He, W.; Lu, S.; Yoshimatsu, Y.; Wang, D.; Ten Dijke, P.; Lin, X.; et al. Smad7-induced β-catenin degradation alters epidermal appendage development. Dev. Cell 2006, 11, 301–312. [Google Scholar]
- Millar, S.E. Smad7: Licensed to kill β-catenin. Dev. Cell 2006, 11, 274–276. [Google Scholar]
- Li, R.; Rosendahl, A.; Brodin, G.; Cheng, A.M.; Ahgren, A.; Sundquist, C.; Kulkarni, S.; Pawson, T.; Heldin, C.H.; Heuchel, R.L. Deletion of exon i of Smad7 in mice results in altered b cell responses. J. Immunol 2006, 176, 6777–6784. [Google Scholar]
- Chen, Q.; Chen, H.; Zheng, D.; Kuang, C.; Fang, H.; Zou, B.; Zhu, W.; Bu, G.; Jin, T.; Wang, Z.; et al. Smad7 is required for the development and function of the heart. J. Biol. Chem 2009, 284, 292–300. [Google Scholar]
- Liu, X.; Chen, Q.; Kuang, C.; Zhang, M.; Ruan, Y.; Xu, Z.C.; Wang, Z.; Chen, Y. A 4.3 kb Smad7 promoter is able to specify gene expression during mouse development. Biochim. Biophys. Acta 2007, 1769, 149–152. [Google Scholar]
- Kollias, H.D.; Perry, R.L.; Miyake, T.; Aziz, A.; McDermott, J.C. Smad7 promotes and enhances skeletal muscle differentiation. Mol. Cell Biol 2006, 26, 6248–6260. [Google Scholar]
- Miyake, T.; Alli, N.S.; McDermott, J.C. Nuclear function of Smad7 promotes myogenesis. Mol. Cell Biol 2010, 30, 722–735. [Google Scholar]
- Kleiter, I.; Pedre, X.; Mueller, A.M.; Poeschl, P.; Couillard-Despres, S.; Spruss, T.; Bogdahn, U.; Giegerich, G.; Steinbrecher, A. Inhibition of Smad7, a negative regulator of TGF-β signaling, suppresses autoimmune encephalomyelitis. J. Neuroimmunol 2007, 187, 61–73. [Google Scholar]
- Monteleone, G.; Kumberova, A.; Croft, N.M.; McKenzie, C.; Steer, H.W.; MacDonald, T.T. Blocking Smad7 restores TGF-β1 signaling in chronic inflammatory bowel disease. J. Clin. Investig 2001, 108, 601–609. [Google Scholar]
- Monteleone, G.; Del Vecchio Blanco, G.; Palmieri, G.; Vavassori, P.; Monteleone, I.; Colantoni, A.; Battista, S.; Spagnoli, L.G.; Romano, M.; Borrelli, M.; et al. Induction and regulation of Smad7 in the gastric mucosa of patients with helicobacter pylori infection. Gastroenterology 2004, 126, 674–682. [Google Scholar]
- Kleeff, J.; Ishiwata, T.; Maruyama, H.; Friess, H.; Truong, P.; Buchler, M.W.; Falb, D.; Korc, M. The TGF-β signaling inhibitor Smad7 enhances tumorigenicity in pancreatic cancer. Oncogene 1999, 18, 5363–5372. [Google Scholar]
- Boulay, J.L.; Mild, G.; Reuter, J.; Lagrange, M.; Terracciano, L.; Lowy, A.; Laffer, U.; Orth, B.; Metzger, U.; Stamm, B.; et al. Combined copy status of 18q21 genes in colorectal cancer shows frequent retention of Smad7. Genes Chromosomes Cancer 2001, 31, 240–247. [Google Scholar]
- He, W.; Cao, T.; Smith, D.A.; Myers, T.E.; Wang, X.J. Smads mediate signaling of the TGFβ superfamily in normal keratinocytes but are lost during skin chemical carcinogenesis. Oncogene 2001, 20, 471–483. [Google Scholar]
- Afrakhte, M.; Moren, A.; Jossan, S.; Itoh, S.; Sampath, K.; Westermark, B.; Heldin, C.H.; Heldin, N.E.; ten Dijke, P. Induction of inhibitory Smad6 and Smad7 mrna by TGF-β family members. Biochem. Biophys. Res. Commun 1998, 249, 505–511. [Google Scholar]
- Bitzer, M.; von Gersdorff, G.; Liang, D.; Dominguez-Rosales, A.; Beg, A.A.; Rojkind, M.; Bottinger, E.P. A mechanism of suppression of TGF-β/Smad signaling by NF-κb/RelA. Genes Devel 2000, 14, 187–197. [Google Scholar]
- Ulloa, L.; Doody, J.; Massague, J. Inhibition of transforming growth factor-β/Smad signalling by the interferon-γ/STAT pathway. Nature 1999, 397, 710–713. [Google Scholar]
- Kim, S.; Han, J.; Lee, S.K.; Koo, M.; Cho, D.H.; Bae, S.Y.; Choi, M.Y.; Kim, J.S.; Kim, J.H.; Choe, J.H.; et al. Smad7 acts as a negative regulator of the epidermal growth factor (EGF) signaling pathway in breast cancer cells. Cancer Lett 2012, 314, 147–154. [Google Scholar]
- Monteleone, G.; Del Vecchio Blanco, G.; Monteleone, I.; Fina, D.; Caruso, R.; Gioia, V.; Ballerini, S.; Federici, G.; Bernardini, S.; Pallone, F.; et al. Post-transcriptional regulation of Smad7 in the gut of patients with inflammatory bowel disease. Gastroenterology 2005, 129, 1420–1429. [Google Scholar]
- Dowdy, S.C.; Mariani, A.; Reinholz, M.M.; Keeney, G.L.; Spelsberg, T.C.; Podratz, K.C.; Janknecht, R. Overexpression of the tgf-beta antagonist Smad7 in endometrial cancer. Gynecol. Oncol 2005, 96, 368–373. [Google Scholar]
- Osawa, H.; Nakajima, M.; Kato, H.; Fukuchi, M.; Kuwano, H. Prognostic value of the expression of Smad6 and Smad7, as inhibitory Smads of the tgf-beta superfamily, in esophageal squamous cell carcinoma. Anticancer Res 2004, 24, 3703–3709. [Google Scholar]
- Boulay, J.L.; Mild, G.; Lowy, A.; Reuter, J.; Lagrange, M.; Terracciano, L.; Laffer, U.; Herrmann, R.; Rochlitz, C. Smad7 is a prognostic marker in patients with colorectal cancer. Int. J. Cancer. J. Int. Du Cancer 2003, 104, 446–449. [Google Scholar]
- Halder, S.K.; Beauchamp, R.D.; Datta, P.K. Smad7 induces tumorigenicity by blocking TGF-β-induced growth inhibition and apoptosis. Experi. Cell Res 2005, 307, 231–246. [Google Scholar]
- Halder, S.K.; Rachakonda, G.; Deane, N.G.; Datta, P.K. Smad7 induces hepatic metastasis in colorectal cancer. Br. J. Cancer 2008, 99, 957–965. [Google Scholar]
- Rizzo, A.; Waldner, M.J.; Stolfi, C.; Sarra, M.; Fina, D.; Becker, C.; Neurath, M.F.; Macdonald, T.T.; Pallone, F.; Monteleone, G.; et al. Smad7 expression in T cells prevents colitis-associated cancer. Cancer Res 2011, 71, 7423–7432. [Google Scholar]
- Kuang, C.; Xiao, Y.; Liu, X.; Stringfield, T.M.; Zhang, S.; Wang, Z.; Chen, Y. In vivo disruption of TGF-β signaling by Smad7 leads to premalignant ductal lesions in the pancreas. Proc. Natl. Acad. Sci. USA 2006, 103, 1858–1863. [Google Scholar]
- Wang, P.; Fan, J.; Chen, Z.; Meng, Z.Q.; Luo, J.M.; Lin, J.H.; Zhou, Z.H.; Chen, H.; Wang, K.; Xu, Z.D.; et al. Low-level expression of Smad7 correlates with lymph node metastasis and poor prognosis in patients with pancreatic cancer. Ann. Surg. Oncol 2009, 16, 826–835. [Google Scholar]
- Leng, A.; Liu, T.; He, Y.; Li, Q.; Zhang, G. Smad4/Smad7 balance: A role of tumorigenesis in gastric cancer. Experi. Mol. Pathol 2009, 87, 48–53. [Google Scholar]
- Kim, Y.H.; Lee, H.S.; Lee, H.J.; Hur, K.; Kim, W.H.; Bang, Y.J.; Kim, S.J.; Lee, K.U.; Choe, K.J.; Yang, H.K. Prognostic significance of the expression of Smad4 and Smad7 in human gastric carcinomas. Ann. Oncol 2004, 15, 574–580. [Google Scholar]
- Liu, X.; Lee, J.; Cooley, M.; Bhogte, E.; Hartley, S.; Glick, A. Smad7 but not Smad6 cooperates with oncogenic ras to cause malignant conversion in a mouse model for squamous cell carcinoma. Cancer Res 2003, 63, 7760–7768. [Google Scholar]
- Javelaud, D.; Delmas, V.; Moller, M.; Sextius, P.; Andre, J.; Menashi, S.; Larue, L.; Mauviel, A. Stable overexpression of Smad7 in human melanoma cells inhibits their tumorigenicity in vitro and in vivo. Oncogene 2005, 24, 7624–7629. [Google Scholar]
- Javelaud, D.; Mohammad, K.S.; McKenna, C.R.; Fournier, P.; Luciani, F.; Niewolna, M.; Andre, J.; Delmas, V.; Larue, L.; Guise, T.A.; et al. Stable overexpression of Smad7 in human melanoma cells impairs bone metastasis. Cancer Res 2007, 67, 2317–2324. [Google Scholar]
- DiVito, K.A.; Trabosh, V.A.; Chen, Y.S.; Chen, Y.; Albanese, C.; Javelaud, D.; Mauviel, A.; Simbulan-Rosenthal, C.M.; Rosenthal, D.S. Smad7 restricts melanoma invasion by restoring n-cadherin expression and establishing heterotypic cell-cell interactions in vivo. Pigment Cell Mel. Res. 2010, 23, 795–808. [Google Scholar]
- Theohari, I.; Giannopoulou, I.; Magkou, C.; Nomikos, A.; Melissaris, S.; Nakopoulou, L. Differential effect of the expression of TGF-β pathway inhibitors, Smad-7 and Ski, on invasive breast carcinomas: Relation to biologic behavior. APMIS 2012, 120, 92–100. [Google Scholar]
- Azuma, H.; Ehata, S.; Miyazaki, H.; Watabe, T.; Maruyama, O.; Imamura, T.; Sakamoto, T.; Kiyama, S.; Kiyama, Y.; Ubai, T.; et al. Effect of Smad7 expression on metastasis of mouse mammary carcinoma JygMC(A) cells. J. Nat. Cancer Inst 2005, 97, 1734–1746. [Google Scholar]
- Papageorgis, P.; Lambert, A.W.; Ozturk, S.; Gao, F.; Pan, H.; Manne, U.; Alekseyev, Y.O.; Thiagalingam, A.; Abdolmaleky, H.M.; Lenburg, M.; et al. Smad signaling is required to maintain epigenetic silencing during breast cancer progression. Cancer Res 2010, 70, 968–978. [Google Scholar]
- Smith, A.L.; Iwanaga, R.; Drasin, D.J.; Micalizzi, D.S.; Vartuli, R.L.; Tan, A.C.; Ford, H.L. The miR-106b-25 cluster targets Smad7, activates TGF-β signaling, and induces EMT and tumor initiating cell characteristics downstream of Six1 in human breast cancer. Oncogene 2012, 31, 5162–5171. [Google Scholar]
- Wang, J.; Zhao, J.; Chu, E.S.; Mok, M.T.; Go, M.Y.; Man, K.; Heuchel, R.; Lan, H.Y.; Chang, Z.; Sung, J.J.; et al. Inhibitory role of Smad7 in hepatocarcinogenesis in mice and in vitro. J. Pathol. 2013, 230, 441–452. [Google Scholar]
- Xia, H.; Ooi, L.L.; Hui, K.M. MicroRNA-216a/217-induced epithelial-mesenchymal transition targets pten and Smad7 to promote drug resistance and recurrence of liver cancer. Hepatology 2013, 58, 629–641. [Google Scholar]
- Landstrom, M.; Heldin, N.E.; Bu, S.; Hermansson, A.; Itoh, S.; ten Dijke, P.; Heldin, C.H. Smad7 mediates apoptosis induced by transforming growth factor β in prostatic carcinoma cells. Curr. Biol 2000, 10, 535–538. [Google Scholar]
- Davoodpour, P.; Landstrom, M. 2-Methoxyestradiol-Induced apoptosis in prostate cancer cells requires Smad7. J. Biol. Chem 2005, 280, 14773–14779. [Google Scholar]
- Ekman, M.; Mu, Y.; Lee, S.Y.; Edlund, S.; Kozakai, T.; Thakur, N.; Tran, H.; Qian, J.; Groeden, J.; Heldin, C.H.; et al. APC and Smad7 link TGFβ type I receptors to the microtubule system to promote cell migration. Mol. Biol. Cell 2012, 23, 2109–2121. [Google Scholar]
- Broderick, P.; Carvajal-Carmona, L.; Pittman, A.M.; Webb, E.; Howarth, K.; Rowan, A.; Lubbe, S.; Spain, S.; Sullivan, K.; Fielding, S.; et al. A Genome-wide association study shows that common alleles of Smad7 influence colorectal cancer risk. Nature Genet 2007, 39, 1315–1317. [Google Scholar]
- Tenesa, A.; Farrington, S.M.; Prendergast, J.G.; Porteous, M.E.; Walker, M.; Haq, N.; Barnetson, R.A.; Theodoratou, E.; Cetnarskyj, R.; Cartwright, N.; et al. Genome-wide association scan identifies a colorectal cancer susceptibility locus on 11q23 and replicates risk loci at 8q24 and 18q21. Nat. Genet 2008, 40, 631–637. [Google Scholar]
- Slattery, M.L.; Herrick, J.; Curtin, K.; Samowitz, W.; Wolff, R.K.; Caan, B.J.; Duggan, D.; Potter, J.D.; Peters, U. Increased risk of colon cancer associated with a genetic polymorphism of Smad7. Cancer Res 2010, 70, 1479–1485. [Google Scholar]
- Thompson, C.L.; Plummer, S.J.; Acheson, L.S.; Tucker, T.C.; Casey, G.; Li, L. Association of common genetic variants in Smad7 and risk of colon cancer. Carcinogenesis 2009, 30, 982–986. [Google Scholar]
- Li, Q.; Zou, C.; Han, Z.; Xiao, H.; Wei, H.; Wang, W.; Zhang, L.; Zhang, X.; Tang, Q.; Zhang, C.; et al. MicroRNA-25 functions as a potential tumor suppressor in colon cancer by targeting Smad7. Cancer Lett 2013, 335, 168–174. [Google Scholar]
- Arnold, N.B.; Ketterer, K.; Kleeff, J.; Friess, H.; Buchler, M.W.; Korc, M. Thioredoxin is downstream of Smad7 in a pathway that promotes growth and suppresses cisplatin-induced apoptosis in pancreatic cancer. Cancer Res 2004, 64, 3599–3606. [Google Scholar]
- Boyer Arnold, N.; Korc, M. Smad7 abrogates transforming growth factor-β1-mediated growth inhibition in COLO-357 cells through functional inactivation of the retinoblastoma protein. J. Biol. Chem 2005, 280, 21858–21866. [Google Scholar]
- Saloman, D.S.; Bianco, C.; Ebert, A.D.; Khan, N.I.; De Santis, M.; Normanno, N.; Wechselberger, C.; Seno, M.; Williams, K.; Sanicola, M.; et al. The EGF-CFC family: Novel epidermal growth factor-related proteins in development and cancer. Endocr. Relat. Cancer 2000, 7, 199–226. [Google Scholar]
- Park, Y.N.; Chae, K.J.; Oh, B.K.; Choi, J.; Choi, K.S.; Park, C. Expression of Smad7 in hepatocellular carcinoma and dysplastic nodules: Resistance mechanism to transforming growth factor-β. Hepato-Gastroenterology 2004, 51, 396–400. [Google Scholar]
- Brodin, G.; ten Dijke, P.; Funa, K.; Heldin, C.H.; Landstrom, M. Increased Smad expression and activation are associated with apoptosis in normal and malignant prostate after castration. Cancer Res 1999, 59, 2731–2738. [Google Scholar]
- Edlund, S.; Bu, S.; Schuster, N.; Aspenstrom, P.; Heuchel, R.; Heldin, N.E.; ten Dijke, P.; Heldin, C.H.; Landstrom, M. Transforming growth factor-β1 (TGF-β)-induced apoptosis of prostate cancer cells involves Smad7-dependent activation of p38 by TGF-β-activated kinase 1 and mitogen-activated protein kinase kinase 3. Mol. Biol. Cell 2003, 14, 529–544. [Google Scholar]
- Riggins, G.J.; Kinzler, K.W.; Vogelstein, B.; Thiagalingam, S. Frequency of Smad gene mutations in human cancers. Cancer Res 1997, 57, 2578–2580. [Google Scholar]
- Hahn, S.A.; Hoque, A.T.; Moskaluk, C.A.; da Costa, L.T.; Schutte, M.; Rozenblum, E.; Seymour, A.B.; Weinstein, C.L.; Yeo, C.J.; Hruban, R.H.; et al. Homozygous deletion map at 18q21.1 in pancreatic cancer. Cancer Res 1996, 56, 490–494. [Google Scholar]
- Massague, J. Tgf-beta signal transduction. Annu. Rev. Biochem 1998, 67, 753–791. [Google Scholar]
- Zhang, B.; Halder, S.K.; Kashikar, N.D.; Cho, Y.J.; Datta, A.; Gorden, D.L.; Datta, P.K. Antimetastatic role of Smad4 signaling in colorectal cancer. Gastroenterology 2010, 138. [Google Scholar]
- Papageorgis, P.; Cheng, K.; Ozturk, S.; Gong, Y.; Lambert, A.W.; Abdolmaleky, H.M.; Zhou, J.R.; Thiagalingam, S. Smad4 inactivation promotes malignancy and drug resistance of colon cancer. Cancer Res 2011, 71, 998–1008. [Google Scholar]
- Zhu, Y.; Richardson, J.A.; Parada, L.F.; Graff, J.M. Smad3 mutant mice develop metastatic colorectal cancer. Cell 1998, 94, 703–714. [Google Scholar]
- Takaku, K.; Oshima, M.; Miyoshi, H.; Matsui, M.; Seldin, M.F.; Taketo, M.M. Intestinal tumorigenesis in compound mutant mice of both dpc4 (Smad4) and apc genes. Cell 1998, 92, 645–656. [Google Scholar]
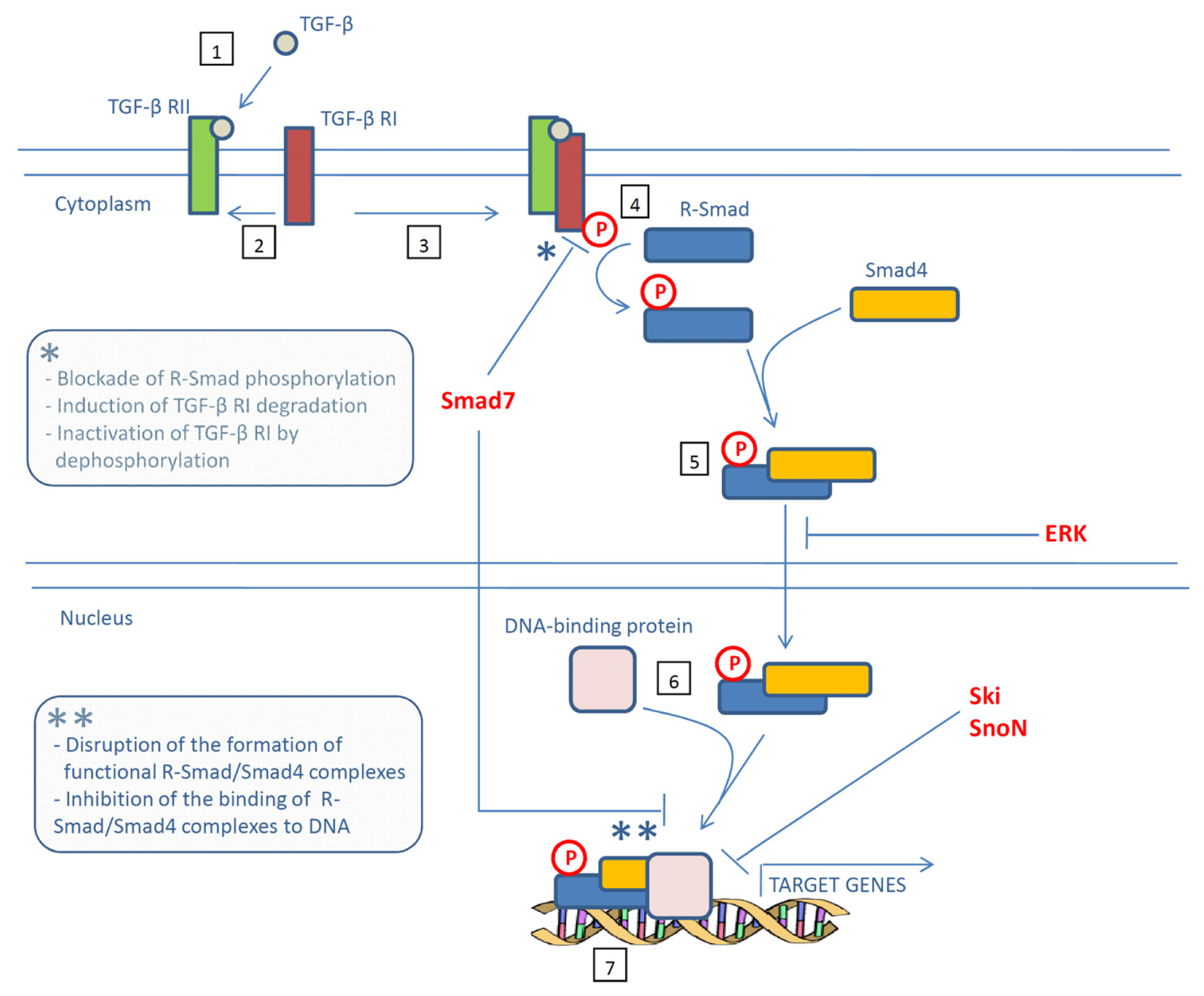

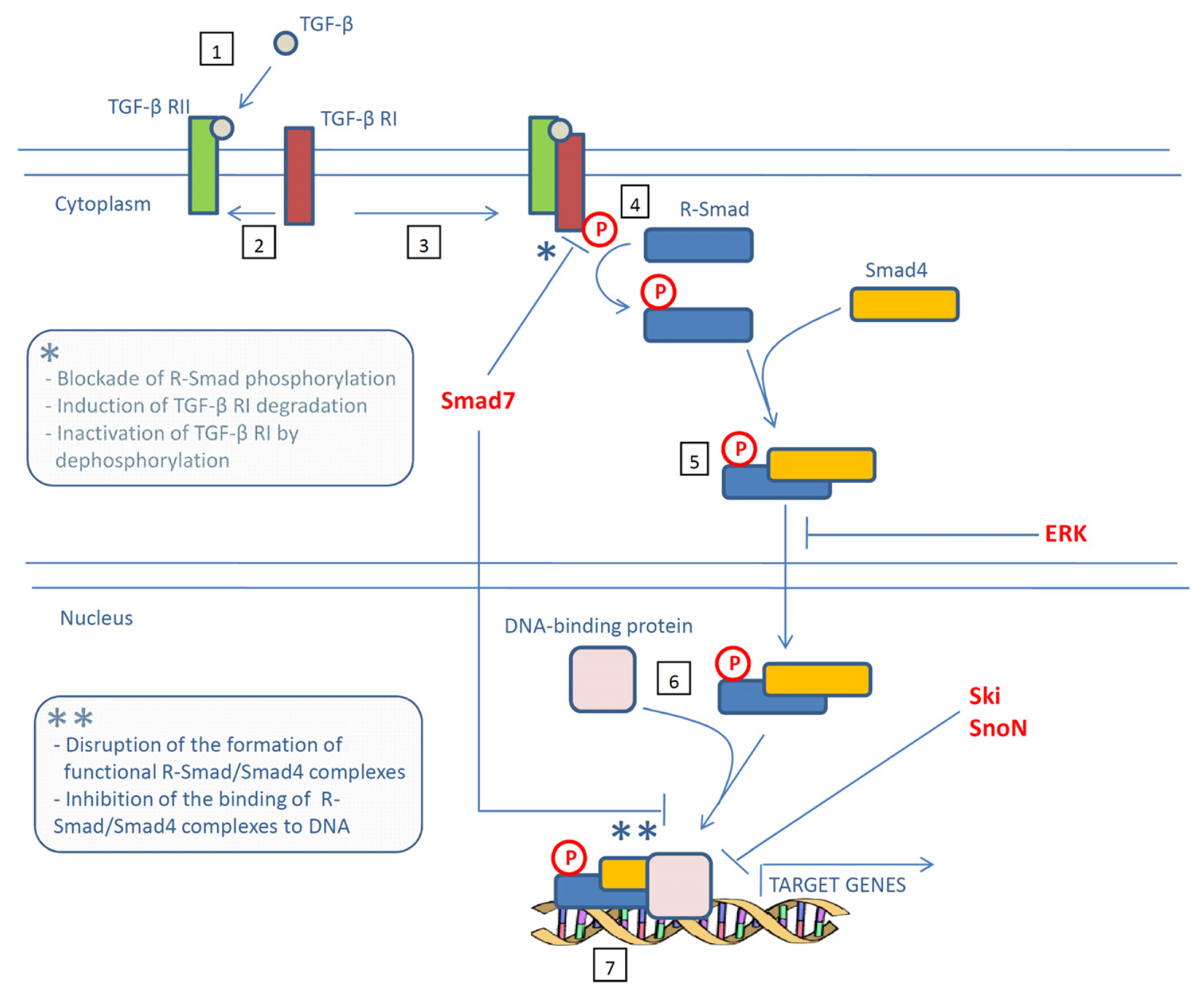{kind=link}
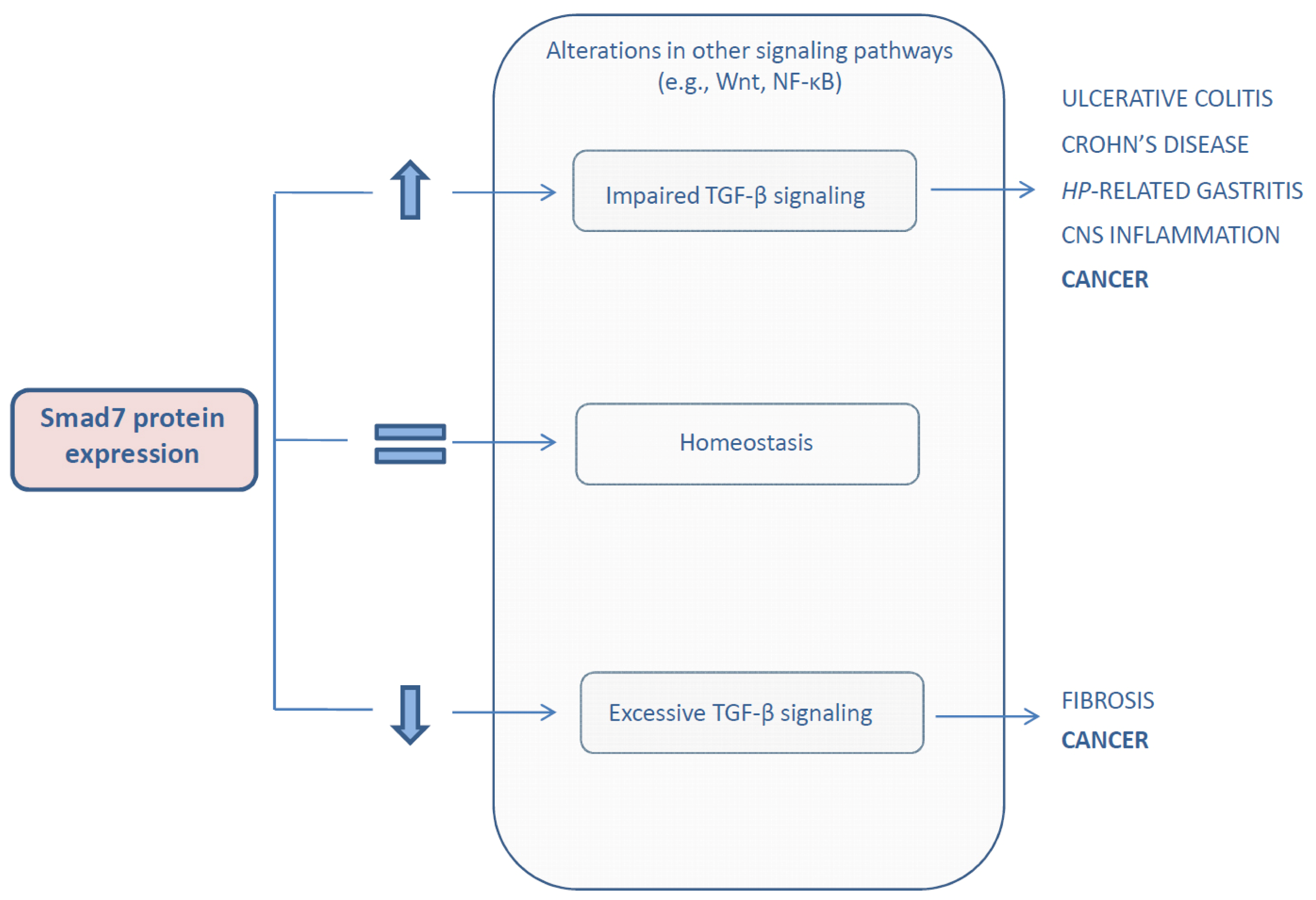
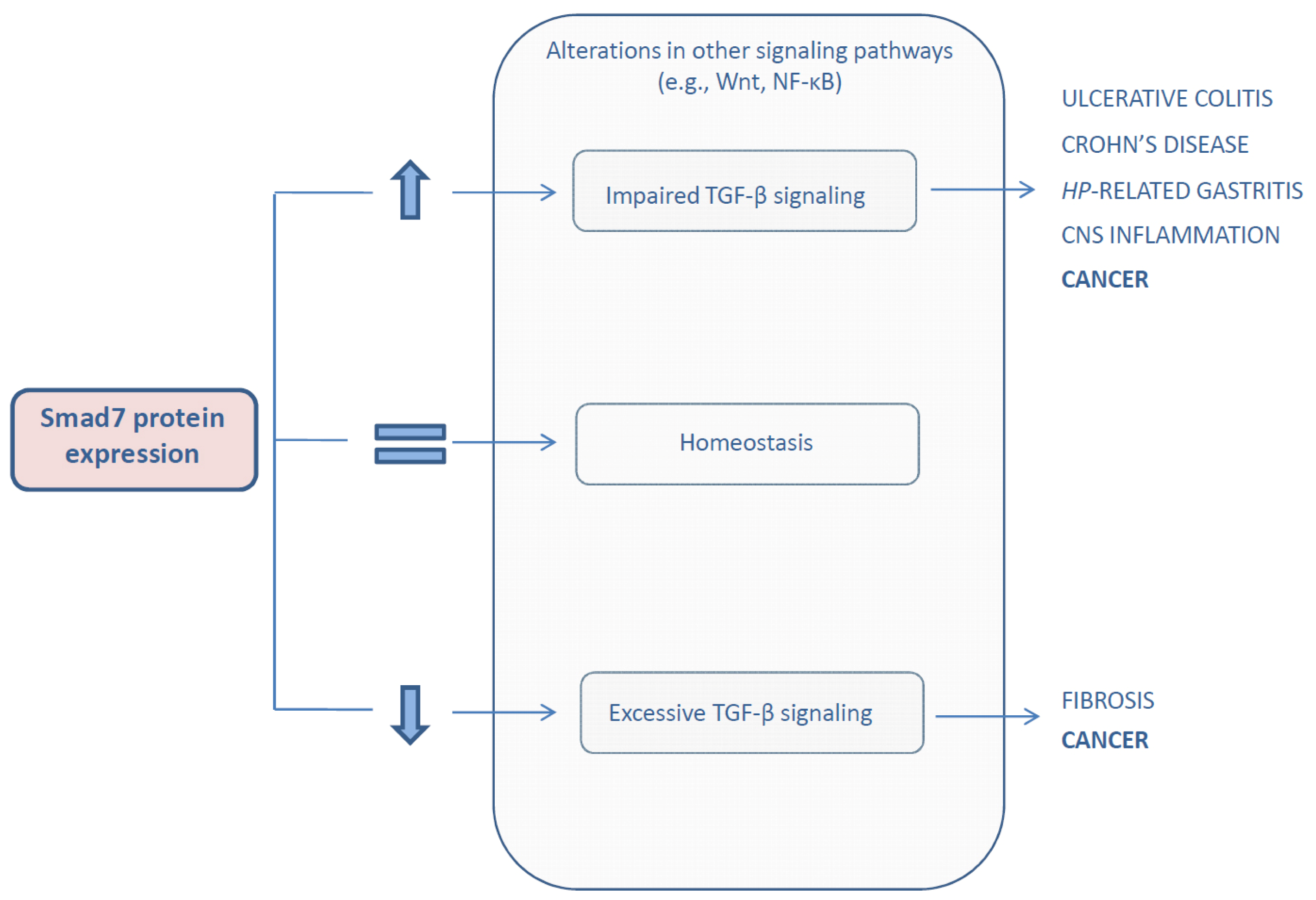{kind=link}
| Cancer type | Method | Observation | Ref. |
|---|---|---|---|
| Endometrial | Observational study | Smad7 upregulation associates with poor survival rate | [42] |
| Esophageal | Observational study | Smad7 upregulation associates with shorter time to recurrence | [43] |
| Colorectal | Observational study | CRC patients with deletion of Smad7 have a favorable clinical outcome compared with patients with Smad7 amplification | [44] |
| Colorectal | Colony formation assay, xenografts induced by FET cells in immunodeficient mice | Smad7-overexpressing FET cells show aggressive colony formation on soft agar and increased tumorigenicity in vivo compared with control FET cells | [45] |
| Colorectal | Metastasis induced by the injection of FET cells in the spleen of immunodeficient mice | Injection of Smad7-overexpressing FET cells results in the development of liver metastasis | [46] |
| Colorectal | AOM + DSS-driven colitis associated CRC | Over-expression of Smad7 in T cells associates with severe colitis and reduces the growth of colitis-associated CRC | [47] |
| Pancreatic | Colony formation assay, xenografts induced by FET cells in immunodeficient mice | Smad7 overexpressing COLO-357 cells are resistant to the TGF-β-driven growth inhibition in vitro and exhibit a marked increase in their capacity to form colonies in soft agar and tumors in nude mice | [34] |
| Pancreatic | Transgenic mouse with pancreatic overexpression of Smad7 | Smad7 blocks TGF-β signaling in the pancreas and induces premalignant ductal lesions with the characteristics of pancreatic intraepithelial neoplasia | [48] |
| Pancreatic | Observational study | Expression of Smad7 associates with a more favorable prognosis compared with patients with lower levels of Smad7 who exhibited increased incidence of lymph node metastasis and liver metastasis after surgery | [49] |
| Gastric | Observational study | Elevated Smad7 levels in tumors with lymphatic metastasis | [50] |
| Gastric | Observational study | Patients bearing tumors with positive Smad7 expression have a poor prognosis | [51] |
| Gastric | Cell culture | Ectopic Smad7 expression increased the survival of SGC7901 gastric cancer cells | [50] |
| Skin | Mouse model of chemically-induced skin carcinogenesis | Smad7 overexpression in H-ras-transduced keratinocytes determines the conversion of benign to malignant epithelial cells and a rapid progression to squamous cell carcinoma | [52] |
| Skin | Xenograft model in which primary keratinocytes mixed with dermal fibroblasts are grafted into nude mice | H-ras/Smad7 but not H-ras keratinocytes progresses to SCC | [52] |
| Skin | Colony formation assay, xenografts induced by 1205Lu cells into immunodeficient mice | Stable over-expression of Smad7 in 1205Lu cells reduces MMP-2 and MMP-9 production, invasive capacity and anchorage-independent growth in vitro as well as subcutaneous tumor formation in nude mice | [53] |
| Skin | Model of bone metastases in which tumor cells are inoculated into the left cardiac ventricle of nude mice | Animals injected with Smad7-transfected 1205Lu cells have significantly less osteolytic metastases and longer survival compared with mice injected with parental and mock-transfected 1205Lu cells | [54] |
| Skin | In vivo human skin grafting system | Smad7-expressing 1205Lu cells position proximal to the dermal-epidermal junction and fail to form tumors, while control cells form tumors within the dermis | [55] |
| Breast | Observational study | Smad7 expression correlates with a poor prognosis in patients with invasive breast carcinoma | [56] |
| Breast | Breast cancer metastasis induced by intravenous injection of mouse mammary carcinoma JygMC(A) cells | Mice injected with Smad7-transfected JygMC(A) cells show fewer lung and liver metastasis and longer survival than mice injected with mock-transfected JygMC(A) cells | [57] |
| Breast | Cell culture | Smad7 sensitizes MCF7 breast cancer cells to TNF-induced cell death | [20] |
| Breast | Cell culture | Ectopic Smad7 expression in SKBR3 cells completely abrogates EGF-induced MMP-9 expression | [40] |
| Breast | Cell culture | Smad7 overexpression suppresses migration and invasion of mesenchymal-like MCF10CA1h cells | [58] |
| Breast | Cell culture | miR-106b-25 cluster negatively regulates Smad7 expression thereby activating TGF-β signaling and inducing EMT in MCF7 cells | [59] |
| Liver | Mouse model of HCC induced by DEN | Smad7-deficient mice have higher tumor incidence and multiplicity than wild-type mice | [60] |
| Liver | Observational study | Low Smad7 expression in HCC samples associates with better disease free survival | [61] |
| Liver | Cell culture | Smad7 restrains EMT and cell migration of HCC cells | [61] |
| Prostate | Cell culture | Ectopic Smad7 expression induces apoptosis in PC-3U human prostate cancer cells | [62] |
| Prostate | Cell culture | Smad7 is required for the induction of apoptosis by the anti-cancer agent 2-Methoxyestradiol in PC-3U cells | [63] |
| Prostate | Cell culture | Smad7 promotes migratory responses in PC-3U cells | [64] |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Stolfi, C.; Marafini, I.; De Simone, V.; Pallone, F.; Monteleone, G. The Dual Role of Smad7 in the Control of Cancer Growth and Metastasis. Int. J. Mol. Sci. 2013, 14, 23774-23790. https://doi.org/10.3390/ijms141223774
Stolfi C, Marafini I, De Simone V, Pallone F, Monteleone G. The Dual Role of Smad7 in the Control of Cancer Growth and Metastasis. International Journal of Molecular Sciences. 2013; 14(12):23774-23790. https://doi.org/10.3390/ijms141223774
Chicago/Turabian StyleStolfi, Carmine, Irene Marafini, Veronica De Simone, Francesco Pallone, and Giovanni Monteleone. 2013. "The Dual Role of Smad7 in the Control of Cancer Growth and Metastasis" International Journal of Molecular Sciences 14, no. 12: 23774-23790. https://doi.org/10.3390/ijms141223774